

Abstract
Background
Biomedical ontologies are increasingly instrumental in the advancement of biological research primarily through their use to efficiently consolidate large amounts of data into structured, accessible sets. However, ontology development and usage can be hampered by the segregation of knowledge by domain that occurs due to independent development and use of the ontologies. The ability to infer data associated with one ontology to data associated with another ontology would prove useful in expanding information content and scope. We here focus on relating two ontologies: the Gene Ontology (GO), which encodes canonical gene function, and the Mammalian Phenotype Ontology (MP), which describes non-canonical phenotypes, using statistical methods to suggest GO functional annotations from existing MP phenotype annotations. This work is in contrast to previous studies that have focused on inferring gene function from phenotype primarily through lexical or semantic similarity measures.
Results
We have designed and tested a set of algorithms that represents a novel methodology to define rules for predicting gene function by examining the emergent structure and relationships between the gene functions and phenotypes rather than inspecting the terms semantically. The algorithms inspect relationships among multiple phenotype terms to deduce if there are cases where they all arise from a single gene function.
We apply this methodology to data about genes in the laboratory mouse that are formally represented in the Mouse Genome Informatics (MGI) resource. From the data, 7444 rule instances were generated from five generalized rules, resulting in 4818 unique GO functional predictions for 1796 genes.
Conclusions
We show that our method is capable of inferring high-quality functional annotations from curated phenotype data. As well as creating inferred annotations, our method has the potential to allow for the elucidation of unforeseen, biologically significant associations between gene function and phenotypes that would be overlooked by a semantics-based approach. Future work will include the implementation of the described algorithms for a variety of other model organism databases, taking full advantage of the abundance of available high quality curated data.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-014-0405-z) contains supplementary material, which is available to authorized users.
Keywords: Gene ontology, Mammalian phenotype ontology, Function prediction, Ontology development
Background
A hallmark of modern biomedical research is the generation of increasingly large amounts of scientific data. Biomedical ontologies have the potential to greatly accelerate biomedical research by enhancing our ability to integrate and access these data. A biomedical ontology is a resource that represents a controlled set of terms for entities in a particular biomedical domain and how those terms are related to one another [1]. Biocurators are scientists who review experimental data, primarily as reported in the biomedical literature, to create empirical connections between different aspects of biological data, that is to say, annotations [2]; .e.g. biocurators annotate, or tag, biological entities (e.g. proteins, functional RNAs) with ontology terms (capturing all relevant metadata as well). One of the most widely used modern bio-ontologies is the Gene Ontology (GO), a resource that describes canonical gene functions in a computable species-independent manner so that they may be used for statistical analysis of gene sets or for comparative genomic analysis [3,4]. Another bio-ontology is the Mammalian Phenotype Ontology (MP) that provides an independently curated set of terms and relationships describing non-canonical phenotypes, primarily in mouse models, and that is used to query the effects of genetic mutations [5].
Genes and alleles (genetic variants) of genes are annotated respectively for both function and phenotype within Mouse Genome Informatics (MGI) system, a comprehensive resource for genomic research of the laboratory mouse [6]. Within the MGI curation workflow, different subsets of biocurators separately process papers identified for function (GO) and phenotype (MP) curation. As a result, although papers may be selected for curation in regards to both GO and MP, they are not processed simultaneously, leading to short-term temporal discrepancies in overall curation coverage in MGI. Because scientific literature is published much faster than biocurators can read and curate the papers, the development of methods to computationally infer annotations from one source to another would greatly add and enhance curation efficiency [7].
Recent years have seen efforts to complement curated annotation data sets with text mined and association-rule mined predicted annotations. Broadly, text mining approaches use natural language processing methods to alleviate the backlog of papers awaiting curation, while association-rule mining uses curated annotation sets to predict new annotations and to assess the validity of automated annotation methods [8-17]. It is worth noting that several of these same approaches have been used to improve and expand the ontology structure and relationships as well [18-21]. These efforts have been used both to predict additional annotations from curated annotations in the same ontology and to predict across ontologies, as we do here.
The prediction efforts mentioned above may include lexical matching [8], semantic similarity measures [9-11], ontology matching [18,19], and so-called ‘guilt-by-association’ methods [12]; several efforts use a combination of these approaches [13]. Some prediction methods are primarily ontology based and others are annotation, or instance, based. Our method uses an extension of ‘guilt-by-association’ and is annotation based.
Lexical matching methods, including text mining and text clustering, have been used to infer gene function from phenotype and vice versa. Semantic matching is facilitated by the use within the OBO community of equivalence axioms and logical definitions and by curated inter-ontology links. Semantic similarity measures based on ontology structure or information theory between phenotype and GO have been used to predict additional GO annotations. In a departure from semantic similarity approaches, various groups have performed network analyses to align GO terms with protein association networks to predict protein function [14-16].
Other efforts follow a more empirical approach such as instance based ontology matching and other so-called ‘guilt-by-association’ methods: annotation co-occurrence pairs, knowledge-based annotation inference based on, for example, protein-protein interactions or pathway term enrichment.
Our approach is strictly empirical and makes no assumptions about lexical matching, semantics, or ontology structure, except to infer annotations according to the true-path rule. The rationale behind our approach is to make a simplifying assumption that in some cases ‘interesting’ biology could be missed by limiting the analysis to include an alignment of ontology structure or by attempting to compare the ‘meaning’ of phenotype versus GO terms. Using this simplified approach there is no underlying assumption that ‘similar’ areas of the MPO and the GO should correlate. Instead, we examine the feasibility of constructing rules based only on conjunctions and disjunctions of high-quality phenotype annotations made by MGI curators to predict GO annotations. A crucial difference in our approach is that, where most empirical methods group annotations based on gene entities, our analysis is allele-specific, and therefore addresses the potential that a given set of varied phenotypes may be the result of a single underlying genetic perturbation. Additionally, mouse phenotypes can vary widely for different alleles of the same gene on different strain backgrounds. Indeed, this is the reason for the detailed study of spontaneous and targeted mutations in specific strains of the laboratory mouse. Consider, for example, the phenotypes of three alleles of the mouse Pax3 gene. One spontaneous allele, Pax3Sp-d manifests phenotypes in many areas: embryogenesis, integument, limbs/digits/tail, mortality/aging, nervous system, pigmentation. This allele is present in the mouse model for the human disease Waardenburg Syndrome, Type 1; WS1 (OMIM:193500) [22,23]. Another targeted allele, Pax3tm1Mrc manifests a different set of phenotypes: craniofacial, growth/size, mortality/aging, muscle, nervous system, respiratory, skeleton, tumorigenesis, vision/eye. This allele is present in the mouse model for another human disease Rhabdomyosarcoma 2; RMS2 (OMIM:268220) [24]. Yet another targeted allele, Pax3tm2.1Joe [25], is present in a mouse in which no abnormal phenotype is observed.
In this work, we describe an original method to predict novel GO annotations for genes associated with alleles that have existing MP annotations. We apply our derived set of rules to a set of papers that have been selected for curation for both MP and GO but that have, as yet, been annotated only for MP, but not for GO, within the MGI system. The algorithms draw inspiration from set and graph theory as they attempt to mathematically analyze relationships between GO and MP term(s). The approach used is as follows: First, gather relevant data and align MP and GO terms based on co-curated literature and shared alleles as our training set; Second, analyze the data such that rules can be made to predict a GO term from MP term(s); And finally, Third, apply the rules to a new set of papers/alleles that are currently annotated for MP and selected for but not yet annotated for GO. The goal of the work is to complement and facilitate the work of biocurators.
Methods
Consolidation of datasets
Both MP and GO are used in MGI, an open source resource that freely publishes its datasets. We collected all data used in this study from the MGI ftp site (ftp://ftp.informatics.jax.org/pub/reports/index.html, retrieved 10 June 2013). First, the set of literature that was annotated for both GO and MP in MGI was gathered and formatted; this set provided our training data set (3662 publications with both GO and MP annotations to the same allele). Then, the set of literature selected for both MP and GO, but annotated only for MP (63,028 publications) was collected from internal reports used by curators in their workflow; this set provided the test set for our derived inference rules.
Data alignment and processing
A base set of MP-GO pairs was generated by matching the set of all collected GO terms to the set of all collected MP terms used to annotate the same allele (we use the allele here as a proxy for full genotype) in the same study, knowledge of which is reflected by shared PubMedID (PMID) and alleleID. The GO terms were filtered to select only those terms from the biological process (BP) subontology, and only those GO annotations with evidence as “inferred from a mutant phenotype” (IMP). These selection criteria provide a defined set of papers restricted formally to those tagged for phenotype-based GO annotation for biological process, provided a first level of quality control on the dataset. There were found to be 81,245 MP-GO pairs sharing PMID/alleleID, and 67,424 unique MP-GO pairs, indicating that many of the MP-GO pairs occurred more than once. The MP, GO and the PMID/alleleID data were modeled as a network with PMID/alleleID nodes connecting MP and GO nodes derived from the same study.
All network visualizations were created using Cytoscape v2.8 [26] and all calculations were performed using Numerical Python (Python 2.7.3, NumPy 1.6.2, SciPy 0.10.1, Matplotlib 1.2) [27]. The network was modeled with an adjacency-like matrix. The term “adjacency matrix” is used loosely here, as the matrices presented in this work are quite different than those traditionally used—the matrices used here are rectangular matrices with the columns representing the annotated terms and rows representing PMID/alleleIDs. This approach is necessary since we wish to track individual studies for which both MP and GO annotations have been made. Two matrices were created, one with all unique PMID/alleleIDs (3662 rows) by all unique GO IDs (2472 columns); the other with all unique PMID/alleleIDs by all unique MP IDs (4978 columns). The network was found to be very sparse—that is, the number of all possible edges far exceeded the number of actual edges (graph density of approximately 6e-04).
Calculation and evaluation of statistical significance
After constructing our networks, we calculated the probability that a connection between an MP and GO node was statistically significant rather than due to chance alone. Some gene function-phenotype connections might be supported by many shared annotations, implying that there is some underlying connection between the gene function and the mutant phenotype, while others might be connected by only a few connections relative to the number other connections and yet also be informative.
From a set of unique integers of size N representing all the PMID/alleleID combinations, the probability that a selection of n at random will include j or more ‘successes’ – that is, a PMID/alleleID that is shared between a particular MP-GO node pair, can be defined as a modification of the cumulative binomial distribution (for values of N large compared to n):
| 1 |
In our case, N = 3662, and n, the maximum number of PMID/alleleID combinations between any MP-GO node pair, is 250, so the condition of equation 1 is satisfied. Applying this definition, the total probability, ptot, that a particular MPi-GOj association is due to chance is given by:
| 2 |
where PM(MPi) or PM(GOj) returns the set of all PMID/alleleID nodes that are connected to the MPi or GOj nodes, respectively, and:
| 3 |
is the set of data confirmed PMID/alleleID connections between the MPi and the GOj nodes. The p-value was calculated in this way for each MP-GO pair found in the data and sorted. The efficacy of the p-value was evaluated by using each MP-GO connection as a prediction, or ‘rule’: MPi → GOj —to wit: “if an annotation of a particular allele has been made to the MP term MPi by curation of a particular study then an IMP annotation corresponding to that allele can be made to the GO term GOj.” The resulting true and false positives (sensitivity and specificity) were plotted on the receiver operating characteristic (ROC) curve (Figure 1). As shown, the calculated p-value shows sufficient discriminatory power (AUC = 0.733) to discriminate MP-GO pairs that are more likely to correctly predict a GO node from a given MP term. An alternative scenario was also addressed, where possible MP terms were predicted from alleles with GO annotations (but no MP annotations); it was found that this prediction route does not carry as much predictive value (AUC = .686), further validating our approach to predict GO from MP annotations. We therefore used the calculated p-values as our measure of statistical significance for the GO function prediction from the simple rule.
Figure 1.
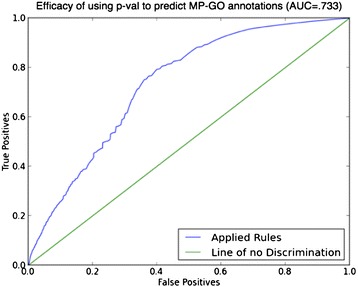
ROC curve. Evaluation of the efficacy of a rule: if MPi then GOj, using the receiver operating characteristic.
Generalized patterns lead to extended rules
While the calculation of the p-value is useful in discovering which MP-GO pairs have stronger connections when run against the training sets, we found that even those with the lowest probabilities of being due to chance returned many false positives. Therefore, the need arose to either better identify the true positives of an MP-GO pair, or to better exclude the false positives: that is, to improve the discriminatory power of our rules.
Two types of generalized patterns arose from the network, which we chose to identify as the + (plus) rule and the - (minus) rule. The plus-rule can be qualitatively described as inspecting the connections of an MP-GO pair and examining if there is another MP node that is connected to the true positives of the pair, but excludes all of the false positives (PMID/alleleID nodes that are connected to MP, but not to GO) (Figure 2a). We can define the plus-rule: MP1 AND MP2 → GO1 —to wit: “if an annotation of a particular allele has been made to the MP term MP1 and to the MP term MP2 by curation of a single particular study then an IMP annotation corresponding to that allele can be made to the GO term GO1.” The conditional statement for finding an MP1 and MP2 that follow the pattern of the plus-rule is as follows:
| 4 |
Figure 2.
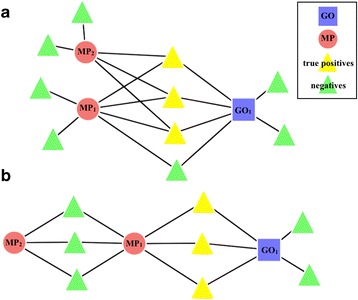
A graphical representation of the plus and minus rule. The highlighted nodes represent the ‘successes’ identified by the corresponding rule. a, The plus rule-a pattern whereby if MP1 and MP2, then GO1. b, The minus rule-another pattern whereby if MP1 and not MP2, then GO1.
The minus-rule takes the somewhat opposite approach from the plus-rule; that is, instead of discerning if the intersection of two MP nodes can define the successes, the minus-rule examines if the set difference of two MP nodes can define the successes (Figure 2b). Similarly, we can define the minus-rule: MP1 AND NOT MP2 → GO1 —to wit: “if an annotation of a particular allele has been made to the MP term MP1 but not to the MP term MP2 by curation of a particular study then an IMP annotation corresponding to that allele can be made to the GO term GO1.” The statement used to find an MP1 and MP2 that follow the pattern of the minus-rule is as follows:
| 5 |
The network was then searched for collections of MP and GO nodes that followed the aforementioned patterns, with 3105 instances of the plus-rule pattern and 234 instances of the minus-rule pattern. Detailed examples of both rules are illustrated in Figure 3. However, the need to differentiate between those rules that were more likely to give results to be due to chance versus those that were less likely was still present. Utilizing the demonstrated efficacy of the cumulative binomial distribution as applied to the undirected graph (equation 1), the p-values for the plus and minus-rules were calculated respectively:
| 6 |
| 7 |
| 8 |
| 9 |
where S in each case is the set of ‘successes’ and Sc is the set complement. As the network is sparse, the p-values are expected to be small. Arising from the basic plus and minus-rule patterns, three other rule patterns were defined, descriptively designated as plus-plus (++) (Figure 4a), minus-minus (--) (Figure 4b) and plus-minus (+-) (Figure 4c). The mathematical statements used to find rules from three additional rule patterns respectively are as follows:
| 10 |
| 11 |
| 12 |
Figure 3.
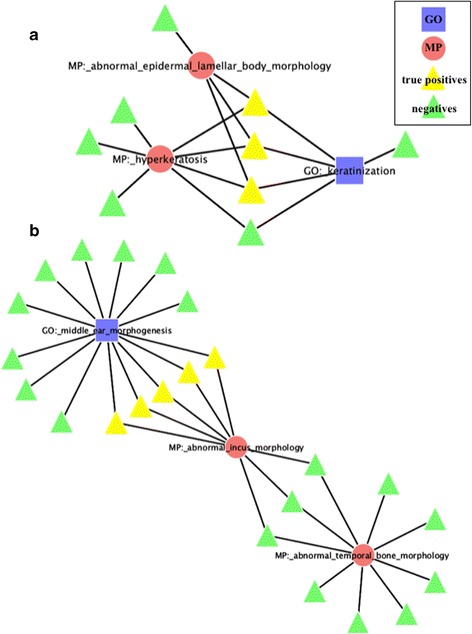
Example of rule instances. The highlighted nodes represent the ‘successes’ identified by the corresponding rule. a, Plus rule instance. b, Minus rule instance.
Figure 4.
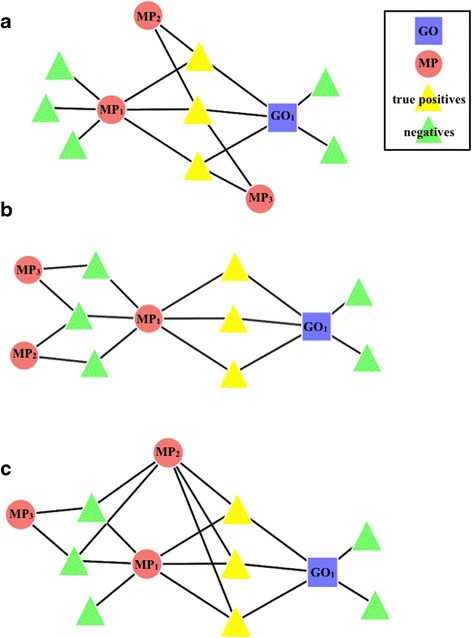
A graphical representation of the three rules emerging from the plus and minus rules. The highlighted nodes represent the ‘successes’ identified by the corresponding rule. a, The plus-plus rule-if MP1 and MP2 or MP3, then GO1. b, The minus-minus rule-if MP1 and not either MP2 or MP3, then GO1. c, The plus-minus rule-if MP1 and MP2 and not MP3, then GO1.
The statements used to calculate the p-values for these rule patterns are expounded in the appendix. The network was again searched for arrangements of nodes that followed the three other patterns, with 3143, 328, and 634 instances of the plus-plus-rule, minus-minus-rule and plus-minus-rule respectively. While the composite plus- and minus-rules can be combined iteratively in theory, there seems to be a point where the statements used to calculate the probabilities for large combinations of nodes become too large to be feasibly evaluated, and where they have the potential of straying further from biological reality. Therefore, we limited our analysis to these five rules as examples. All rule instances are compiled in Additional file 1.
Evaluation of extended rules
All rules were applied to the literature set of PMID/alleleIDs that are annotated to MP term(s) but not yet annotated to a GO term to predict which GO term would be annotated to the PMID/alleleID (set may be found in Additional file 2). Validation of predictions was performed by the selection of a set of 20 papers for each rule that represented a range of the p-values. The papers were read by a GO scientific curator and the curatorial predictions for functional GO annotation were examined in the context of our rule structures. Companion software for the prediction of GO annotations is provided as Additional file 3.
Results and discussion
Evaluation of predicted rules
Many of the rules and annotation predictions are immediately intuitive in nature. Table 1 shows a selection of rules of each type (Plus, Minus, …) along with p-values and a preliminary assessment of whether the rule is biologically ‘obvious’ (O) or ‘subtle/surprising’ (S). The inclusion of depth of GO terms as a proxy for specificity provided no additional information. The average depths for various rule sets are: ‘plus’ 7.25; ‘minus’ 7.28; ‘plus-plus’ 7.30; ‘plus-minus’ 7.15; ‘minus-minus’ 7.60. The reviewed annotations showed no trend according to GO depth.
Table 1.
Selection of proposed rules
| Assessment | |||||||
|---|---|---|---|---|---|---|---|
| Rule type | Rule number | Phenotype terms included or excluded in rule | Implied GO term | p-value | O = obvious | ||
| Plus-rule | MP 1 + | MP 2 + | GO | S = subtle | |||
| Plus-rule | 0 | impaired ovarian folliculogenesis | absent mature ovarian follicles | ovarian follicle development | 1.47E-36 | O | |
| Plus-rule | 5 | absent Peyer's patches | abnormal spleen morphology | lymph node development | 6.82E-30 | S | |
| Plus-rule | 21 | abnormal direction of heart looping | situs inversus | determination of left/right symmetry | 1.53E-23 | O | |
| Minus-rule | MP 1 + | MP 2 - | GO | ||||
| Minus-rule | 5 | abnormal sperm flagellum morphology | asthenozoospermia | fertilization | 2.70E-26 | S | |
| Minus-rule | 6 | abnormal incus morphology | abnormal temporal bone morphology | middle ear morphogenesis | 1.45E-25 | O | |
| Minus-rule | 227 | failure of vascular branching | embryonic growth retardation | regulation of angiogenesis | 2.49E-10 | S | |
| Plus-Plus-rule | MP 1 + | MP 2 + | MP 3 + | GO | |||
| Plus-Plus-rule | 109 | increased kidney apoptosis | dilated renal tubules | increased kidney cell proliferation | negative regulation of apoptotic process | 1.83E-20 | O |
| Plus-Plus-rule | 1188 | enlarged liver | hepatic necrosis | increased glycogen level | glycogen metabolic process | 1.95E-14 | S |
| Plus Minus-rule | MP 1 + | MP 2 + | MP 3 - | GO | |||
| Plus Minus-rule | 0 | globozoospermia | male infertility | absent acrosome | spermatogenesis | 4.21E-39 | O |
| Plus Minus-rule | 309 | abnormal sperm principal piece morphology | male infertility | abnormal sperm axoneme morphology | spermatid development | 5.38E-16 | O |
| Minus-Minus-rule | MP 1 + | MP 2 - | MP 3 - | GO | |||
| Minus-Minus-rule | 0 | absent mature ovarian follicles | postnatal growth retardation | abnormal female reproductive system morphology | ovarian follicle development | 1.10E-31 | O |
| Minus-Minus-rule | 8 | abnormal lysosome morphology | decreased embryo size | abnormal neuron morphology | lysosome organization | 1.91E-29 | O |
For example, the first Plus-rule #0: If a study shows that an allele has both phenotypes “impaired ovarian folliculogenesis” [MP:0001129] and “absent mature ovarian follicles” [MP:0001132], that allele is predicted be curated to function annotation “ovarian follicle development” [GO:0001541]. This rule has a very low p-value of 1.47E-36 and is assessed as ‘obvious.’ As another example, consider Plus-rule #21: If a PMID/alleleID has both the MP terms “abnormal direction of heart looping” [MP:0004252] and “situs inversus” [MP:0002766], then it should be annotated to the GO term “determination of left/right symmetry” [GO:0007368]. Both phenotypes are almost always associated with a defect in the biological process of “determination of left/right symmetry”, and it makes sense that the occurrence of both phenotypes simultaneously would more strongly indicate a defect in the aforementioned process.
Perhaps the more interesting results of our analysis are in the identification of plus-rule statements that are not necessarily obvious, but make sense biologically. For example, Plus-rule #5: If a PMID/alleleID has both the MP terms “absent Peyer’s patches” [MP:0002831] and “abnormal spleen morphology” [MP:0000689] that allele is predicted to have function “lymph node development” [GO:0048535] and is assessed as ‘subtle.’ Many of these rule statements predict relationships that would not result from a purely semantic approach. Plus-plus-rule #1188 predicts that if a PMID/alleleID has the MP terms “enlarged liver” [MP:0000599] and either “hepatic necrosis” [MP:0001654] or “increased glycogen levels” [MP:0005440], then the allele should be annotated to the GO term “glycogen metabolic process” [GO:0005977]. This is interesting because while an “enlarged liver” may not be intuitively or semantically linked to “glycogen metabolic processes”, the liver is instrumental in the storing and metabolism of glycogen, reflecting the overall physiology. This result illustrates another potential use of our correlative analysis in that a perturbation of glycogen metabolism might result in an enlarged liver phenotype or that in an animal with an enlarged liver, but no other phenotype reported, the underlying defect might be in glycogen metabolism. If our method took into consideration the structure of the ontology or a semantic link between terms, the liver correlation would likely have been missed because there is nothing in the ontology itself that states glycogen metabolism and the liver are linked.
Evaluation of predicted annotations
The next step in our prediction pipeline is the application of our rules to particular allele instances. As a result of this work, 4818 unique potential annotations associated with 1796 genes have been predicted.
Plus-rule #21, discussed above, is predicted to apply to mouse gene Zic3 [MGI:106676] based on the paper titled “Zic3 is required in the extracardiac perinodal region of the lateral plate mesoderm for left-right patterning and heart development” [PMID:23184148]. The study describes a variety of congenital defects primarily due to defects in the development of embryonic left-right patterning in Zic3tm1Bca mice [28]. Our prediction is that since the paper has been used to make annotations to the MP terms “abnormal direction of heart looping” [MP:0004252] and “situs inversus” [MP:0002766], then it would be annotated, or should be annotated, to the GO term “determination of left/right symmetry” [GO:0007368]. In this case, curatorial review confirmed that the rule held true.
Tables 2, 3, 4, 5, and 6 show the curator’s evaluation of the annotations derived from the Plus, Minus, Plus-plus, Minus-minus, Plus-minus-rules respectively for the 20 papers that were reviewed for each rule type.
Table 2.
Validation of annotations derived from plus-rules
| Rule number | Gene | Phenotype terms included in rule | Implied GO term | |||
|---|---|---|---|---|---|---|
| MP 1 + | MP 2 + | GO | p-value | Valid? | ||
| 0 | Foxl2 | ovarian follicle development | impaired ovarian folliculogenesis | absent mature ovarian follicles | 1.47E-36 | yes |
| 1 | Mbd4 | determination of left/right symmetry | abnormal left-right axis patterning | situs inversus | 6.13E-36 | yes |
| 2 | Ar | spermatogenesis | small testis | decreased testis weight | 3.47E-33 | yes |
| 2 | Ar | spermatogenesis | small testis | decreased testis weight | 3.47E-33 | yes |
| 2 | Ar | spermatogenesis | small testis | decreased testis weight | 3.47E-33 | yes |
| 2 | Hsp90aa1 | spermatogenesis | small testis | decreased testis weight | 3.47E-33 | yes |
| 3 | Psip1 | anterior/posterior pattern specification | thoracic vertebral transformation | abnormal rib morphology | 3.76E-33 | yes |
| 4 | Fen1 | lung development | pulmonary hypoplasia | decreased lung weight | 2.48E-30 | yes |
| 5 | Rag2 | lymph node development | absent Peyer's patches | abnormal spleen morphology | 6.82E-30 | yes |
| 6 | Tapt1 | anterior/posterior pattern specification | thoracic vertebral transformation | abnormal rib-sternum attachment | 3.01E-29 | yes |
| 3072 | Mir140 | decreased body size | decreased length of long bones | collagen fibril organization | 2.36E-08 | no |
| 3087 | Fancm | premature death | abnormal ovary morphology | cell proliferation | 4.05E-08 | yes |
| 3091 | Flnb | abnormal angiogenesis | decreased body weight | angiogenesis | 5.29E-08 | yes |
| 3096 | Slc6a8 | abnormal spatial learning | decreased body weight | learning or memory | 6.56E-08 | yes |
| 3098 | Tgfb1 | abnormal extraembryonic tissue morphology | decreased body size | gastrulation with mouth forming second | 8.17E-08 | no |
| 3099 | Sirt1 | decreased embryo size | ventricular septal defect | neural tube closure | 8.29E-08 | yes |
| 3100 | Kcnq1 | deafness | decreased body weight | neuromuscular process controlling balance | 1.06E-07 | yes |
| 3100 | Slc12a7 | deafness | decreased body weight | neuromuscular process controlling balance | 1.06E-07 | no |
| 3101 | Sall4 | open neural tube | premature death | neural tube closure | 1.09E-07 | yes |
| 3103 | Pkhd1 | respiratory failure | postnatal growth retardation | lung development | 1.31E-07 | no |
Table 3.
Validation of annotations derived from minus-rules
| Rule number | Gene | Phenotype Terms included in rule | Implied GO term | |||
|---|---|---|---|---|---|---|
| MP 1 + | MP 2 - | GO | p-value | Valid? | ||
| 0 | Fn1 | abnormal vitelline vascular remodeling | complete embryonic lethality during organogenesis | angiogenesis | 2.86E-41 | yes |
| 1 | Gja1 | absent mature ovarian follicles | postnatal growth retardation | ovarian follicle development | 8.95E-32 | yes |
| 1 | Stra8 | absent mature ovarian follicles | postnatal growth retardation | ovarian follicle development | 8.95E-32 | yes |
| 2 | Ppp1cc | globozoospermia | absent acrosome | spermatogenesis | 8.65E-28 | yes |
| 4 | Tert | thoracic vertebral transformation | cervical vertebral transformation | anterior/posterior pattern specification | 2.45E-26 | yes |
| 4 | Kat2a | thoracic vertebral transformation | cervical vertebral transformation | anterior/posterior pattern specification | 2.45E-26 | yes |
| 5 | Krt19 | abnormal sperm flagellum morphology | asthenozoospermia | fertilization | 2.70E-26 | no |
| 5 | Akap4 | abnormal sperm flagellum morphology | asthenozoospermia | fertilization | 2.70E-26 | no |
| 6 | Emx2 | abnormal incus morphology | abnormal temporal bone morphology | middle ear morphogenesis | 1.45E-25 | yes |
| 7 | Lmx1a | head tossing | circling | auditory receptor cell stereocilium organization | 1.02E-24 | no |
| 8 | Bmp7 | palatal shelves fail to meet at midline | cleft secondary palate | thyroid gland development | 1.37E-23 | no |
| 214 | Sctr | decreased urine sodium level | decreased body weight | negative regulation of blood pressure | 3.25E-11 | no |
| 217 | Fbn1 | abnormal intercostal muscle morphology | respiratory failure | somitogenesis | 5.27E-11 | no |
| 218 | Krt19 | short sperm flagellum | asthenozoospermia | cilium morphogenesis | 5.46E-11 | yes |
| 220 | Rab38 | abnormal pulmonary acinus morphology | respiratory distress | lung development | 5.84E-11 | no |
| 224 | Ube2b | abnormal double-strand DNA break repair | small testis | synapsis | 1.33E-10 | yes |
| 227 | Adam10 | failure of vascular branching | embryonic growth retardation | regulation of angiogenesis | 2.49E-10 | no |
| 229 | Casr | decreased circulating calcium level | postnatal growth retardation | calcium ion transport | 4.98E-10 | no |
| 230 | Hes7 | abnormal sacral vertebrae morphology | complete neonatal lethality | embryonic skeletal system development | 5.26E-10 | yes |
| 231 | Bhlhe22 | abnormal corticospinal tract morphology | complete neonatal lethality | adult walking behavior | 5.26E-10 | no |
Table 4.
Validation of annotations derived from plus-plus-rules
| Rule number | Gene | Phenotype terms included in rule | Implied GO term | ||||
|---|---|---|---|---|---|---|---|
| MP 1 + | MP 2 + | MP 3 + | GO | p-value | Valid? | ||
| 0 | Foxl2 | absent mature ovarian follicles | impaired ovarian folliculogenesis | decreased uterus weight | ovarian follicle development | 4.15E-37 | yes |
| 0 | Fads2 | absent mature ovarian follicles | impaired ovarian folliculogenesis | decreased uterus weight | ovarian follicle development | 4.15E-37 | yes |
| 0 | Kiss1 | absent mature ovarian follicles | impaired ovarian folliculogenesis | decreased uterus weight | ovarian follicle development | 4.15E-37 | yes |
| 6 | Mapk6 | decreased lung weight | small lung | pulmonary hypoplasia | lung development | 9.35E-31 | yes |
| 17 | Kiss1r | decreased uterus weight | decreased ovary weight | decreased uterus weight | ovarian follicle development | 1.11E-27 | yes |
| 17 | Esr1 | decreased uterus weight | decreased ovary weight | decreased uterus weight | ovarian follicle development | 1.11E-27 | no |
| 22 | Ndrg2 | sacral vertebral transformation | lumbar vertebral transformation | sacral vertebral transformation | anterior/posterior pattern specification | 3.92E-27 | yes |
| 27 | Gmcl1 | abnormal acrosome morphology | abnormal sperm flagellum morphology | arrest of spermiogenesis | acrosome assembly | 2.48E-24 | yes |
| 28 | Gdf6 | abnormal middle ear ossicle morphology | abnormal middle ear morphology | abnormal stapes morphology | middle ear morphogenesis | 7.47E-24 | yes |
| 48 | Gucy2d | abnormal olfactory system physiology | abnormal odor adaptation | abnormal olfactory system physiology | sensory perception of smell | 4.15E-22 | yes |
| 83 | Tnfrsf11b | abnormal middle ear ossicle morphology | abnormal middle ear morphology | abnormal incus morphology | middle ear morphogenesis | 6.33E-21 | yes |
| 109 | Bag6 | increased kidney apoptosis | dilated renal tubules | increased kidney cell proliferation | negative regulation of apoptotic process | 1.83E-20 | yes |
| 150 | Mns1 | kinked sperm flagellum | oligozoospermia | hairpin sperm flagellum | spermatid development | 4.96E-20 | yes |
| 719 | Pou1f1 | abnormal cerebellar foliation | tremors | abnormal cerebellar granule layer | neuron migration | 5.59E-16 | yes |
| 763 | Prdm9 | abnormal double-strand DNA break repair | abnormal ovary morphology | decreased oocyte number | double-strand break repair | 8.50E-16 | yes |
| 812 | Camk4 | increased bone mineral density | decreased osteoclast cell number | failure of tooth eruption | ossification | 1.27E-15 | no |
| 814 | Ddr2 | decreased circulating testosterone level | absent corpus luteum | Leydig cell hypoplasia | spermatogenesis | 1.31E-15 | yes |
| 816 | Bmper | curly tail | short tail | abnormal rib-vertebral column attachment | skeletal system morphogenesis | 1.33E-15 | yes |
| 1373 | Sec61a1 | enlarged liver | increased circulating triglyceride level | increased glycogen level | glycogen metabolic process | 6.61E-14 | no |
| 1709 | Ctsc | decreased circulating interleukin-6 level | decreased susceptibility to endotoxin shock | decreased circulating interleukin-6 level | response to lipopolysaccharide | 4.38E-13 | no |
Table 5.
Validation of annotations derived from plus-minus-rules
| Rule number | Gene | Phenotype terms included in rule | Implied GO term | ||||
|---|---|---|---|---|---|---|---|
| MP 1 + | MP 2 + | MP 3 - | GO | p-value | Valid? | ||
| 0 | M1ap | globozoospermia | male infertility | absent acrosome | spermatogenesis | 4.21E-39 | yes |
| 0 | Ing2 | globozoospermia | male infertility | absent acrosome | spermatogenesis | 4.21E-39 | yes |
| 1 | Cdk16 | abnormal sperm flagellum morphology | male infertility | teratozoospermia | cilium morphogenesis | 1.71E-28 | yes |
| 8 | Adipor2 | abnormal glucose homeostasis | decreased circulating insulin level | increased insulin sensitivity | positive regulation of insulin secretion | 1.49E-24 | yes |
| 10 | Steap3 | polychromatophilia | decreased hemoglobin content | anisocytosis | skeletal system morphogenesis | 4.46E-24 | no |
| 13 | Ptges3 | abnormal production of surfactant | atelectasis | cyanosis | lung alveolus development | 8.94E-24 | yes |
| 19 | Mbl1 | increased IgG2a level | increased IgG1 level | abnormal T cell physiology | negative regulation of B cell proliferation | 5.82E-23 | no |
| 47 | Kat2a | decreased body size | exencephaly | domed cranium | neural tube closure | 3.40E-20 | yes |
| 77 | Siglec1 | decreased tumor necrosis factor secretion | decreased interleukin-6 secretion | abnormal macrophage physiology | cellular response to lipopolysaccharide | 2.95E-19 | yes |
| 96 | Chga | dilated heart left ventricle | heart left ventricle hypertrophy | absent caveolae | heart morphogenesis | 1.56E-18 | no |
| 118 | Mark3 | improved glucose tolerance | decreased circulating insulin level | abnormal glucose homeostasis | response to insulin | 3.83E-18 | no |
| 124 | Gpr64 | kinked sperm flagellum | teratozoospermia | hairpin sperm flagellum | fertilization | 5.57E-18 | no |
| 139 | Pick1 | abnormal acrosome morphology | asthenozoospermia | absent acrosome | spermatogenesis | 8.55E-18 | yes |
| 153 | Col2a1 | abnormal long bone metaphysis morphology | abnormal long bone epiphyseal plate proliferative zone | protruding tongue | bone morphogenesis | 1.35E-17 | yes |
| 167 | Odf1 | male infertility | impaired acrosome reaction | impaired fertilization | acrosome reaction | 1.91E-17 | yes |
| 177 | Cftr | absent estrous cycle | small ovary | increased follicle stimulating hormone level | spermatogenesis | 2.26E-17 | no |
| 262 | Zfpm1 | abnormal common myeloid progenitor cell morphology | extramedullary hematopoiesis | decreased B cell number | T cell differentiation | 1.81E-16 | no |
| 309 | Gpx4 | abnormal sperm principal piece morphology | male infertility | abnormal sperm axoneme morphology | spermatid development | 5.38E-16 | yes |
| 331 | Pank1 | increased body weight | hepatic steatosis | abnormal abdominal fat pad morphology | lipid metabolic process | 7.25E-16 | yes |
| 391 | Cd36 | increased circulating triglyceride level | abnormal glucose homeostasis | insulin resistance | glycogen metabolic process | 2.72E-15 | no |
Table 6.
Validation of annotations derived from minus-minus-rules
| Rule number | Gene | Phenotype terms included in rule | Implied GO term | ||||
|---|---|---|---|---|---|---|---|
| MP 1 + | MP 2 + | MP 3 - | GO | p-value | Valid? | ||
| 0 | Esr1 | absent mature ovarian follicles | postnatal growth retardation | abnormal female reproductive system morphology | ovarian follicle development | 1.10E-31 | yes |
| 0 | Tex12 | absent mature ovarian follicles | postnatal growth retardation | abnormal female reproductive system morphology | ovarian follicle development | 1.10E-31 | no |
| 0 | Zglp1 | absent mature ovarian follicles | postnatal growth retardation | abnormal female reproductive system morphology | ovarian follicle development | 1.10E-31 | no |
| 0 | Syce3 | absent mature ovarian follicles | postnatal growth retardation | abnormal female reproductive system morphology | ovarian follicle development | 1.10E-31 | no |
| 0 | Gja1 | absent mature ovarian follicles | postnatal growth retardation | abnormal female reproductive system morphology | ovarian follicle development | 1.10E-31 | yes |
| 1 | Stra8 | absent mature ovarian follicles | postnatal growth retardation | decreased compact bone thickness | ovarian follicle development | 1.15E-31 | yes |
| 8 | Idua | abnormal lysosome morphology | decreased embryo size | abnormal neuron morphology | lysosome organization | 1.91E-29 | yes |
| 8 | Sort1 | abnormal lysosome morphology | decreased embryo size | abnormal neuron morphology | lysosome organization | 1.91E-29 | yes |
| 12 | Zbtb20 | abnormal hippocampus development | complete neonatal lethality | abnormal forebrain development | hippocampus development | 5.56E-26 | yes |
| 15 | Runx1 | abnormal embryonic hematopoiesis | complete embryonic lethality during organogenesis | anemia | spongiotrophoblast layer development | 2.53E-22 | no |
| 15 | Pdgfrb | abnormal embryonic hematopoiesis | complete embryonic lethality during organogenesis | anemia | spongiotrophoblast layer development | 2.53E-22 | no |
| 119 | Jak2 | polychromatophilia | partial postnatal lethality | increased lactate dehydrogenase level | erythrocyte differentiation | 1.15E-11 | no |
| 120 | Gjb2 | abnormal mesenchyme morphology | embryonic growth retardation | internal hemorrhage | neural tube closure | 1.82E-11 | no |
| 129 | T | abnormal limb bud morphology | complete perinatal lethality | abnormal digit morphology | anterior/posterior pattern specification | 2.93E-11 | yes |
| 140 | Stk4 | decreased spleen white pulp amount | postnatal growth retardation | arrested B cell differentiation | lymph node development | 3.39E-11 | no |
| 173 | Mapk7 | abnormal head mesenchyme morphology | abnormal blood vessel morphology | abnormal heart tube morphology | neural tube closure | 6.48E-11 | no |
| 188 | Rtel1 | enlarged allantois | no abnormal phenotype detected | small otic vesicle | gastrulation | 1.67E-10 | no |
| 212 | Smad4 | abnormal proximal-distal axis patterning | embryonic growth arrest | failure to gastrulate | proximal/distal pattern formation | 1.94E-10 | no |
| 219 | Adam10 | failure of vascular branching | embryonic growth retardation | enlarged pericardium | regulation of angiogenesis | 2.53E-10 | no |
| 228 | Rapgef2 | abnormal embryonic hematopoiesis | complete embryonic lethality during organogenesis | abnormal liver morphology | spongiotrophoblast layer development | 3.70E-10 | no |
In 16 out of 20 cases, the Plus-rule generated an annotation that was considered correct by the curator. In all cases, the incorrect inferences were associated with poorer p-values (Table 2), validating its utility as a score to measure confidence level. In one negative case shown here, Plus-rule 3072: “decreased body size” plus “decreased length of long bones” phenotypes implies “collagen fibril organization,” the curator specifically noted that a curator could not make this annotation based on the paper since the authors did not look at the collagen fibrils directly although they did show that the cartilage in the developing bone is affected Another interesting negative case is the application of Plus-rule #3100: “deafness” plus “decreased body weight” phenotypes implies “neuromuscular process controlling balance” to two different genes. For Kcnq1, based on the paper [PMID:15498462], the annotation could be made; while for Slc12a7, based on the paper [PMID:11976689], it could not.
In the case of the Minus-rule, only 10 of the 20 predicted annotations were correct. In the case of the Minus-rule, better p-value scores gave more reliable predictions (Table 3). The Plus-plus (16 correct of 20) and Minus-minus (7 correct of 20) rules displayed similar behavior to the plus and minus-rules respectively (Tables 4 and 5), while the Plus-minus-rule (12 correct of 20) unsurprisingly seemed at almost act as an intermediate between the Plus and Minus-rules (Table 6).
Testing the validation of the various rules showed that, in general, the Plus-rule statements are more reliable that the minus-rule statements in generating valid annotations. This is not surprising for two reasons: (1) combined phenotypes often give ‘additive’ evidence to support the GO annotation and (2) the ‘closed world’ assumption limits the predictive power of the absence of annotation. The value of ‘additive’ evidence is mentioned above for Plus-rule #0, “absent mature ovarian follicles” [MP:0001132] plus “impaired ovarian folliculogenesis” [MP:0001129] additively support that a gene product would be directly involved in the GO process “ovarian follicle development” [GO:0001541]. The ‘closed world assumption’ is the assumption that what is not currently known to be true is assumed to be false. In our case, the Minus-rules rely on the lack of phenotype annotation as if it the phenotype was tested and found not to obtain.
We did a rather simple comparison of the predictive power of our rules based on the reviewed annotation predictions. We calculated the Positive Predictive Value from the true positives (TP) and false positives (FP) in the reviewed annotation sets for each of the composite rules: PPV = TP/(TP + FP). Figure 5 shows a comparison of the PPV for various p-value cutoffs of the composite rules for the reviewed annotation predictions. The PPV for the first three composite rules is markedly better for a given p-value than for the last two rules.
Figure 5.
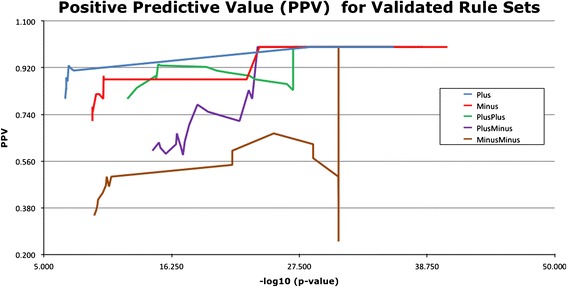
Comparison of the Positive Predictive Value for various p-value cutoffs of the composite rules for the reviewed annotation predictions. The PPV for the first three composite rules is markedly better for a given p-value than for the last two rules.
Our analysis shows that the Minus-rule statements are not reliable at predicting specific GO annotations. However, these results are very interesting because although the rules failed to accurately predict correct GO terms, the general area of biology of the predictions was often accurate. In several cases we found that the processes predicted might lead to downstream phenotypic similarities that would fit the phenotypes given in the rules. For example, the terms that predicted the process of ‘fertilization’ (Minus-rule 5) were not entirely correct, we noted, because the gene products identified did affect sperm motility and would create defective sperm, but we could not conclude from the papers [PMID:16015579; PMID:12167408] that the predicted gene products contributed to “fertilization” [GO:0009566] which in GO is specifically defined as the union of the two gametes. Likewise, the prediction of “regulation of angiogenesis” [GO:0045765] would have been correct if the prediction were “vasculogenesis” [GO:0001570], both of which can lead to abnormal branching of blood vessels (Minus-rule 227). These types of predictions may still prove useful for curators in that they point to relevant branches of biology for annotation consideration.
Our results show that the use of correlation modeling can be used to infer biological knowledge about the processes that underlie phenotypic expression. We show that the use of the Plus-rule statements has the potential to accurately predict GO annotations, and that while the Minus-rule generally predicts an incorrect specific term, it often predicts a correct area of biology. Our results show that curators can use our correlative rules to guide manual curation however, individual instances should not necessarily be annotated without the review of a biocurator. The rules can be used to help curators decide about a general subcategorization of GO terms from which to choose when curating GO data based on mutant phenotypes. The method could also be used when biocurators are trying to identify literature that covers an area of biology of interest. As the rules are further tested and more papers are cocurated for phenotype and GO, the rules will become further refined and accurate p-value cutoffs for reliability can be determined.
Future work will use these methods to create and test more complex rules based on this strategy. Our methodology could be used in future work to help predict the biological process that underlies a given disease by correlating the disease-phenotypes with GO biological processes. This methodological approach can be adapted to any species with rich phenotypic data. Lastly, we could similarly reverse the method to use multiple GO terms to predict likely phenotypic outcomes such as the disruption of specific pathways and then test those predictions as potential new mouse models of human disease.
Conclusions
Our correlative analysis for predicting GO annotations can be used to assist biocurators in the curation of papers with no previous GO annotations while also giving potential insight into complex phenotype-gene function relationships. We believe this is the first attempt to predict a GO term from its composite relationship with MP terms independently of semantic analysis but rather through a shared allele, and to apply that method to predict GO annotations to a set of papers that have not yet been annotated for GO. Our method has the advantage in that since it does not take semantic similarity into account, it can potentially find correlations between phenotypes and biological processes that are not intuitively obvious or that are not explicitly states in either ontology. Of course we are in a good position to perform this type of analysis since there are large independent efforts in the MGI resource to annotate both phenotypes and GO. The independent annotation serves as an internal control in that the annotations that are in the current corpus are essentially ‘blind’ with respect to one another. Co-curation of phenotype and underlying biological processes could potentially change our results dramatically since a single curator could be swayed to search for a semantic similarity in terms chosen from each ontology. One interesting experiment would be to perform this same analysis on a data set from another group in which individual curators annotate to both ontologies. Since our methodology is purely correlative and does not rely on any other metric, it could potentially be used with other data sets such a GO terms and disease ontologies, or even in combination with several ontologies such as GO, phenotype and expression. The five rules analyzed in this work can be combined in various manners to create many more possible derived rule structures—our results here serve as a ‘proof of principal’ for this type of analysis and pave the way for future iterations.
In practice, we have developed a script and methodology that, given a gene/PubMed ID, will suggest a GO term(s) to a curator if it meets the requirements of one of our rule statements. We intend to integrate this immediate approach into the workflow of MGI curators and hope that others will use our methodology to explore correlative algorithms in the context of diverse data sets.
Acknowledgments
This research was supported by NIH NHGRI grant HG-002273 for the Gene Ontology Consortium. JAA internship funding was provided by NIH NHGRI grant HG-007053. We would like to thank the Jackson Laboratory Summer Student Program for their outstanding support.
Abbreviations
- GO
Gene ontology
- MP
Mammalian phenotype ontology
- MGI
Mouse genome informatics
- PMID
PubMed identification number
- IMP
Inferred from mutant phenotype
- BP
Biological process
Additional files
5 files, where [RuleName] is either Plus, Minus, Plus-Plus, Minus-Minus, or Plus-Minus, corresponding to the rules described in this work. The files include the instances of all the rules and the calculated p-value.
5 files, where [RuleName] follows the same pattern as described above. The files include all of the PMIDs predicted from the rules described, along with the corresponding p-value.
Contains a simple companion software with documentation that uses data updated in real time to infer GO annotations given an MGI ID. Inputs: MGI ID, PMID (optional); Outputs: Rule #, Inferred GO, PMID, p-val.
Footnotes
Joao A Ascensao and Mary E Dolan contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JAA and MED designed the algorithms and produced the figures. DPH conducted the analysis of the results. JAB initiated the study and provided constructive input. All of the authors contributed to the preparation and approval of the final manuscript.
Contributor Information
Joao A Ascensao, Email: Joao.Ascensao@rice.edu.
Mary E Dolan, Email: Mary.Dolan@jax.org.
David P Hill, Email: David.Hill@jax.org.
Judith A Blake, Email: Judith.Blake@jax.org.
References
- 1.Gruber TR. A Translation Approach to Portable Ontology Specifications. Knowledge Acquisition. 1993;5(2):199–220. doi: 10.1006/knac.1993.1008. [DOI] [Google Scholar]
- 2.Hill DP, Smith B, McAndrews-Hill MS, Blake JA. Gene Ontology annotations: what they mean and where they come from. BMC Bioinformatics. 2008;9(Suppl 5):S2. doi: 10.1186/1471-2105-9-S5-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gene Ontology Consortium Gene Ontology annotations and resources. Nucl Acids Res. 2013;41:D530–D535. doi: 10.1093/nar/gks1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gene Ontology Consortium Gene Ontology: tool for the unification of biology. Nature Genetics. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith CL, Eppig JT. The Mammalian Phenotype Ontology as a unifying standard for experimental and high-throughput phenotyping data. Mamm Genome. 2012;23(910):653–668. doi: 10.1007/s00335-012-9421-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE, Mouse Genome Database Group The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucl Acids Res. 2014;42(1):D810–D817. doi: 10.1093/nar/gkt1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashurst JL, Collins JE. Gene Annotation: Prediction and Testing. Annu Rev Genomics Hum Genet. 2003;4:69–88. doi: 10.1146/annurev.genom.4.070802.110300. [DOI] [PubMed] [Google Scholar]
- 8.Groth P, Weiss B, Pohlenz HD, Leser U. Mining phenotypes for gene function prediction. BMC Bioinformatics. 2008;9:136. doi: 10.1186/1471-2105-9-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tao Y, Li J, Friedman C, Lussier YA. Information theory applied to the sparse gene ontology annotation network to predict novel gene function. Bioinformatics. 2007;23(13):i529–i538. doi: 10.1093/bioinformatics/btm195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Azuaje F, Bodenreider O. Incorporating Ontology-Driven Similarity Knowledge into Functional Genomics: An Exploratory Study. Proc BIBE. 2004;4:317–324. doi: 10.1109/BIBE.2004.1317360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoehndorf R, Hardy NW, Osumi-Sutherland D, Tweedie S, Schofield PN, Gkoutos GV. Systematic analysis of experimental phenotype data reveals gene functions. PLOS One. 2013;8(4):e60847. doi: 10.1371/journal.pone.0060847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.King OD, Lee JC, Dudley AM, Janse DM, Church GM, Roth FP. Predicting phenotype from patterns of annotation. Bioinformatics. 2003;19(Suppl 1):i183–i189. doi: 10.1093/bioinformatics/btg1024. [DOI] [PubMed] [Google Scholar]
- 13.Kemmeren P, Kockelkorn TT, Bijma T, Donders R, Holstege FC. Predicting gene function through systematic analysis and quality assessment of high-throughput data. Bioinformatics. 2005;21(8):1644–1652. doi: 10.1093/bioinformatics/bti103. [DOI] [PubMed] [Google Scholar]
- 14.Gómez A, Cedano J, Amela I, Planas A, Piñol J, Querol E. Gene Ontology function prediction in Mollicutes using protein-protein association networks. BMC Systems Biology. 2011;5:49. doi: 10.1186/1752-0509-5-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng M, Zhang K, Mehta S, Chen T, Sun F. Prediction of Protein Function Using Protein–Protein Interaction Data. J Comput Biol. 2004;10(6):947–960. doi: 10.1089/106652703322756168. [DOI] [PubMed] [Google Scholar]
- 16.Deng M, Tu Z, Sun F, Chen T. Mapping gene ontology to proteins based on protein–protein interaction data. Bioinformatics. 2003;20(6):895–902. doi: 10.1093/bioinformatics/btg500. [DOI] [PubMed] [Google Scholar]
- 17.Wood V, private communication
- 18.Joslyn C, Baddeley B, Blake J, Bult C, Dolan M, Riensche R, Rodland K, Sanfilippo A, White A: Automated Annotation-Based Bio-Ontology Alignment with Structural Validation.Nature Proceedings 2009<http://dx.doi.org/10.1038/npre.2009.3518.1>
- 19.Lambrix P, Tan H. Ontology Alignment and Merging. In: Burger A, Davidson D, Baldock R, editors. Anatomy Ontologies for Bioinformatics. London: Springer; 2008. pp. 133–149. [Google Scholar]
- 20.Manda P, McCarthy F, Bridges SM. Interestingness measures and strategies for mining multi-ontology multi-level association rules from gene ontology annotations for the discovery of new GO relationships. J Biomed Informat. 2013;46(5):849–856. doi: 10.1016/j.jbi.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 21.Myhre S, Tveit H, Mollestad T, Lægreid A. Additional Gene Ontology structure for improved biological reasoning. Bioinformatics. 2006;22(16):2020–2027. doi: 10.1093/bioinformatics/btl334. [DOI] [PubMed] [Google Scholar]
- 22.Dickie MM. New Splotch Alleles in Mouse. J Hered. 1964;55:97–101. doi: 10.1093/oxfordjournals.jhered.a107317. [DOI] [PubMed] [Google Scholar]
- 23.Moase CE, Trasler DG. Spinal ganglia reduction in the splotch-delayed mouse neural tube defect mutant. Teratology. 1989;40(1):67–75. doi: 10.1002/tera.1420400109. [DOI] [PubMed] [Google Scholar]
- 24.Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA, Capecchi MR. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004;18(21):2614–2626. doi: 10.1101/gad.1244004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Degenhardt KR, Milewski RC, Padmanabhan A, Miller M, Singh MK, Lang D, Engleka KA, Wu M, Li J, Zhou D, Antonucci N, Li L, Epstein JA. Distinct enhancers at the Pax3 locus can function redundantly to regulate neural tube and neural crest expressions. Dev Biol. 2010;339(2):519–527. doi: 10.1016/j.ydbio.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics. 2010;27(3):431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oliphant TE. Python for scientific computing. Comput Sci Eng. 2007;9(3):10–20. doi: 10.1109/MCSE.2007.58. [DOI] [Google Scholar]
- 28.Jiang Z, Zhu L, Hu L, Slesnick TC, Pautler RG, Justice MJ, Belmont JW. Zic3 is required in the extracardiac perinodal region of the lateral plate mesoderm for left-right patterning and heart development. Hum Mol Genet. 2013;22(5):879–889. doi: 10.1093/hmg/dds494. [DOI] [PMC free article] [PubMed] [Google Scholar]