

Abstract
Phosphorylation can modulate the activities of enzymes. The phosphoryl donor in the catalytic cleft of α-D-phosphohexomutases is transiently dephosphorylated while the reaction intermediate completes a 180° reorientation within the cleft. The phosphorylated form of 52 kDa bacterial phosphomannomutase/phosphoglucomutase is less accessible to dye or protease, more stable to chemical denaturation, and widely stabilized against NMR-detected hydrogen exchange across the core of domain 3 to juxtaposed domain 4 (each by ≥1.3 kcal/mol) and parts of domains 1 and 2. However, phosphorylation accelerates hydrogen exchange in specific regions of domains 1 and 2, including a metal-binding residue in the active site. Electrostatic field lines reveal attraction across the catalytic cleft between phosphorylated Ser-108 and domain 4, but repulsion when Ser-108 is dephosphorylated. Molecular dynamics (MD) simulated the dephosphorylated form to be expanded due to enhanced rotational freedom of domain 4. The contacts and fluctuations of the MD trajectories enabled correct simulation of more than 80% of sites that undergo either protection or deprotection from hydrogen exchange due to phosphorylation. Electrostatic attraction in the phosphorylated enzyme accounts for 1) domain 4 drawing closer to domains 1 and 3; 2) decreased accessibility; and 3) increased stability within these domains. The electrostriction due to phosphorylation may help capture substrate, whereas the opening of the cleft upon transient dephosphorylation allows rotation of the intermediate. The long-range effects of phosphorylation on hydrogen exchange parallel reports on protein kinases, suggesting a conceptual link among these multidomain, phosphoryl transfer enzymes.
Introduction
Phosphorylation regulates enzymes with diverse consequences ranging from long-range allosteric activation of glycogen phosphorylase to million-fold inhibition of isocitrate hydrogenase (1). Phosphorylation of the activation loop of many eukaryotic protein kinases triggers electrostatic rearrangement of the loop and assembly of the internal regulatory spine of hydrophobic residues in the N- and C-lobes of these proteins, thereby supporting interaction with substrate and activity (2). Phosphorylation has been proposed to stabilize proteins by decreasing the range of motions, typically with global conformational adjustments of <2 Å (3); however, exceptions to this stabilization have been noted (4).
α-D-phospho-hexomutases are also sensitive to phosphorylation. Their reaction cycles oscillate between serine phosphorylated and dephosphorylated forms (5–7) (Fig. 1). The serine donates a phosphoryl group to the phosphosugar substrate. While it is transiently dephosphorylated, the serine accepts a phosphoryl group from the bisphosphorylated intermediate (Fig. 1). The phosphorylated enzyme binds and unbinds the phosphorylated sugar, and the bisphosphorylated intermediate reorients by 180° while it is associated with the dephosphorylated enzyme (6,8,9). An attractive member of the enzyme superfamily for biophysically characterizing the effects of phosphorylation on catalytic switching is phosphomannomutase/phosphoglucomutase (PMM/PGM) from Pseudomonas aeruginosa.
Figure 1.
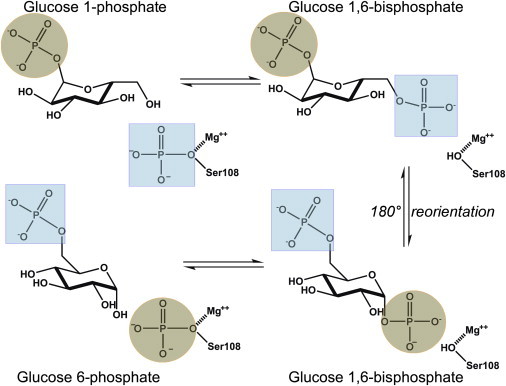
Catalytic mechanism of PMM/PGM. The enzyme transforms G1P to G6P via G1,6P, which undergoes rotation in the active site. To see this figure in color, go online.
The phosphomutase activity of PMM/PGM contributes to the infectivity of P. aeruginosa in ventilator-assisted pneumonia, chronic obstructive pulmonary disease, and cystic fibrosis (10–12) through the biosynthesis of sugar precursors of lipopolysaccharide, the exopolysaccharides Pel and Psl, rhamnolipids, and alginate (13–16). Ser-108 of PMM/PGM is phosphorylated at the base of a deep, positively charged catalytic cleft formed by its four domains (see Fig. 3 B). Upon binding of a phosphosugar, domain 4 (D4) rotates to close the catalytic cleft (9,17). Structures containing a bisphosphorylated intermediate suggested a half-open state of the enzyme (9), which may accommodate the 180° reorientation of the intermediate required by the catalytic cycle (8). 15N NMR relaxation indicates partial independence of the rotational mobility of D4 (18), consistent with crystallographic snapshots of varying conformers of D4 (9). Amide NMR spectra suggest a mixture of phosphorylated and dephosphorylated enzyme forms (18). The catalytic cleft was widened by the S108A mutation (6), suggesting a role for phosphorylation of Ser-108 in conformational adjustment.
Figure 3.
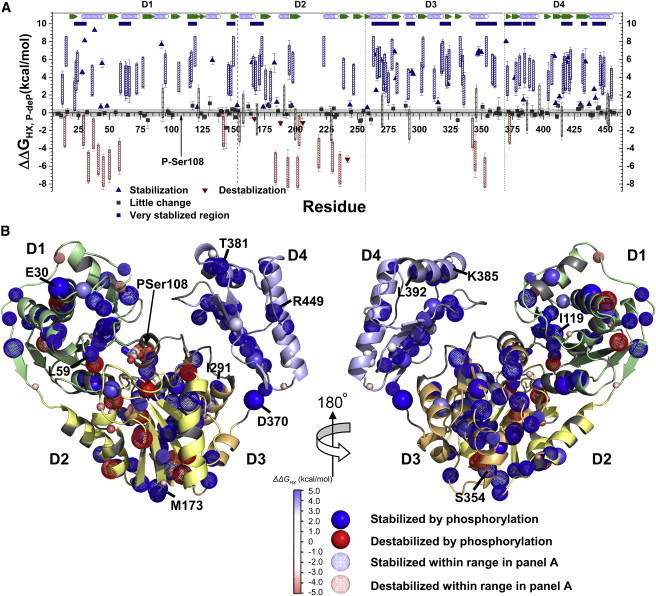
Phosphorylation stabilizes or slows HX at more sites than it accelerates. (A) Rate constants of HX, kex, measured either as HDX by NMR or on the subsecond scale by CLEANEX-PM (29), were transformed to ΔGHX using Eq. 2 and the ΔΔGHX,P-deP differences are plotted. Blue indicates where phosphorylation is significantly stabilizing, and red indicates where it is destabilizing. Triangles represent cases in which kex was measured in both Apo-P and Apo-deP forms. Hatched bars mark cases in which kex in one phosphorylation state lay in the intermediate, unmeasured time range where 1 s−1 > kex > 4 × 10−3 s−1; the height of the bar indicates ΔΔGHX,P-deP lies within this range. Sequence segments with more than four of 10 residues protected by phosphorylation >2 SD above average are marked with navy blue bars. Locations of strands and helices are given in green and open blue symbols, respectively. (B) Amide groups protected from HX by phosphorylation of Ser-108 are marked by spheres with shades of blue, and amide groups destabilized by phosphorylation are in red. Dotted spheres correspond to residues with the uncertainty ranges marked in (A). The amplitude of the phosphorylation-dependent change, ΔΔGHX, is symbolized by the color intensity and radii of the spheres. To see this figure in color, go online.
The conformational effects of phosphorylation can be investigated by hydrogen exchange (HX, broadly referring to fast to slow timescales), which depends on the stability of the structural environment and the longevity of the hydrogen bonds (H-bonds). Transient openings of H-bonds facilitate access of hydroxide ions to amide protons to catalyze exchange. Previously, hydrogen-deuterium exchange mass spectrometry (HDX-MS) revealed percentage changes in the deuteration of peptide fragments of PMM/PGM, suggesting that its dephosphorylated form (Apo-deP) is more flexible than the phosphorylated form (Apo-P), consistent with a role in facilitating the 180° rotation of the intermediate (19). However, limitations inherent to HDX-MS precluded characterization of important aspects of HDX in this enzyme, including regions critical for function. Much of the active site could not be characterized because 1) the peptide containing catalytic phosphoSer-108 was missing from mass spectra; and 2) affected residues in four other peptides encompassing the active site could not be pinpointed due to peptide lengths of 24–35 residues. Residue-specific information was also missing elsewhere. Moreover, HDX-MS data were not available for timescales of seconds or days (20). Consequently, the HDX-MS results did not provide the detailed kinetic and thermodynamic insights required for a biophysical understanding (21).
We implemented NMR to measure HDX and millisecond-scale HX of 463-residue PMM/PGM to pinpoint the effects of phosphorylation on HX as well as its rate constants and free energies. We found that phosphorylation causes both slowing and acceleration of HX around the active site and in D1 and D2, little change at many residues, and slowing of HX of ∼100 residues widely and remotely. The sites that are most protected against HDX, as identified by NMR, report the global folding stability (22). This provides a lower limit for the phosphoryl stabilization of the fold of PMM/PGM and its domains, where traditional denaturation experiments fail for lack of reversible folding. Based on changes in the free energy of HX and the density of protected residues, the core of D3 appears to be most protected by phosphorylation, followed by the proximal face of D4. A biophysical accounting for the latter remote pattern of protection by phosphorylation is provided by electrostatic fields and molecular dynamics (MD) trajectories. Comparative calculations for Apo-P and Apo-deP suggest that compaction results from electrostatic attraction introduced by phosphorylation of catalytic Ser-108, i.e., electrostriction. The use of surrounding contacts and either H-bonding or root mean-square fluctuation (RMSF)−1 from the MD trajectories (23,24) proved to be qualitatively highly consistent with the locations of altered HX protection accompanying dephosphorylation. The solution and computational lines of evidence reveal what x-ray diffraction constrained by crystal packing (19) could not, i.e., that electrostatic changes due to phosphorylation appear to narrow the active site of PMM/PGM, compact the fold, and slow HX in D3 and D4. These changes may facilitate the capture and accommodation of phosphohexose substrates during the multistep reaction of the enzyme.
Materials and Methods
Enzyme preparation
His-tagged PMM/PGM from P. aeruginosa was prepared (18). Samples were labeled with 2H and 15N for NMR. The enzyme was phosphorylated to 85–90% or dephosphorylated to 90–95% by incubation with excess glucose 1,6-bisphosphate (G16P) or glucosamine 1-phosphate (Sigma-Aldrich, St. Louis, MO), respectively, at 4°C for 18 h (19).
Accessibility measurements
Apo-P and apo-deP (10 μM), with 90% and 0% levels of phosphorylation, respectively, were incubated with 1 mM of 1-anilinonaphthalene-8-sulfonate (ANS, Fisher, Pittsburgh, PA) for 1 h in 50 mM MOPS (pH 7.4, Fisher) at 25°C. Fluorescence was excited at 365 nm. The fluorescence emission of free ANS in buffer was subtracted away (25).
Then, 30 μM Apo-P and apo-deP, at 87% and 10% levels of phosphorylation, respectively, were treated with proteinase K in 50 mM MOPS (pH 7.4) at a 300:1 (w/w) ratio at 25°C until the reaction was terminated with 3 mM phenylmethylsulfonyl fluoride (MP Biomedicals, Santa Ana, CA). Bands on SDS-PAGE (25) were quantified using ImageJ.
NMR spectroscopy
Band-selective excitation short-transient transverse relaxation-optimized spectroscopy (BEST-TROSY) (26) and other NMR spectra were collected at 35°C on a Bruker Avance III 800 MHz spectrometer with TCI cryoprobe. Samples were incubated in 50 mM MOPS (pH 7.4 at 25°C), 1 mM MgCl2 (Fisher), and 10 mM dithiothreitol (DTT, Gold Biotechnology, St. Louis, MO). The spectra were processed with NMRPipe (27) and peaks were interpreted using Sparky (28) with the reported assignments (18). An in-house-written Python script mapped Sparky-assigned peak shifts to NMRPipe peak tables and is available in the Supporting Material. NMRPipe provided the volume and height of each peak.
Rapid HX by NMR
HX occurring on the millisecond scale was observed using the phase-modulated clean chemical exchange (CLEANEX-PM) pulse sequence enhanced with TROSY detection (29,30) but with interleaved acquisition (see Supporting Material). The solvent-exchange rates were fitted to the equation
| (1) |
where I/I0 represents normalized peak heights, R1A is the relaxation rate constant during the spinlock, k is the rate constant of HX, and R1B is the relaxation rate constant for the water (Fig. S2). Radiation damping complicated measurement of R1B. Since trial values of R1B from 0.1 to 0.001 s−1 provide the same fitted rate constants (31), a value of 0.01 s−1 was used.
Quantification of rates of HDX
Ensuring the uniformity of phosphorylation and dephosphorylation, and keeping the cysteine residues reduced for a week prevented mixtures and biexponential changes. To initiate HDX, 2H/15N-labeled samples were concentrated to ∼5 mM in H2O-based buffer and then diluted 5-fold in D2O-based buffer at 22°C. Addition of 10 mM DTT and Ar to the samples (then sealed in Shigemi tubes) maintained reproducible TROSY spectra for 1 week (judged in H2O). The 20 min of dead time for these handling steps limited measurable kobs to < 5 × 10−2 min−1. A series of at least 57 BEST-TROSY spectra (26) were acquired at 35°C, continuously during the first 24 h and then several per day until ∼8000 min had elapsed. After the first 24 h, the samples were maintained at 35°C in a water bath between acquisitions. Exponential decay rates, kobs, were fitted to the peak heights (Fig. 2 D), with correction for the residual 20% H2O described by Eq. S1 in the Supporting Material. Monitoring of gradual concurrent dephosphorylation and correction of the affected rate constants of HDX (kex) are detailed in the Supporting Material.
Figure 2.
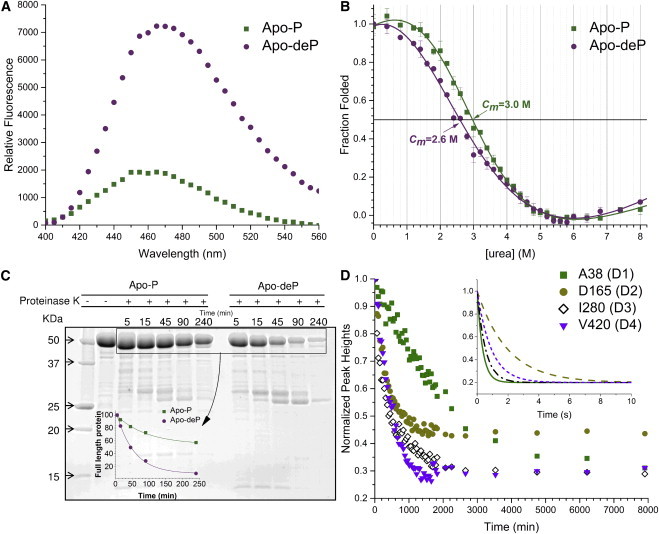
Phosphorylation effects on the accessibility and stability of PMM/PGM. (A) ANS emission spectra with Apo-P and Apo-deP. (B) Apo-P and Apo-deP were denatured by incubation in urea for 12 h at 25°C in 50 mM MOPS, pH 7.4, and 0.1 mM DTT. The intrinsic Trp fluorescence at each point was measured in duplicate. (C) Effect of proteinase K on Apo-P and Apo-deP. Aliquots were removed at the indicated times and monitored with SDS-PAGE. The inset plots the decay of the intact enzyme. (D) Examples of an amide group in each domain with HX slowed by phosphorylation. The disappearance of an amide peak of each domain from TROSY spectra upon a change of Apo-P to D2O solution is plotted. The inset shows subsecond components of HX simulated using the rate constants fitted to CLEANEX time courses for the same four residues in the Apo-deP form (Fig. S1). To see this figure in color, go online.
Each intrinsic rate constant of random coil HX (krc) was obtained using the SPHERE server (http://www.fccc.edu/research/labs/roder/sphere/sphere.html). The free energy of amide protection from HX was calculated as
| (2) |
The folding stabilities of each domain and the entire enzyme were estimated from the highest values of ΔGHX in each domain and corrected for proline isomerization (22).
Electrostatic field calculations
Atomic charges and sizes around the metal-binding site were parameterized by the Amber tools module MCPB/MTKPP, which invokes Gaussian 2009 (32,33). In the structure of Apo-P (6) (Protein Data Bank (PDB): 1K35), Zn2+ is bound by phosphoSer-108 and Asp-242, Asp-244, and Asp-246. In Apo-deP (PDB: 4MRQ) (19), a bound water replaces Ser-108 in coordinating Zn2+. Other atomic sizes and charges were defined by the Amber ff99SB force field (34). The electrostatic potentials were then computed using the Adaptive Poisson-Boltzmann Solver (35) at 310 K with protein and solvent dielectric constants of 2.0 and 78.5, respectively, and ionic strength of 30 mM. Electrostatic field lines were visualized using VMD (36) as described previously (37,38).
Modeling HX protection from MD simulations
The logarithm of the simulated HX protection factor, , of an amide group (of residue n) can be approximated by linear combination of its number of contacts and a second protection factor element (PFE2) measured from a dynamics simulation, e.g., either the number of H-bonds or the RMSF−1 at that site:
| (3) |
where the generic term can be either , with the weights of the terms being and either or (23,24). For application below, two independent pairs of weight parameters were optimized for slower and faster regimes of HX:
The modeling of protections improved using the alternative sets of coefficients (Table S3; Fig. S11). The performance of candidate sets of parameters with PMM/PGM is given in Table S4. Simulated protection factors were converted into free energies using Eq. 2: . For details of HX protection simulated from the MD trajectories, see Supporting Materials and Methods.
Results
We investigated phosphorylation-induced changes between the Apo-P and Apo-deP states of PMM/PGM from P. aeruginosa in solution and computationally to better understand the interplay of these enzyme forms with the catalytic cycle.
Phosphorylation decreases accessibility to probes and enhances stability
Several approaches were used to compare the overall structural flexibility, openness, and stability of Apo-P and Apo-deP in solution. ANS dye has long been used to probe the conformational changes of proteins because it fluoresces little in water but much more upon binding to a protein (25,39). ANS has a significantly greater fluorescence emission when mixed with Apo-deP than when mixed with Apo-P (Fig. 2 A). This suggests that phosphorylation decreases ANS access to binding pockets. We compared the folding stabilities of Apo-P and Apo-deP using urea denaturation. The unfolding curves with [urea] of 3.0 M needed for 50% denaturation (Cm) of Apo-P suggest that it has greater stability than Apo-deP with a Cm of 2.8 M (Fig. 2 B). (Cm values are compared here because PMM/PGM cannot be refolded after denaturation, indicating a lack of the reversibility needed to obtain ΔGfold.) Limited proteolysis by proteinase K degrades Apo-deP 6.3-fold faster than it degrades Apo-P, based on rate constants fitted to the disappearance of the intact enzyme band on gels of 2.4 × 10−3 ± 0.4 × 10−3 min−1 for Apo-P and 1.5 × 10−2 ± 0.2 × 10−2 min−1 for Apo-deP (Fig. 2 C). Binding of the small dye, proteolysis by the large enzyme, and urea denaturation suggest that phosphorylation curbs the accessibility of PMM/PGM and enhances its folding stability.
Effects of dephosphorylation on NMR spectra
Time-dependent changes in NMR spectra and mass spectra (loss of the mass of a phosphoryl group) revealed that PMM/PGM undergoes spontaneous dephosphorylation at physiological temperatures (Fig. S2). We performed a residue-by-residue structural comparison of the phosphorylation-dependent changes in accessibility and stability using NMR. Amide peaks representing active Apo-P and Apo-deP forms proved distinguishable for 98 residues with good sensitivity (Fig. S2; Table S1). At 35°C, the NMR peaks of Apo-deP appeared and those of Apo-P disappeared (Fig. S2, C and D) with a time constant of 19.0 ± 1.1 h (1140 ± 70 min) in the joint fits of the pairs of peaks of 43 residues.
Most of the amide NMR peaks that were shifted by dephosphorylation were distributed from D1 to D3 (Fig. S3, A and B). Lys-219 of D2, and Arg-262 and Trp-336 of D3 underwent the largest peaks shifts (with ΔωHN > 0.2 ppm) despite being >19 Å distant from the phosphoryl group of Ser-108 (Fig. S3 B). In addition, dephosphorylation was found to broaden at least 33 amide peaks beyond detection, with other amide peaks of Apo-deP also being broader than their counterparts in Apo-P, especially in D2 and D3, but also D4 (Figs. S3 and S4). The line broadening probably arises from exchange among structural environments in these domains enabled by dephosphorylation, one manifestation of its long-range effects.
To investigate whether phosphorylation affects the backbone flexibility of PMM/PGM on the scale of picoseconds to nanoseconds, we measured 15N{1H} nuclear Overhauser effect (NOE) relaxation on multiple freshly phosphorylated or dephosphorylated samples. Twenty or more loops and termini showed locally decreased NOE ratios indicating fast, localized fluctuations (Fig. S5). The high similarity of the NOEs of Apo-P and Apo-deP, without differences beyond the standard deviation (SD) of the triplicate measurements, suggests no convincing phosphorylation-dependent change on the subnanosecond scale. Coupled with the much slower millisecond dynamics in Apo-deP implied by its NMR line broadening (Figs. S3 and S4), this suggests the need to test whether the phosphorylation state instead affects more rigidly structured regions, where the conformational resistance may influence dynamics on slow timescales accessible by HX methods.
Slowing of HX by phosphorylation, especially in D3 and the adjacent face of D4
To scrutinize the working hypothesis that dephosphorylation globally mobilizes and increases the accessibility of the structure of Apo-deP (19) (Fig. 2), we performed residue-specific and thermodynamic quantification of HX using NMR. A pH of 7.4, which is needed for solubility and activity of PMM/PGM, and a temperature of 35°C, which enhances its TROSY NMR spectra, promote HX that is intrinsically comparatively rapid. The majority of amide protons were replaced by deuterons within 20 min after exposure to D2O, especially in Apo-deP (Movie S1). After 1–3 h of exchange, however, more than 100 amide groups across Apo-P appeared to be slowed in HX (Movie S2) relative to Apo-deP, suggesting that multiple locales are stabilized by phosphorylation of the enzyme (Figs. 2 D, 3, and S6).
Comparisons of the effects of phosphorylation status on the thermodynamics of HX, i.e., ΔGHX, are newly furnished by these NMR studies of PMM/PGM, for both its individual amide groups and entire domains, provided that its HX occurs in the bimolecular (EX2) limit. In the EX2 regime, the rate of closing of the H-bond (kcl) greatly exceeds the intrinsic rate of HX of a free amide of that residue, krc (40), in the Linderström-Lang model of HX (41):
| (4) |
EX2 behavior is recognizable from ΔGHX values shared across a wide range of krc, and from a slope of 1 among such residues in log-log plots of kex versus krc (42,43). Groups of PMM/PGM residues that share similar ΔGHX,apparent values do indeed have slopes of 1 in such plots, for both the Apo-P and Apo-deP forms (Fig. S7). This strongly suggests that EX2 behavior prevails for both states of the enzyme, from the slowest-exchanging residues with ΔGHX > 10 kcal/mol through sites of faster exchange with ΔGHX of ∼6 kcal/mol (Fig. S7). The EX2 behavior validates the application of Eq. 2 to PMM/PGM to estimate its ΔGHX values from HDX experiments and the stability of H-bonding implied.
Phosphorylation induces stabilization of HX that is significant in segments of D1 and D2, and throughout D3 and D4, as depicted by differences in ΔGHX in Fig. 3. Phosphorylation stabilizes HX by 1.5–9.3 kcal/mol at 35°C (with slowings of 12-fold to 3.8 million-fold) in ∼23 residues in D1, 12 residues in D2, 32 residues in D3, and 28 residues in D4 (Fig. 3 A). The greatest density of phosphorylation-stabilized residues occurs in the core of D3 throughout its central helix (Leu-263–Ser-273) and the two flanking β-strands (Ala-278–Phe-282 and Leu-322–Gly-324, with 10 phosphorylation-stabilized residues between them). In D4, two long, central β-strands that face D3 (Gly-416–Ala-422 and Arg-432–Glu-434) are similarly stabilized (Fig. 3 B). This suggests that phosphorylation may promote closer packing of D4 against D3.
Additional phosphorylation-dependent stabilization ΔΔGHX,P-deP appears to radiate outward from the most stabilized cores of D3 and D4. Next to the stabilized core helix of D3 are two helices spanning Ile-291–Ser-295 and Ala-347–Ser-354 that also show phospho-stabilization (Fig. 3 B). Adjacent to the latter helix, a loop from D2 (Asp-165–Met-173) is similarly stabilized. In D4, additional stabilized residues are found in a stretch from Asp-370 to Thr-381 that contains two β-strands, and in the solvent-exposed helices from Lys-385–Leu-392 and Leu-441–Arg-449 (Fig. 3 B). D1 manifests phosphorylation-dependent stabilization ΔΔGHX,P-deP in a β-strand that faces the active site and phosphoSer-108 (Asn-115–Ile-119), in the adjoining loops, and at the N-terminal ends of two helices within the Val-23–Glu-30 and Leu-59–Lys-66 segments (Fig. 3 B). In total, ∼15 residues of D1 within 20 Å of the phosphoryl group of phosphoSer-108 are protected by phosphorylation (Fig. 3 B).
Twenty amide groups of Apo-P, mainly in D1 and D3, were found to exchange so slowly that the full amplitude of their ΔGHX values could not be determined due to the gradual, concurrent dephosphorylation of the sample (τ = 19.0 h; see above and Fig. S2). Consequently, it is the lower bound for ΔGHX that is estimated for these 20 groups (open squares in Fig. S6) after correction for the slow dephosphorylation (Fig. S8 A). Since the HX of Apo-P and Apo-deP lies in the EX2 regime, the most slowly exchanging sites with the largest ΔGHX values can be used to estimate folding stability (44). Applying this method to PMM suggests at the very least a 0.4 kcal/mol greater folding stability of Apo-P (≥11.0 kcal/mol) compared with Apo-deP (10.6 kcal/mol). Local stabilizations by phosphorylation are much greater (Fig. 3 A), especially in D3 and D4, where Apo-P appears to be stabilized by at least 1.3 kcal/mol over Apo-deP (Fig. S9). The increased stability from phosphorylation may accompany increases in the conformational rigidity of the protein. This is consistent with the dozens of peaks of Apo-deP that are lost or weakened by line broadening (Fig. S3). These observations may be attributable to chemical exchange among conformers at locations in D3 and D1 in the absence of phosphorylation.
In contrast to the above behaviors, we find sites in D1 and D2 that are locally destabilized by phosphorylation. Specifically, nine residues in D1 and 11 in D2 show HX accelerated by phosphorylation. At least five of these residues in D1 and seven in D2 are destabilized by >1.5 kcal/mol (Fig. 3 A). The bulk of the phospho-destabilization in D1 is found within the first helix and the subsequent two loops (Fig. 3). Phosphorylation-destabilized residues are scattered throughout D2.
The NMR studies presented here provide a new opportunity to assess HX around the active site of PMM/PGM. One of the D2 residues destabilized by phosphorylation is Asp-242, which coordinates the divalent cation in the active site. HDX by NMR shows that Asp-242 is well protected in Apo-deP, with ΔGHX,deP = 7.3 ± 0.3 kcal/mol (Fig. 3 A). However, it is strongly destabilized in Apo-P, where its HX is rapid and detected by CLEANEX-PM. This might suggest partial repulsion between the phosphoryl and aspartyl carboxylate groups, which could perturb the metal-binding loop. Ala-184 of D2 and Ile-343 of D3, both within 5 Å of the metal-binding loop, are destabilized in Apo-P as well. In contrast, phosphorylation of Ser-108 stabilizes against HX the amide groups of neighboring residues Thr-106, Asn-115, and Gly-116 of D1, as well as Val-183 and Arg-247 of D2, each within 8 Å of Ser-108.
Phosphorylation attracts D4, whereas dephosphorylation repels D4
The pattern of increased protection from HX due to phosphorylation led to the hypothesis that the negative charge of phosphoSer-108 at the base of the catalytic cleft could 1) attract the positively charged walls of D1 and D4 surrounding the cleft; and 2) draw movable D4 closer to phosphoSer-108 and D3. We tested this by calculating electrostatic field lines and MD trajectories for structures of both Apo-P and Apo-deP. First, we carefully simulated the partial charges on the atoms around the divalent cation-binding site, and near Ser-108 with and without phosphorylation. Many of the electrostatic interactions between domains revealed by the field lines are similar between Apo-P and Apo-deP. However, the phosphorylation of Ser-108 in D1 profoundly alters at long range the charge on the face of D4 and the negative charge in D1 and D2, as evidenced by field lines that converge to the negative potential near the metal-binding site near Val-248 and Gly-249 in D2 (Fig. 4 C). In contrast, Apo-deP switches to electrostatic repulsion between these sites in D2 and D4 (Fig. 4 D) due to the increased positive charge of its catalytic cleft and metal-binding region. This gives rise to the diverging field lines that are characteristic of repulsion (Fig. 4 D). Several other charged residues on the surface of Apo-P appear to form favorable long-range interactions between D1 and D4 or between D2 and D4, which are absent in Apo-deP. Such favorable field lines in Apo-P, which are lost in Apo-deP, include those between Ala-11 (+) and Asp-383 (−), between Thr-426 (+) and both Val-22 and Asp-113 (−), and between Thr-426 (+) and both Phe-210 and Glu-221 (−) (Fig. 4, A and B). Since D4 is attracted to D1 and D2 in Apo-P, it is likely to contact the intervening D3 more snugly when it rotates toward D1 and D2.
Figure 4.
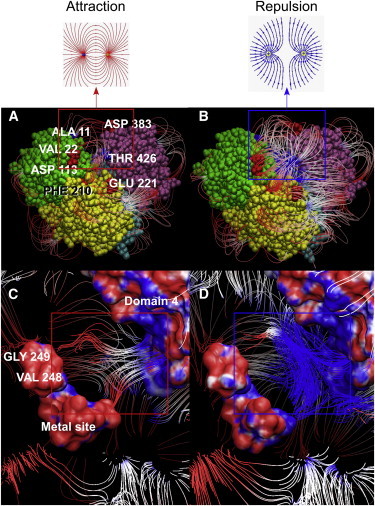
(A–D) D1 and D4 are electrostatically attracted when Ser-108 is phosphorylated (A and C), but repelled when it is dephosphorylated (B and D). (A and B) D1–D4 are colored green, yellow, cyan, and pink, respectively. (A–D) Electrostatic field lines of Apo-P (A and C) and Apo-deP (B and D). (C and D) Expansions of the catalytic cleft highlight attraction and repulsion between D1 and D4 in the red and blue boxes, respectively. To see this figure in color, go online.
MD suggests compaction due to phosphorylation and more freedom without it
To test the working hypotheses that 1) D3 and proximal areas of the other domains are stabilized in their H-bonding (higher ΔGHX); and 2) phosphorylation draws D4 with D3, we compared Apo-P and Apo-deP forms by MD. The force field integrates the long-range electrostatics with van der Waals and covalent interactions. The crystal structures of Apo-P and Apo-deP share identical angles of orientation (73.2°) and distances of separation (28.2 Å) of D4 as defined by Fig. 5, A and D. The energies and root mean-square deviations from the starting structures stabilized in 5 ns. The distributions of angles and distances were tabulated from this equilibration through the end of the 120 ns trajectories (Fig. 5).
Figure 5.
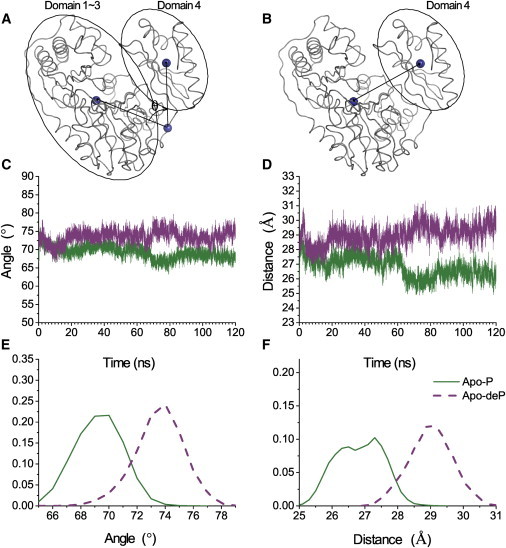
D4 opens wider in Apo-deP than in Apo-P in MD simulations. (A, C, and E) Angle (θ) between the centers of mass (spheres in A) of D1–D3 and D4. (B, D, and F) Distances between the center of mass of D4 and the metal site (spheres in B). (C) The instantaneous angle between the centers is plotted. (D) Plot showing the instantaneous separation between the centers of mass in panel D. (E) Histogram marking the frequency with which the angle occurs over the equilibrated portions of the MD trajectories from 5 to 120 ns. (F) Histogram plotting the frequency with which the separation occurs between 5 and 120 ns. Green represents the Apo-P simulation and purple represents Apo-deP. To see this figure in color, go online.
After 18 ns and 60 ns in the simulation, Apo-deP increased in separation of D4 from D1–D3 and from Apo-P (Fig. 5). In Apo-P, the angle defined between the centers of mass of D4 and D1–D3 with a vertex at the D3-D4 hinge (Fig. 5 A) averaged 69.3° ± 1.7° (Fig. 5, C and E). In Apo-deP, this angle averaged 73.6 ± 1.7° (i.e., more than 4° wider). In Apo-P, the distances between the catalytic metal site and the center of mass of D4 (Fig. 5 B) averaged 26.8 ± 0.7 Å, with double maxima around 26.5 and 27.5 Å in the histogram and greater compaction after 60 ns (Fig. 5, D and F). The distance of separation in Apo-deP averaged 29 ± 0.7 Å (i.e., often more than 2 Å greater than that in Apo-P; Fig. 5, D and F). D4 of Apo-deP also manifested greater movement relative to D3 by an orthogonal mode not shown. The radius of gyration was also larger in Apo-deP (Fig. S10). Although the crystal structures of Apo-P and Apo-deP differ very little (19), the MD simulations imply that Apo-deP samples a wider range of conformations in solution than does Apo-P. We also reproduced the MD results for Apo-deP by starting with the Apo-P crystal structure, after removing its phosphoryl group and recalculating the surrounding partial charges. Further simulations of the S108E mutant (a partial phosphomimetic modeled from the Apo-deP coordinates of PDB 4MRQ) resulted in a compact form like that of Apo-P, although with a smaller angle of separation of D4 of 66–67°. The greater separation and freedom of D4 in Apo-deP simulations strongly support the hypothesis that phosphorylation of Ser-108 electrostatically attracts D4, limiting its mobility. This electrostatic compaction by phosphorylation may be called electrostriction, a behavior of dielectric materials that is well known in materials science and acknowledged in biomacromolecules (45).
Phosphorylation effects on simulated backbone mobility and HX
The backbone RMSFs in MD simulations of Apo-P and Apo-deP are generally similar (Fig. 6 A) but for the following exceptions: phosphorylation enhances the simulated backbone rigidity in D2 near phosphoSer-108, the core of D3, part of the D3 interface with D4, and the face of D4 toward the catalytic cleft and D3. Surprisingly, phosphorylation appears to subtly increase backbone mobility in parts of D1.
Figure 6.
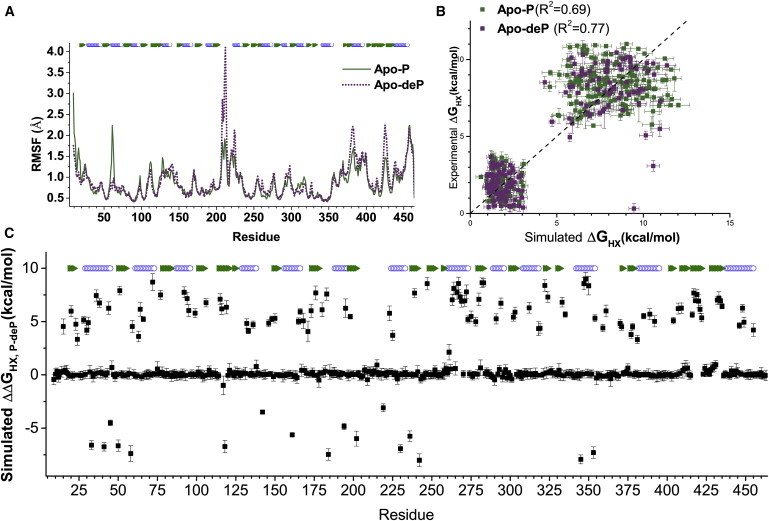
Simulated phosphorylation-dependent changes in free energies of HX, derived from the MD trajectories, agree with the locations of experimentally measured changes. (A) Backbone RMSFs of Apo-P and Apo-deP are compared. (B) Measured ΔGHX is plotted against ΔGHX simulated from each MD trajectory using values simulated using Eq. 3 and the pairs of βc and β1/RMSF coefficients in the text and at the bottom of Table S4. (C) The differences in the simulated ΔGHX modeled from the MD trajectories of Apo-P and Apo-deP are plotted against the sequence position for comparison with Fig. 3A. To see this figure in color, go online.
Previous studies have correlated HX protection factors with dynamics simulations via a linear combination of the fluctuating contacts with either H-bonds or RMSF−1 (23,24). Using three of the same proteins studied previously, we found that we could obtain improved correlations by optimizing and applying separate sets of coefficients of Eq. 3 for rapid and slow regimes of HX (Table S3), as shown by a comparison of the previous and new analyses of the HX data for staphylococcal nuclease (Fig. S11). We optimized the coefficients of Eq. 3 for the number of contacts and RMSF−1 as described previously (24) for agreement with the HX rates of PMM/PGM across the 120 ns MD trajectories, but with the enhancement of two sets of coefficients. The coefficients obtained are βc = 0.22 and β1/RMSF = 0.28 for the less protected amide groups. The coefficients are βc = 0.56 and β1/RMSF = 3.94 for the more protected residues (Table S4). We used these coefficients to simulate the protection factors and free energies of HX from the MD trajectories for both the rapid and slow cases of HX (Fig. 6 B). Differences in these simulated free energies between the phosphorylated and dephosphorylated states are plotted in Fig. 6 C. Significant increases in and in Apo-P are based on increased contacts and H-bonding, and lower RMSFs at those sites in the MD trajectory (see Eq. 3 above). Although the magnitudes of the simulated large protection factors (free energies in the EX2 regime) obtained by this approach are not quantitatively reliable (23) (Fig. 6 B), the locations and patterns of protection and deprotection by phosphorylation (Fig. 6 C) are in a qualitative sense remarkably consistent with the locations measured to have a strong dependence of HX on phosphorylation status (Fig. 3 A). The simulated increases of ΔΔGHX,P-deP identify more than 80% of the sites with measured increases (Figs. 3 A and 6 C), i.e., 85% of the amide groups with increased protection in D1, 87% in D2, 83% in D3, and 81% in D4. The simulated decreases identify all 14 residues in D1, D2, and D3, with ΔΔGHX,P-deP being decreased by >2 kcal/mol (Figs. 3 A and 6 C). Thus, the differences between the MD trajectories of Apo-P and Apo-deP agree with the distribution of not only the HX slowed by phosphorylation (densest between D4 and D3) but also the phosphorylation-hastened HX in D1–D3.
Discussion
The studies described herein reveal the extent of the increased rigidity of PMM/PGM due to phosphorylation, revising a recent qualitative assessment (19) with residue-specific and quantitative detail. The computational studies agree with the sites of phosphorylation-increased and -decreased protection from HX, and furnish strong evidence that the structural compaction is electrostatic in nature.
Structural compaction by phosphorylation
Dephosphorylation partially frees D4 in MD simulations, typically increasing its separation from the center of PMM/PGM by 1 Å to 4 Å (Fig. 5, D and F). This is consistent with the 1 Å larger radius of gyration of the dephosphorylated form suggested by small-angle x-ray scattering (19). Expansion of Apo-deP agrees with the increase of simulated backbone fluctuations at the D3-D4 interface (Fig. 6), faster HX at this interface, destabilization of D3 and two β-strands of D4 facing D3 (Fig. 3), and NMR line-broadening evidence of a conformational exchange in these regions (Fig. S3). The lack of expansion of Apo-deP in its crystal structure (19) was attributed to restriction of the crystal lattice. Without this constraint, all of the solution measurements and simulations agree with Apo-deP being loosened and expanded. The compaction of Apo-P may now be attributed to the electrostatic attraction of the positive face of D4 for phosphoSer-108 and adjoining electronegative surfaces of D2 (Fig. 4 C), i.e., electrostriction. The closer packing of D4 against D3 and superficially with D1 (Fig. 7 B) must result from this, accounting for the longer-lived H-bonds and enhanced stability in these regions.
Figure 7.
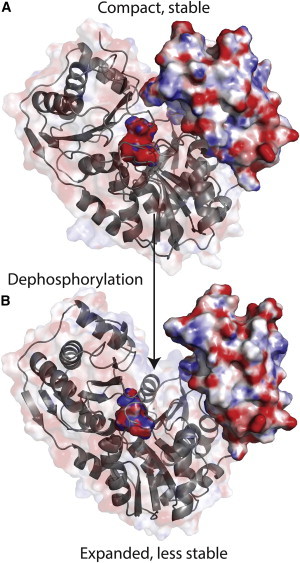
Expansion of PMM/PGM accompanying its dephosphorylation. The surfaces colored by electrostatics are plotted for D4 and around Ser-108 at the base of the cleft. (A) Phosphorylation of Ser-108 stabilizes the domains of PMM/PGM through electrostatic attraction of D4. Apo-P at 82 ns in the MD trajectory is plotted. (B) Transient dephosphorylation during the catalytic cycle appears to free D4 partially, resulting in an expanded structure with less rigidity in the D3–D4 region. Apo-deP at 117 ns in its MD trajectory is plotted. To see this figure in color, go online.
Catalytic relevance of compaction by phosphorylation and loosening by dephosphorylation
The phosphoryl group at Ser-108 of PMM/PGM forms H-bonds with hydroxyl groups of substrates (17) and is transferred to and from the hexose phosphate substrates during catalysis (5,6) (Fig. 1). The evidence presented here suggests that the phosphoryl group also serves to narrow the catalytic cleft of the enzyme (Fig. 7). This may help position phosphoSer-108 and the substrate for H-bond formation and subsequent phosphoryl transfer to the 1- or 6-hydroxyl group of the substrate.
The unusual 180° rotation of G16P and mannose 1,6-bisphosphate intermediates, which occurs in the middle of the catalytic cycle during association with PMM/PGM (8), may also be facilitated by changes in the dynamics of the transiently dephosphorylated enzyme. Measurements of accessibility to probes (Fig. 2), HX (Figs. 3 and S6), radius of gyration (19), electrostatics (Fig. 4), and MD simulations (Figs. 5, 6, and 7) establish that Apo-deP has a wider catalytic cleft and is looser overall, except for small portions of D1 and D2 that may gain rigidity upon dephosphorylation (Figs. 3 and 6; Table S2). Expansion and opening of the cleft could help accommodate the catalytically required 180° reorientation of the bisphosphorylated intermediate.
The stabilization of the active site of PMM/PGM by phosphorylation is less than that at distal sites (Fig. 3). Despite coordination of Mg2+ by three aspartate side chains in the active site, phosphorylation of Ser-108 nearby might introduce a degree of repulsion from these three carboxylate groups. This may account for the phospho-destabilization of Mg2+ ligand Asp-242, a few neighboring residues, and other residues in D2. Locally diminished stability near the active site due to phosphorylation required for enzyme activity could be reminiscent of observed trends of trade-offs between stability and function, in which clefts and their key residues for catalysis tend to destabilize the protein fold (46,47).
Insights into HX by NMR beyond qualitative HDX-MS
The NMR studies reveal that the HX behavior within PMM/PGM is more complex and varied than was previously appreciated. The most obvious trend of phosphorylation-increased rigidity and stability to HX is clearly long in range, with the distant core of D3 and proximal face of D4 being especially shielded by Ser-108 phosphorylation (Fig. 3). However, the residue-by-residue accounting provided by NMR also implicates ∼99 residues with HX unaffected by phosphorylation and 24 residues with HX increased by phosphorylation (Fig. 3). This is corroborated by comparison with the changes in HX protection factors (free energies) simulated from the MD trajectories (Fig. 6 C). Among the 237 residues for which ΔΔGHX,P-deP comparisons of HX are available, 48% represent significant slowing by phosphorylation, 10% undergo acceleration by phosphorylation, and 42% have little dependence on phosphorylation (Fig. 3 A). The computational results are consistent with this, but suggest that a much higher proportion of residues are unaffected by phosphorylation (Fig. 6 C). Thus, in light of the NMR and computational data, the HDX-MS-based proposal that flexibility increases of PMM/PGM upon dephosphorylation are global (19) should not be construed as universal or comprehensive. Six of the peptides monitored by HDX-MS are shown by NMR to have at least two residues each with increased HX upon Ser-108 phosphorylation (Table S2). Four more peptides have single residues accelerated in HX by NMR. Most of the regions with HX expedited by phosphorylation map to portions of D1 and D2, including Asp-242 at the active site (Figs. 3 and 6 C; Movie S1). Observation of the varying and nuanced HX behaviors, including local destabilizations within this multidomain enzyme, required the residue resolution and quantification of NMR (enhanced by subsecond and long timescales). The phosphorylation-induced changes in rigidity are long in range, with widespread increases and localized decreases in stability.
To our knowledge, the NMR methods have also provided the first detailed HX insight into functionally important loops within the active site of PMM/PGM, where HDX-MS did not contribute. For example, HDX by NMR identified the residues flanking Ser-108 in sequence as being stabilized by its phosphorylation (Fig. 3), whereas MS failed to detect the peptide containing Ser-108 when it was phosphorylated (92–117 in Table S2). Neighboring residues in D2 and D3 that were destabilized by phosphoSer-108 were already noted (Fig. 3). NMR also revealed key ligand-binding residues protected by phosphorylation in two other peptides: 1) the C-terminal half of HDX-MS peptide 14–27 (containing key Arg-20 at the active site); and 2) the middle third of peptide 407–429 near Arg-421 and Ser-423 that binds the phosphate group of ligands.
Using NMR, we were able to quantify the rates and thermodynamics of HX of specific amide groups, which allowed us to examine the stabilities of individual H-bonds, domains, and the enzyme as a whole. This revealed phosphorylation-dependent increases in stability that are locally large for individual H-bonds (>4 kcal/mol at many sites in D1, D3, and D4), moderate in the folding stability of D3 and D4 (≥1.3 kcal/mol), and more subtle for the enzyme as a whole (≥0.4 kcal/mol) (Figs. 3, S6, and S9). Along with localized variations in stability (Figs. 3 and 6; Movies S1 and S2), these findings provide a complex but unified biophysical portrayal. The combination of HX methods and dynamics simulations (Fig. 3 and 6) may have wider applicability for appreciating the varied effects of phosphorylation on proteins.
Long-range effects of phosphorylation on HDX of enzymes
Phosphorylation of Ser-108 slowed HDX to the farthest reaches of D2–D4 (Fig. 3). Moreover, the addition of two negative charges at the site of phosphorylation attracted D4 in both static and dynamic simulations, resulting in domain rotation about the flexible hinge between D3 and D4 (Figs. 5, 6, and 7). These results are reminiscent of those due to phosphorylation in other systems. For example, phosphorylation of the activation segment of p38 MAP kinase at two sites or protein kinase A (PKA) at a single site analogously slowed HDX of loops at long range and locally in the activation segment, and increased the folding stability of PKA (48,49). In the protein kinase ERK2, dual phosphorylation of the activation segment also induced long-range changes in HDX (mostly small decreases) as well as increases in four peptides, suggesting functionally relevant mobility in its ATP-binding loop and activation segment (50). The results for PMM/PGM suggest that the trend of phosphorylation restricting motions at long range while enabling some motions closer to the active site may be shared among some members of the protein kinase and α-D-phosphohexomutase superfamilies, which are mechanistically very different phosphoryl transfer enzymes. Mobility appears to modulate substrate access to protein kinases and the 180° reorientation of the intermediate in dephosphorylated PMM/PGM.
Conclusions
Our calculations strongly suggest that phosphorylation of Ser-108 in D1 electrostatically attracts D4, narrowing the catalytic cleft and breadth of PMM/PGM. This electrostriction accounts for the properties of the phosphorylated form (Apo-P) demonstrated above, i.e., less accessibility to probes, enhanced folding stability, stabilization of many H-bonds by >4 kcal/mol, much slowing of HX with stability increases of >1.3 kcal/mol for D3 and the adjacent face of D4, and increased rigidity in the stabilized D3-D4 interface. Narrowing of the active-site cleft due to phosphorylation probably aids capture of phosphohexose substrates, whereas opening upon dephosphorylation aids rotation of the bisphosphorylated intermediate. NMR and MD simulations paint a more complex, varied, and detailed landscape of HX changes than does peptide-resolved HDX-MS. The proposal that flexibility increases in the dephosphorylated form (Apo-deP) are global (19) must be tempered by the observation of a large number of unaffected sites and some sites with dephosphorylation-induced slowing of HX, especially in D1 and D2, which is also supported by MD trajectories. The rigidity increases upon phosphorylation are widespread, being mostly concentrated in D3 and D4, which draw closer upon phosphorylation. The ∼100 sites with phosphorylation-promoted stability increases, as well as the stability decreases in D1 and D2, are truly long-range effects that reach the most distant flanks of the enzyme. Such behaviors parallel HDX-MS characterizations of protein kinases, suggesting analogous long-range effects of phosphorylation in both superfamilies of multidomain enzymes.
Acknowledgments
We thank A. Hopkins for enzyme preparations and S. Jiang for assistance with interleaving of the CLEANEX pulse sequence.
This work was supported by National Science Foundation grants MCB 0918389 and 1409898. National Institutes of Health grant S10RR022341 contributed to the purchase of the 800 MHz NMR spectrometer.
Supporting Material
Time course of hydrogen-deuterium exchange of Apo-deP. Yellow is >90% exchanged. Orange is 10–90% exchanged. Red is <10% exchanged.
Time course of hydrogen-deuterium exchange of Apo-P. Yellow is >90% exchanged. Orange is 10–90% exchanged. Red is <10% exchanged.
References
- 1.Johnson L.N., Lewis R.J. Structural basis for control by phosphorylation. Chem. Rev. 2001;101:2209–2242. doi: 10.1021/cr000225s. [DOI] [PubMed] [Google Scholar]
- 2.Taylor S.S., Keshwani M.M., Kornev A.P. Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012;367:2517–2528. doi: 10.1098/rstb.2012.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xin F., Radivojac P. Post-translational modifications induce significant yet not extreme changes to protein structure. Bioinformatics. 2012;28:2905–2913. doi: 10.1093/bioinformatics/bts541. [DOI] [PubMed] [Google Scholar]
- 4.Johnson L.N., O’Reilly M. Control by phosphorylation. Curr. Opin. Struct. Biol. 1996;6:762–769. doi: 10.1016/s0959-440x(96)80005-4. [DOI] [PubMed] [Google Scholar]
- 5.Naught L.E., Tipton P.A. Kinetic mechanism and pH dependence of the kinetic parameters of Pseudomonas aeruginosa phosphomannomutase/phosphoglucomutase. Arch. Biochem. Biophys. 2001;396:111–118. doi: 10.1006/abbi.2001.2618. [DOI] [PubMed] [Google Scholar]
- 6.Regni C., Tipton P.A., Beamer L.J. Crystal structure of PMM/PGM: an enzyme in the biosynthetic pathway of P. aeruginosa virulence factors. Structure. 2002;10:269–279. doi: 10.1016/s0969-2126(02)00705-0. [DOI] [PubMed] [Google Scholar]
- 7.Ray W.J., Jr., Roscelli G.A. A kinetic study of the phosphoglucomutase pathway. J. Biol. Chem. 1964;239:1228–1236. [PubMed] [Google Scholar]
- 8.Naught L.E., Tipton P.A. Formation and reorientation of glucose 1,6-bisphosphate in the PMM/PGM reaction: transient-state kinetic studies. Biochemistry. 2005;44:6831–6836. doi: 10.1021/bi0501380. [DOI] [PubMed] [Google Scholar]
- 9.Regni C., Schramm A.M., Beamer L.J. The reaction of phosphohexomutase from Pseudomonas aeruginosa: structural insights into a simple processive enzyme. J. Biol. Chem. 2006;281:15564–15571. doi: 10.1074/jbc.M600590200. [DOI] [PubMed] [Google Scholar]
- 10.Ballok A.E., O’Toole G.A. Pouring salt on a wound: Pseudomonas aeruginosa virulence factors alter Na+ and Cl− flux in the lung. J. Bacteriol. 2013;195:4013–4019. doi: 10.1128/JB.00339-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X.J., Li Q., Yuan Q.Y. Bacteriological differences between patients with acute exacerbation of COPD and community-acquired pneumonia. Respir. Care. 2011;56:1818–1824. doi: 10.4187/respcare.00915. [DOI] [PubMed] [Google Scholar]
- 12.Govan J.R.W., Deretic V. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 1996;60:539–574. doi: 10.1128/mr.60.3.539-574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye R.W., Zielinski N.A., Chakrabarty A.M. Purification and characterization of phosphomannomutase/phosphoglucomutase from Pseudomonas aeruginosa involved in biosynthesis of both alginate and lipopolysaccharide. J. Bacteriol. 1994;176:4851–4857. doi: 10.1128/jb.176.16.4851-4857.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.King J.D., Kocíncová D., Lam J.S. Review: lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate Immun. 2009;15:261–312. doi: 10.1177/1753425909106436. [DOI] [PubMed] [Google Scholar]
- 15.Olvera C., Goldberg J.B., Soberón-Chávez G. The Pseudomonas aeruginosa algC gene product participates in rhamnolipid biosynthesis. FEMS Microbiol. Lett. 1999;179:85–90. doi: 10.1111/j.1574-6968.1999.tb08712.x. [DOI] [PubMed] [Google Scholar]
- 16.Remminghorst U., Rehm B.H. Bacterial alginates: from biosynthesis to applications. Biotechnol. Lett. 2006;28:1701–1712. doi: 10.1007/s10529-006-9156-x. [DOI] [PubMed] [Google Scholar]
- 17.Regni C., Naught L., Beamer L.J. Structural basis of diverse substrate recognition by the enzyme PMM/PGM from P. aeruginosa. Structure. 2004;12:55–63. doi: 10.1016/j.str.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 18.Sarma A.V., Anbanandam A., Van Doren S.R. Solution NMR of a 463-residue phosphohexomutase: domain 4 mobility, substates, and phosphoryl transfer defect. Biochemistry. 2012;51:807–819. doi: 10.1021/bi201609n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee Y., Villar M.T., Beamer L.J. Promotion of enzyme flexibility by dephosphorylation and coupling to the catalytic mechanism of a phosphohexomutase. J. Biol. Chem. 2014;289:4674–4682. doi: 10.1074/jbc.M113.532226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coales S.J., E S.Y., Hamuro Y. Expansion of time window for mass spectrometric measurement of amide hydrogen/deuterium exchange reactions. Rapid Commun. Mass Spectrom. 2010;24:3585–3592. doi: 10.1002/rcm.4814. [DOI] [PubMed] [Google Scholar]
- 21.Mayne L., Kan Z.-Y., Englander S.W. Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method. J. Am. Soc. Mass Spectrom. 2011;22:1898–1905. doi: 10.1007/s13361-011-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huyghues-Despointes B.M., Scholtz J.M., Pace C.N. Protein conformational stabilities can be determined from hydrogen exchange rates. Nat. Struct. Biol. 1999;6:910–912. doi: 10.1038/13273. [DOI] [PubMed] [Google Scholar]
- 23.Vendruscolo M., Paci E., Karplus M. Rare fluctuations of native proteins sampled by equilibrium hydrogen exchange. J. Am. Chem. Soc. 2003;125:15686–15687. doi: 10.1021/ja036523z. [DOI] [PubMed] [Google Scholar]
- 24.Kieseritzky G., Morra G., Knapp E.-W. Stability and fluctuations of amide hydrogen bonds in a bacterial cytochrome c: a molecular dynamics study. J. Biol. Inorg. Chem. 2006;11:26–40. doi: 10.1007/s00775-005-0041-1. [DOI] [PubMed] [Google Scholar]
- 25.Cellini B., Montioli R., Voltattorni C.B. Molecular defects of the glycine 41 variants of alanine glyoxylate aminotransferase associated with primary hyperoxaluria type I. Proc. Natl. Acad. Sci. USA. 2010;107:2896–2901. doi: 10.1073/pnas.0908565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lescop E., Schanda P., Brutscher B. A set of BEST triple-resonance experiments for time-optimized protein resonance assignment. J. Magn. Reson. 2007;187:163–169. doi: 10.1016/j.jmr.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 28.Goddard T.D., Kneller D.G. University of California; San Francisco, San Francisco: 2000. SPARKY. [Google Scholar]
- 29.Hwang T.L., van Zijl P.C., Mori S. Accurate quantitation of water-amide proton exchange rates using the phase-modulated CLEAN chemical EXchange (CLEANEX-PM) approach with a Fast-HSQC (FHSQC) detection scheme. J. Biomol. NMR. 1998;11:221–226. doi: 10.1023/a:1008276004875. [DOI] [PubMed] [Google Scholar]
- 30.Hernández G., LeMaster D.M. Relaxation compensation in chemical exchange measurements for the quantitation of amide hydrogen exchange in larger proteins. Magn. Reson. Chem. 2003;41:699–702. [Google Scholar]
- 31.Bertini I., Ghosh K., Vasos P.R. A high-resolution NMR study of long-lived water molecules in both oxidation states of a minimal cytochrome c. Biochemistry. 2003;42:3457–3463. doi: 10.1021/bi0272961. [DOI] [PubMed] [Google Scholar]
- 32.Peters M.B., Yang Y., Merz K.M., Jr. Structural survey of zinc containing proteins and the development of the zinc AMBER force field (ZAFF) J. Chem. Theory Comput. 2010;6:2935–2947. doi: 10.1021/ct1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frisch M.J., Trucks G.W., Fox D.J. Gaussian, Inc.; Wallingford, CT: 2009. Gaussian 09, revision B.01. [Google Scholar]
- 34.Hornak V., Abel R., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baker N.A., Sept D., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–38. [DOI] [PubMed] [Google Scholar]
- 37.Yamasaki K., Daiho T., Suzuki H. Roles of long-range electrostatic domain interactions and K+ in phosphoenzyme transition of Ca2+-ATPase. J. Biol. Chem. 2013;288:20646–20657. doi: 10.1074/jbc.M113.482711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Craddock T.J., Tuszynski J.A., Hameroff S. Cytoskeletal signaling: is memory encoded in microtubule lattices by CaMKII phosphorylation? PLOS Comput. Biol. 2012;8:e1002421. doi: 10.1371/journal.pcbi.1002421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schonbrunn E., Eschenburg S., Amrhein N. Structural basis for the interaction of the fluorescence probe 8-anilino-1-naphthalene sulfonate (ANS) with the antibiotic target MurA. Proc. Natl. Acad. Sci. USA. 2000;97:6345–6349. doi: 10.1073/pnas.120120397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bai Y., Sosnick T.R., Englander S.W. Protein folding intermediates: native-state hydrogen exchange. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Linderstrøm-Lang K. Deuterium exchange between peptides and water. Chem. Soc. (Lond.). Spec. Publ. 1955;2:1–20. [Google Scholar]
- 42.Yan S., Kennedy S.D., Koide S. Thermodynamic and kinetic exploration of the energy landscape of Borrelia burgdorferi OspA by native-state hydrogen exchange. J. Mol. Biol. 2002;323:363–375. doi: 10.1016/s0022-2836(02)00882-3. [DOI] [PubMed] [Google Scholar]
- 43.Liang X., Lee G.I., Van Doren S.R. Partially unfolded forms and non-two-state folding of a beta-sandwich: FHA domain from Arabidopsis receptor kinase-associated protein phosphatase. J. Mol. Biol. 2006;364:225–240. doi: 10.1016/j.jmb.2006.08.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huyghues-Despointes B.M., Langhorst U., Scholtz J.M. Hydrogen-exchange stabilities of RNase T1 and variants with buried and solvent-exposed Ala —> Gly mutations in the helix. Biochemistry. 1999;38:16481–16490. doi: 10.1021/bi9919450. [DOI] [PubMed] [Google Scholar]
- 45.Marky L.A., Kupke D.W. Enthalpy-entropy compensations in nucleic acids: contribution of electrostriction and structural hydration. Methods Enzymol. 2000;2000:419–441. doi: 10.1016/s0076-6879(00)23376-4. [DOI] [PubMed] [Google Scholar]
- 46.Beadle B.M., Shoichet B.K. Structural bases of stability-function tradeoffs in enzymes. J. Mol. Biol. 2002;321:285–296. doi: 10.1016/s0022-2836(02)00599-5. [DOI] [PubMed] [Google Scholar]
- 47.Liang X., Arunima A., Van Doren S.R. Apparent tradeoff of higher activity in MMP-12 for enhanced stability and flexibility in MMP-3. Biophys. J. 2010;99:273–283. doi: 10.1016/j.bpj.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sours K.M., Kwok S.C., Ahn N.G. Hydrogen-exchange mass spectrometry reveals activation-induced changes in the conformational mobility of p38α MAP kinase. J. Mol. Biol. 2008;379:1075–1093. doi: 10.1016/j.jmb.2008.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steichen J.M., Iyer G.H., Taylor S.S. Global consequences of activation loop phosphorylation on protein kinase A. J. Biol. Chem. 2010;285:3825–3832. doi: 10.1074/jbc.M109.061820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoofnagle A.N., Resing K.A., Ahn N.G. Changes in protein conformational mobility upon activation of extracellular regulated protein kinase-2 as detected by hydrogen exchange. Proc. Natl. Acad. Sci. USA. 2001;98:956–961. doi: 10.1073/pnas.98.3.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time course of hydrogen-deuterium exchange of Apo-deP. Yellow is >90% exchanged. Orange is 10–90% exchanged. Red is <10% exchanged.
Time course of hydrogen-deuterium exchange of Apo-P. Yellow is >90% exchanged. Orange is 10–90% exchanged. Red is <10% exchanged.