

Abstract
Total internal reflection fluorescence microscope has often been used to study the molecular mechanisms underlying vesicle exocytosis. However, the spatial occurrence of the fusion events within a single cell is not frequently explored due to the lack of sensitive and accurate computer-assisted programs to analyze large image data sets. Here, we have developed an image analysis platform for the nonbiased identification of different types of vesicle fusion events with high accuracy in different cell types. By performing spatiotemporal analysis of stimulus-evoked exocytosis in insulin-secreting INS-1 cells, we statistically prove that individual vesicle fusion events are clustered at hotspots. This spatial pattern disappears upon the disruption of either the actin or the microtubule network; this disruption also severely inhibits evoked exocytosis. By demonstrating that newcomer vesicles are delivered from the cell interior to the surface membrane for exocytosis, we highlight a previously unappreciated mechanism in which the cytoskeleton-dependent transportation of secretory vesicles organizes exocytosis hotspots in endocrine cells.
Introduction
In secretory cells such as neurons and endocrine cells, transient depolarization induces Ca2+ entry, followed by the rapid fusion of secretory vesicles with the plasma membrane, thus releasing neurotransmitters and hormones to mediate important physiological processes (1). Electrophysiological techniques, such as membrane capacitance measurements and amperometric recordings, can detect fusion of single vesicles with high temporal resolution (2). By using a combination of flash photolysis, electron microscopy, and genetic manipulation, many aspects of the molecular mechanism of regulated vesicle exocytosis have been revealed (3). However, electrophysiological methods provide little spatial information about vesicle fusion and cannot observe motions of secretory vesicles before exocytosis. Fluorescent imaging methods can map the spatial profile of discrete exocytic events. Using fluorescent dyes such as acidic orange and FM1-43, exocytosis of acidic vesicles are observed in endocrine and neuronal cells (4,5). By imaging pancreatic islets in extracellular solution containing nonpermeable fluorescence dextrans under two-photon microscopy, secretions buried deep within the pancreatic islets can be detected (6). However, the specificity of these labeling protocols remains doubtful. For example, acidic orange has been found to localize in the acidic compartment not colocalized with granules (7), and extracellular labeling cells with fluorescence dextrans cannot distinguish between exocytosis and endocytosis. Specific labeling of secretory vesicle exocytosis can be achieved by tagging the vesicle luminal cargos or vesicular membrane proteins with genetic-coded fluorescent proteins that change fluorescence intensity at a pH ranged from 5.5 to 7.0, such as pHluorin and Venus (8–10). They are quenched in the acidic vesicular lumen, and become dequenched and brightening in the neutral extracellular solution once the vesicle fusion pore opens, which improves the contrast of secretion signal. Although confocal, spinning-disc confocal, or two-photon microscopy can be used to detect discrete vesicle fusion events (11), the signal/noise ratio (SNR) of such a fluorescence imaging method is compromised due to the relatively large excitation volume along the axial dimension. To further confine the focal illumination volume, total internal reflection fluorescence (TIRF) microscopy was developed (12) and used to study the dynamic behaviors of secretory vesicles before and during exocytosis with excellent contrast and better temporal resolution (4). Subsequently, TIRF microscopy becomes the gold standard method to study both regulated and constitutive vesicle exocytosis in a variety of cell types (13–16).
Despite the widespread application of TIRF microscopy, quantitative analysis of the large amount of data generated by time-lapse imaging poses a challenge. It is almost impossible to manually detect and analyze the hundreds of vesicle fusion events recorded from single cells upon stimulation under a TIRF microscope. Most researchers rely on the manual annotation of a limited number of fusion events. Such analysis is prone to the biases of selection and does not always lead to a statistically supported conclusion. Recently, a few groups have started to develop algorithms that facilitate the identification of vesicle fusion from time-lapse images. For example, Bai et al. and Huang et al. reported programs that enable direct analysis of the docking and fusion kinetics of glucose transporter 4 (GLUT4) storage vesicles (GSVs) (13,17). However, these methods are semiautomatic and require extensively manual inspection and revision of individual events. Sebastian et al. (18) implemented an automated algorithm that extracts the spatial location and onset time of each fusion by a forward subtraction method. Such an algorithm does not fully use the time-sequential information from image stacks. Therefore, although it could detect ∼86% of the true fusion events, the specificity was only 65%. Based on particles tracking and statistical testing of the similarity between candidate events and true fusion events, two other algorithms were proposed, but the rate of false positive events was even higher with noisy images (19,20). Hence, none of these methods is widely used. Furthermore, except for one (18), none of these works take full advantage of the spatial information available to conduct spatial analysis of all vesicle fusion events.
The release of synaptic vesicles in synaptic transmission is spatially confined to presynaptic terminals. Abundant synaptic vesicles cluster at the densely packed presynaptic region (active zone), which is organized around scaffolding proteins, such as ELKS and Rab3-interacting molecule (RIM), and these proteins contribute to the spatial preference (21). Isoforms of these proteins also exist in endocrine cells such as pancreatic β-cells. Although no active zone-like physical structures have been observed in these cells under electron microscopy, secretory vesicles have been found to sequentially fuse at the same spot, and this behavior is proposed to depend on ELKS proteins (22). Nevertheless, whether vesicles repeatedly fuse at preferred locations on the plasma membrane has not been statistically proven. Cytoskeletons may play a role in the spatial coordination of the release of secretory vesicles. By acting either as tracks for the transportation of secretory vesicles (23) or as barriers that prevent collapse of vesicles with the plasma membrane (24), cortical actin has been proposed to facilitate or inhibit regulated exocytosis. Similarly, microtubule cytoskeleton may also participate in secretion processes. Cortical microtubules extend within 100 nm underneath the plasma membrane, and vesicles move along microtubules beneath the plasma membrane until fusing with the plasma membrane (25). Consistently, spontaneous neurotransmitter release in excitatory synapse was increased or inhibited in neuron overexpressing or knock-down of cytoplasmic-linker-associated protein2 (CLASP2), a microtubule-associated protein that targets microtubules to the cell cortex (26). Despite these experiments, how disruption of cytoskeleton network quantitatively affects the spatial preference of fusion events remained to be explored.
Here, by combining two different fusion detection paradigms, we have developed an algorithm to detect vesicle fusion based on a sudden increase in the fluorescence signal after a fusion pore opens. We have demonstrated the superior sensitivity and accuracy of our algorithm in detecting vesicle fusion from different cell types. Using this platform, we revealed a tightly orchestrated cortical cytoskeleton network that delivers secretory vesicles for fusion at spatially confined regions in endocrine cells.
Methods
Materials
VAMP2-pHluorin, IRAP-pHluorin, and VAMP2-EGFP plasmids were kindly provided by Prof. Tao Xu. INS-1 and 3T3-L1 cells were cultured as previously described (27,28). Cells were transfected using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Cells were detached using trypsin-EDTA 24 h posttransfection, plated onto poly-L-lysine-coated coverslips, and cultured for 20–28 h before experiments in a 37°C, 5% CO2 incubator. Cytochalasin D (C8273) and nocodazole (M1404) were from Sigma-Aldrich (St. Louis, MO).
Live-cell imaging
All experiments were performed at 37°C. Before an imaging experiment, INS-1 cells were washed and incubated on glass coverslips in a bath solution containing (in mM) 136 NaCl, 4.2 KCl, 2.4 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 4 glucose, 10 HEPES, and 1 L-glutamine (pH 7.4), and the coverslip was placed in a metal chamber mounted on a heated stage. Throughout the study, INS-1 cells were stimulated with a high glucose and high K+ solution containing (in mM) 70 NaCl, 70 KCl, 2.4 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 15 glucose, 10 HEPES, and 1 L-glutamine (pH 7.4) to trigger exocytosis. 3T3-L1 cells were kept in KRBB solution containing (in mM) 129 NaCl, 4.7 KCl, 1.2 KH2PO4, 5 NaHCO3, 10 HEPES, 3 glucose, 2.5 CaCl2, 1.2 MgCl2, and 0.1% bovine serum albumin (pH 7.2); 100 nM insulin was applied later to trigger exocytosis (13).
For actin filament disruption, 20 μM cytochalasin D (CytoD) was added to the culture medium for 20 min before the imaging experiments. For microtubule disruption, 10 μM nocodazole (Nocod) was added to the culture medium for 1 h before the imaging experiments.
The TIRF microscope
For Figs. 1–4, a TIRF microscope equipped with a Sensicam EM682KX (PCO AG, Kelheim, Germany) and a 100×, 1.49 numerical aperture, oil-immersion TIRF objective (Olympus) was used. Images were acquired at 2 Hz. Each pixel was 67 × 67 nm. For Fig. 5, a different TIRF setup was used. We used an Olympus IX81 inverted microscope equipped with a 150 ×, 1.45 numerical aperture, oil-immersion TIRF objective (Olympus). The fluorescence signals were acquired by an electron-multiplying charge-coupled device camera (Andor iXon3 888) at a sampling rate of 6 Hz that was controlled using MetaMorph (Molecular Devices). Each pixel from the Andor camera was 87 × 87 nm. A 473 nm laser was used to excite EGFP/pHluorin.
Figure 1.
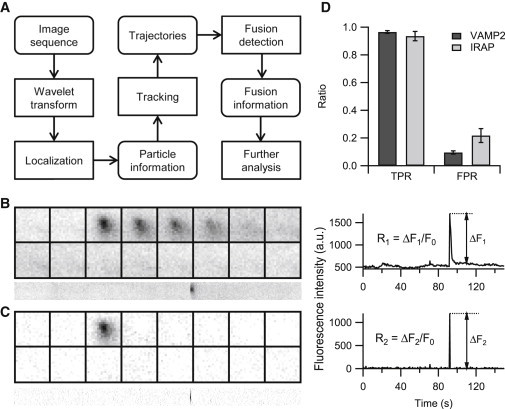
Automatic identification and analysis of fusion events. (A) The framework of the algorithm, comprising five steps (non-rounded rectangles). (B) An example of a typical fusion in an INS-1 cell labeled by VAMP2-pHluorin (left) and its central intensity profile (right). The rapid increase in the central intensity indicates the opening of a fusion pore; thus, the peak/background ratio (parameter R1) can be used to detect candidate events. (C) Consecutive image subtraction of (B) and its central intensity profile; only those peak increases that are greater than the background fluctuation by R2 will be considered candidate events. The montages are 19 × 19 pixels shown at 0.5-s intervals, and the corresponding kymographs (below) are 150 s. (D) The performance of the method for pHluorin-labeled fusion events (n = 1701 events from 6 cells for VAMP2 and n = 531 events from 7 cells for IRAP). R1 and R2 were set to 1.3 and 3 in both situations. Error bars are ±SE.
Figure 2.
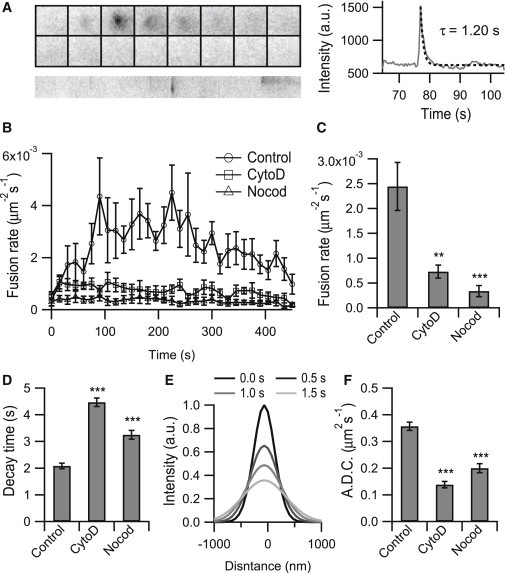
Disruption of the cytoskeleton inhibits secretion and changes the fusion kinetics. (A) An example of a fusion event and the time-dependent fluorescence intensity profile. The montages are 19 × 19 pixels at time intervals of 0.5 s, and the corresponding kymograph (below) is 150 s. The decay time τ of the central intensity was obtained by fitting with an exponential function. (B) Time-dependent changes in the exocytosis rate in control cells and in cells pretreated with either CytoD or Nocod. A KCl and glucose solution was applied to the tested cells at time 0. (C) Average fusion rate for each condition. (n = 1701 events from 6 cells for control, n = 824 events from 6 cells for CytoD treatment, and n = 577 events from 7 cells for Nocod treatment). (D) Average decay time for each condition. (n = 911 events from 6 cells for control, n = 622 events from 6 cells for CytoD treatment, and n = 331 events from 7 cells for Nocod treatment). (E) Intensity profiles of an example fusion event at different times fitted by Gaussian functions. (F) Average apparent diffusion coefficients for each condition. (n = 1629 events from 6 cells for control, n = 826 events from 6 cells for CytoD treatment, and n = 467 events from 7 cells for Nocod treatment). Error bars are ±SE, and all statistical significance values are relative to the control. p values were determined using Student’s t-test. ∗∗p < 0.01; ∗∗∗p < 0.001.
Figure 3.
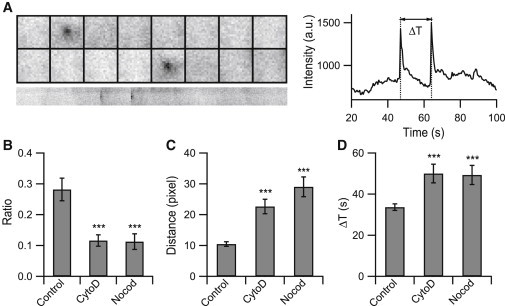
Disruption of the actin or microtubule network reduces the ratio of sequential fusions. (A) An example of sequential fusion and the central intensity profile. The interval between fusions was calculated as ΔT. The montages are 19 × 19 pixels at time intervals of 0.5 s, and the corresponding kymograph (below) is 150 s. (B) The ratio of sequential fusion for each condition. (C) Average minimal distance between each fusion event. In control cells, the minimum distance between each event was 10 pixels (n = 1701 events from 6 cells for control, n = 824 events from 6 cells for CytoD treatment, and n = 577 events from 7 cells for Nocod treatment). (D) Average ΔT for each condition (in B and D, n = 478 events from 6 cells for control, n = 101 events from 6 cells for CytoD treatment, n = 65 events from 7 cells for Nocod treatment). Error bars are ±SE. p values were determined using Student’s t-test. ∗∗∗p < 0.001.
Figure 4.
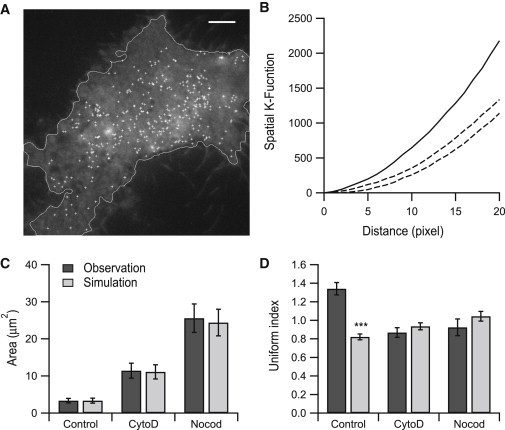
Actin and microtubule networks regulate the spatial pattern of cell secretion. (A) A control cell with all fusion events from a time-lapse series marked on the first frame (crosses). Scale bar, 5 μm. (B) Ripley’s K function of (A). Solid line, observed spatial K-function of fusion events; dashed lines, upper and lower envelopes of 100 simulation results with the same number of random events within the cell boundary as in (A). (C) Average area occupied by each fusion for each condition. (D) Average uniform index for each condition (in C and D, n = 1701 events from 6 cells for control, n = 824 events from 6 cells for CytoD treatment, and n = 577 events from 7 cells for Nocod treatment). Error bars are ±SE. p values were determined using Student’s t-test. ∗∗∗p < 0.001.
Figure 5.
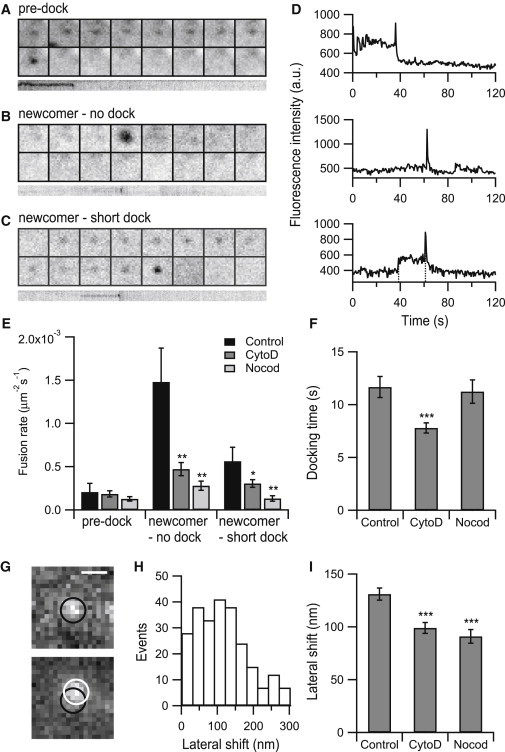
Disruption of the actin or microtubule network reduces the fusion of newcomer vesicles. (A–C) Example images of predock fusion, newcomer-no dock fusion, and newcomer-short dock fusion. (D) Central intensity profiles of (A–C). For newcomer-short dock fusion, the interval between the two dashed lines in (D) was calculated as the docking time. (E) Average fusion rate of each type of fusion for each condition. (n = 1016 events from 6 cells for control, n = 476 events from 9 cells for CytoD treatment, and n = 285 events from 8 cells for Nocod treatment). (F) Average docking time for each condition (n = 131 events from 3 cells for control, n = 150 events from 9 cells for CytoD treatment, and n = 79 events from 8 cells for Nocod treatment). (G) Example of lateral shift of a fusing vesicle. The displacement from the last position before fusion (black circle) to the final fusion site (white circle) was 114 nm. Scale bar, 500 nm. (H) Distribution of the lateral shift in control cells. (I) Average lateral shift for each condition. (n = 262 events from 3 cells for control, n = 223 events from 9 cells for CytoD treatment, and n = 129 events from 8 cells for Nocod treatment). Error bars are ±SE. p values were determined using Student’s t-test. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Results
Automatic identification and analysis of fusion events
Fusion of fluorescently labeled vesicles with the plasma membrane observed with TIRF microscopy has been described as a sudden brightening and rapid spreading of the fluorescence signal (29). The sudden increase in the fluorescence signal occurs because evanescent excitation is more efficient closer to the interface and because of the dequenching of the pHluorin fluorescence once the acidic granule lumen has been exposed to a neutral environment when the fusion pore opens (9). The sudden increase in the fluorescence signal is followed by an exponential decrease in fluorescence intensity, which is the result of the rapid diffusion of vesicular membrane proteins or contents out of exocytic regions. We used a combination of these features (fluorescence increase, decrease, and ensuing diffusion of fluorophore) to detect vesicle fusion. The framework of the algorithm consisted of five major steps (Fig. 1 A). First, the image sequence was denoised, and the background was subtracted using the à trous wavelet transform algorithm (30) (step 1). Here, a hard threshold parameter (the selected wavelet component) after wavelet transformation ultimately determines the sensitivity and the accuracy of the spot detection algorithm. Therefore, when the imaging condition changes, we always manually adjust this parameter at the first image from a series of image stacks to ensure that proper numbers of fluorescence spots were detected. Once determined, the parameter can be used for the rest of the images with similar quality without much adjustment. The localization of all fluorescent particles (representative of vesicles) was then extracted with either a local maxima or a weighted centroid method (within a range of 5 × 5 pixels) at each frame (step 2). After particle detection, time-lapse trajectories of these vesicles were constructed using a multiple particle tracking software based on the linear assignment problem algorithm (31) (step 3). Here, our software pops out four parameters that may need to be adjusted according to vesicle behavior, including the gap closing factor, the minimum and maximum search radius, and the particle motion type (linear or not). Zero gap closing will increase the true positive rate (TPR) of the fusion detector; conversely, increasing the gap closing factor reduces the false positive rate (FPR) at the expense of sensitivity. The start and the end of a trajectory represents the appearance and the disappearance of a vesicle. Because vesicle fusion led to the end of a trajectory, we probed a possible vesicle fusion event within one track (step 4), which considerably reduced the time required for fusion detection (19,20). This step also helps to eliminate false positive fusion detection due to the lateral movement of vesicles. Finally, we performed a two-dimensional Gaussian fit and spatiotemporal analysis to extract additional information (step 5).
Fusion detection is the most difficult step and we used a protocol that combined advantages of previous algorithms. First, we selected vesicles that once exhibited fluorescence intensity (F1) that was greater than the background fluorescence (F0) by a certain ratio (R1) within its lifetime as possible fusion events (Fig. 1 B). The background was calculated as the mean intensity of the area (within a range of 5 × 5 pixels) at the center of the punctum (probable vesicle) and was measured for >5 frames before the appearance or >5 frames after the disappearance of the punctum. The method was basically a time-lapse one-dimensional intensity analysis (19,20), prone to error when local SNR near fusion sites decreased after extensive exocytosis. In addition, we used consecutive image subtraction to normalize the background fluorescence intensity across the entire field of view (18), and detected possible fusion events as a local increase in the fluorescence intensity over a preset ratio (R2) (Fig. 1 C). The detection accuracy and efficiency were much better with a combination of both R1 and R2 than using only one of these parameters (Fig. S1 in the Supporting Material).
In insulin-secreting cells, insulin is stored in secretory granules and is released upon an elevation in glucose levels. We used INS-1 cells containing secretory vesicles labeled with VAMP2-pHluorin, and detected massive vesicle fusion triggered by stimulation with 15 mM glucose and 70 mM KCl. By tuning R1 and R2 to proper values (1.3 and 3 in this set of data), we detected vesicle fusion events with a TPR of 96.5% and a FPR of 9.5% (Fig. 1 D); this FPR is much lower than that of previous methods (18–20). Next, we tested whether our algorithm could be used to detect other types of vesicle fusion. Insulin released into the bloodstream acts on the insulin receptors on adipocytes and muscle cells. This triggers the GSVs to fuse with the plasma membrane, leading to the insertion of the GLUT4 protein into the plasma membrane and potentiating the glucose uptake by these cells (32). Unlike Ca2+-dependent insulin secretion, GSV fusion depends on an insulin-stimulated signaling pathway but not on the cytosolic Ca2+ concentration. We labeled GSVs with pHluorin that was tagged to the luminal side of insulin-regulated aminopeptidase (IRAP, IRAP-pHluorin), a transmembrane protein that colocalizes with GLUT4 on GSVs. In adipocyte-like 3T3-L1 cells, insulin stimulation (100 nM) slowly triggers the fusion of GSVs (33). We still achieved a TPR of 93.5% for GSV fusion (Fig. 1 D). However, the FPR from the IRAP-pHluorin experiments was higher than that from the VAMP2-pHluorin experiments, possibly due to the inferior image quality with high levels of noise and background fluorescence. When setting the parameters, there was always a compromise between TPR and FPR. With high FPR, we could increase the values of R1 and R2 to eliminate false positive nonfusion events. With low TPR, we selected a fusion event that was missed in previous detection because of low local SNR, and the program would automatically update current R1 and R2 to the lowest values between values of this fusion event and preexisting values. These updated parameters could be used to detect fusion events from the same image stack thereafter. By optimizing in this manner for a few rounds, it is easy for the researcher to reach the best compromise between TPR and FPR for the image stack tested. The other parameters in the software package (including denoising and tracking) would not vary much for images acquired under similar conditions. Subsequently, the rest of the images from the same batch of experiments could be processed with little adjustment in a batch-processed manner with high sensitivity and accuracy. This procedure ensures our program to be adaptive and robust in detecting different types of exocytic events from different cells.
Disruption of the cytoskeleton inhibits regulated exocytosis and changes the fusion kinetics
Next, we evaluated whether our software could be used to quantitatively study exocytosis in INS-1 cells. When vesicles were labeled with VAMP2-pHluorin, a typical fusion event exhibited an abrupt increase in fluorescence followed by a rapid decrease, which was fitted with an exponential function (Fig. 2 A). In six cells stimulated with high concentrations of KCl and glucose, we identified up to ∼1700 fusion events. Using the results, we assembled the average time course of the change in exocytosis after stimulation (Fig. 2 B), the average fusion events per μm2 per s (Fig. 2 C) and the average decay in fluorescence after fusion (Fig. 2 D). Disruption of either the actin filaments using CytoD or the microtubule network using Nocod significantly inhibited the time-dependent fusion of vesicles upon stimulation (Fig. 2 B). Overall, average fusion rates were reduced to one-third in the CytoD-treated cells and one-sixth in the Nocod-treated cells, respectively (Fig. 2 C), indicating that both actin and microtubules play an indispensable role in transporting secretory vesicles to the cell surface before exocytosis. Unexpectedly, both the CytoD and Nocod treatments also caused a dramatic increase in the fusion decay time (Fig. 2 D), suggesting that the cytoskeleton affects either the fusion pore dilation or the protein diffusion on the plasma membrane. After performing a two-dimensional Gaussian fit of the fluorescence intensities at different times after the fusion pore opened (Fig. 2 E), we directly plotted the squared widths of these Gaussian functions versus time and determined the apparent diffusion coefficient of VAMP2-pHluorin on the plasma membrane (34). Consistent with the change in the decay time, the apparent diffusion coefficient decreased in both the CytoD- and Nocod-treated cells (Figs. 2 F). Therefore, disruption of the cytoskeleton networks slowed down the diffusion of the vesicle membrane proteins on the plasma membrane after the fusion pore opened.
The cytoskeleton is essential for spatially clustered exocytosis in INS-1 cells
In addition to the temporal occurrence, a detailed spatial map of exocytosis initiation is also provided from these images. It has been reported that some vesicle fusion sites were more likely to harbor another fusion event within a granule diameter (∼300 nm) in chromaffin cells (29). The phenomenon, called sequential exocytosis, has been found in many other cells (35–39). Therefore, we studied the spatial distribution of fusion events in INS-1 cells. It was apparent that some vesicles fused with the plasma membrane at similar locations (within a 5 × 5 pixel area) at different times (sequential exocytosis, Fig. 3 A). The sequential fusions constituted 28% of all fusion events in control cells, but this proportion dramatically decreased to 10% of the events in cells treated with either CytoD or Nocod (Fig. 3 B). In agreement with the selective reduction of repeated exocytosis at the same location, the average distance between two nearest fusion events increased as the actin or microtubules were disrupted (Fig. 3 C). The average time interval between two sequential fusions was longer in either the CytoD- or Nocod-treated cells compared with that in the control (Fig. 3 D), reflecting a reduced fusion efficiency even for vesicles undergoing sequential exocytosis. Overall, vesicle fusion becomes more sparsely distributed when the cellular cytoskeleton is disrupted.
Even for the vesicles undergoing sequential exocytosis, the spatial localizations of different fusion sites did not always precisely overlap. In this regard, we wonder whether randomly diffusing vesicles could hit and fuse at the same area in the plasma membrane multiple times by chance and contribute to the sequential fusion. To address this question, we used Ripley’s K-function (40) to statistically examine the spatial distribution of all fusion events within a cell (Fig. 4 A). The spatial K-function was determined by counting the number of fusion events within a specific distance from one event (18). From 100 Monte Carlo simulations using the same number of fusion events randomly distributed within the cell boundary, we constructed a range of K-function variations at specific distances. The K-function trace of the fusion locations from the real experiment were well beyond that range, indicating that the exocytosis was not randomly distributed in INS-1 cells (Fig. 4 B). Disruption of either actin or microtubules eliminated the clustering of fusion sites and spatially randomized the fusion events (Fig. S2, A and B). Next, we introduced a parameter, the uniform index, to quantitatively evaluate the degree of spatial aggregation of exocytosis. By creating a Voronoi diagram to separate each fusion event, we determined the average area occupied by each fusion (Fig. 4 C and Fig. S2 C). The CytoD or Nocod treatment increased the average area occupied by each fusion, in agreement with the severe inhibition of sequential exocytosis. The uniform index was calculated as the standard deviation of the fusion area divided by the mean of the fusion area. Monte Carlo simulation experiments were performed by randomly spreading the fusion events over the cell surface area using the number of fusion events within the same cell boundary (Fig. S2 D). Compared with the simulation, the vesicle fusion in real experiments exhibited a 63% higher uniform index in the control cells but was not significantly different in the cells treated with CytoD or Nocod (Fig. 4 D). Considering these findings, we conclude that the vesicle exocytosis occurs at spatially preferred regions in INS-1 cells (hotspots) and that this preference is dependent on the presence of an intact cellular cytoskeleton network.
Disruption of the cytoskeleton selectively reduces the fusion of newcomer vesicles
Despite the establishment of the presence of cytoskeleton-dependent exocytosis hotspots in INS-1 cells, it was unclear how the cytoskeleton mediated this process. To address this question, we changed the vesicle marker from VAMP2-pHluorin to VAMP2-EGFP, which is used to observe the trafficking, tethering, docking, and priming of vesicles before fusion. Using TIRF microscopy, we found that fusion events can be classified into three distinct categories (41) (Fig. 5, A–D). Some vesicles were already docked on the plasma membrane for a period of time before stimulation and then fused, called predock fusions (Fig. 5 A). Some vesicles newly entered the evanescent field and fused with the plasma membrane immediately without evident docking, called newcomer-no docking (Fig. 5 B). Some vesicles newly entered the TIRF zone and underwent a short period of docking and then fused, called newcomer-short docking (Fig. 5 C). Disruption of the cytoskeleton did not inhibit the predock fusions but significantly inhibited the fusion rates of newcomer-no docking and newcomer-short docking (Fig. 5 E). In addition, the density of stably docked vesicles did not change after cytoskeleton disruption (Fig. S3). Therefore, rather than mediating the accumulation of secretory vesicles under the plasma membrane, the cytoskeleton network delivers secretory vesicles from the cell interior to specific regions on the cell surface membrane.
Next, we analyzed the docking latency in newcomer-short docking events by fitting the intensity profile before fusion with a step-finding approach (Fig. 5 D). The docking time was shortened in the CytoD-treated cells, consistent with previous works (25), but not in the Nocod-treated cells (Fig. 5 F). This finding suggests that cortical actin filaments prolong the physical docking process of secretory vesicles.
Vesicles can undergo a lateral displacement under the plasma membrane before fusion in chromaffin cells (42,43). We also found such behavior in INS-1 cells, as shown in an example in Fig. 5 G, in which the distance from the position before fusion to the final fusion site was 114 nm. Because our method contains a robust multiple particle tracking algorithm, we were able to detect and analyze such behaviors systematically. The lateral shift varied from tens to hundreds of nanometers with a mean value of 131 ± 5.8 nm in control cells (Fig. 5 H). However, the value was reduced to 99 ± 5.2 nm in CytoD-treated and 91 ± 6.4 nm in Nocod-treated cells (Fig. 5 I). This finding again supported a role of the cortical cytoskeleton in arranging the final spatial redistribution of vesicle fusion events.
Discussion
Here, we have developed a computer-assisted identification and analysis software to detect pHluorin- and EGFP-labeled fusion events in INS-1 cells and 3T3-L1 cells, which provides superior sensitivity and accuracy as compared to previous methods (18–20). Our software can also be used to detect vesicle fusion events labeled by other markers, such as VGLUT1-mOrange2 (44), GFP-insulin (45), and NPY-pHluorin (46), which all share similar fluorescence characteristics during fusion. Prolonged acquisition always poses a challenge to automated image processing, as morphologic changes in the cell shape might result in changes in focus and reduction in image contrast. Our algorithm is capable of analyzing image stacks with a long acquisition time with relative robustness (Fig. S4). Because the algorithm is based on trajectories construction, it will effectively reduce false positive events exhibiting as transient, local disappearance of fluorescence intensity, which are possibly a result of the changes of focus or fluorophore blinking. The increase in FPR during prolonged experiments was mainly due to the accumulation of VAMP2-pHluorin on the plasma membrane after extensive exocytosis, which increased the cellular background fluorescence and reduced the image contrast during exocytosis (Fig. S4). This could be resolved by targeting pHluorin to luminal cargos that diffuse into the extracellular solution after exocytosis. Overall, after simple adjustment, our program is robust and adaptive to detect different types of vesicle fusion under different experimental conditions. In addition, the tracking algorithm can be used to track endocytic markers, such as clathrin and dynamin from time-lapse images (31,47). Therefore, it is also possible to modify our algorithm to study the coupling mechanisms between exocytosis and endocytosis in the future.
Using this tool, we statistically prove that stimulated INS-1 cells release secretory vesicles at spatially preferred hotspots. In principal, our data agree with sequential exocytosis found in chromaffin cells (29), pancreatic acinar cells (48), and islets (39). Different ratios of sequential exocytosis to the total exocytosis may be due to different cell types and preparation methods. Different microscopic methods may also contribute to the differences. For example, two-photon microscopy data were collected at a sampling frequency of 0.3∼2 s per frame (6,39), which is slower as compared to the sampling speed of current study and may miss some of the fast fusion events. Distinct from the active zone in synapses, the exocytosis hotspots in these endocrine cells are not formed by the fusion of densely packed vesicles that are predocked under the plasma membrane. In contrast, secretory vesicles that fuse at hotspots are delivered to the plasma membrane from the cell interior via microtubular and actin filaments. Disruption of either the actin or microtubular network severely reduced the overall exocytosis in addition to randomizing the vesicle fusion sites, suggesting that spatially confined exocytosis plays a major role in stimulation-triggered hormone release from insulin-secreting cells. The molecular mechanism underlying the cytoskeleton-mediated targeted delivery of vesicles remains unknown. Microtubules interact with the active zone protein ELKS through its binding protein CLASP (49) and to recruit vesicles to specific membrane locations, such as focal adhesions (50). Interactions between actin and vesicle membrane proteins, such as synapsin, have also been proposed (51). Future investigations will determine whether one or all of these mechanisms participate in the hotspot secretion of endocrine cells.
It was interesting to find that the disruption of the cytoskeleton changed the lateral shift of vesicles during the final fusion process (Fig. 5 I). This result showed the existence of a cortical cytoskeleton network and its role in precisely interacting with components on the plasma membrane. In agreement with this hypothesis, the diffusion of VAMP2 on the plasma membrane was slowed down in the absence of cytoskeleton filaments (Fig. 2, D and F). This was unlikely to be caused by the facilitatory effects of the cytoskeleton on fusion pore dilation because actin has been proposed to negatively regulate fusion pore expansion (52) and stabilize Ω-shaped structures during vesicle fusion (48). Alternatively, cytoskeleton filaments may interact with lipid domains on the plasma membrane to facilitate the diffusion of membrane proteins on the plasma membrane (47).
In summary, we have developed a software platform for the identification and spatiotemporal analysis of vesicle fusion events obtained by TIRF microscopy. Using this tool, we revealed that newcomer vesicles are delivered from the cell interior to the plasma membrane via cytoskeleton filaments to fuse at hotspots, and these vesicles make a major contribution to insulin-secretion from ΙΝS-1 cells stimulated with glucose and depolarization.
Acknowledgments
Y.Z. worked out the algorithm and implementation; T.Y. and J.L. cultured cells; T.Y. and J.Z. performed imaging; T.Y., Y.Z., and L.C. analyzed the data; T.Y., Y.Z., and L.C. designed the research and wrote the paper.
This work was supported by grants from the National Science Foundation of China (81222020, 31221002, 31327901, and 31301186), the Beijing Municipal Science & Technology Commission (7121008), the Major State Basic Research Program of P.R. China (2013CB531200), and the National Key Technology R&D Program (SQ2011SF11B01041).
Contributor Information
Yongdeng Zhang, Email: zyd0903@gmail.com.
Liangyi Chen, Email: lychen@pku.edu.cn.
Supporting Material
References
- 1.Südhof T.C., Rizo J. Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol. 2011;3:3. doi: 10.1101/cshperspect.a005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.von Gersdorff H., Matthews G. Electrophysiology of synaptic vesicle cycling. Annu. Rev. Physiol. 1999;61:725–752. doi: 10.1146/annurev.physiol.61.1.725. [DOI] [PubMed] [Google Scholar]
- 3.de Wit H., Walter A.M., Verhage M. Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell. 2009;138:935–946. doi: 10.1016/j.cell.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 4.Steyer J.A., Horstmann H., Almers W. Transport, docking and exocytosis of single secretory granules in live chromaffin cells. Nature. 1997;388:474–478. doi: 10.1038/41329. [DOI] [PubMed] [Google Scholar]
- 5.Zenisek D., Steyer J.A., Almers W. Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature. 2000;406:849–854. doi: 10.1038/35022500. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi N., Kishimoto T., Kasai H. Fusion pore dynamics and insulin granule exocytosis in the pancreatic islet. Science. 2002;297:1349–1352. doi: 10.1126/science.1073806. [DOI] [PubMed] [Google Scholar]
- 7.Moreno A., SantoDomingo J., Alvarez J. A confocal study on the visualization of chromaffin cell secretory vesicles with fluorescent targeted probes and acidic dyes. J. Struct. Biol. 2010;172:261–269. doi: 10.1016/j.jsb.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 8.Sankaranarayanan S., Ryan T.A. Real-time measurements of vesicle-SNARE recycling in synapses of the central nervous system. Nat. Cell Biol. 2000;2:197–204. doi: 10.1038/35008615. [DOI] [PubMed] [Google Scholar]
- 9.Miesenböck G., De Angelis D.A., Rothman J.E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 10.Tsuboi T., Rutter G.A. Multiple forms of “kiss-and-run” exocytosis revealed by evanescent wave microscopy. Curr. Biol. 2003;13:563–567. doi: 10.1016/s0960-9822(03)00176-3. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez N.A., Liang T., Gaisano H.Y. Live pancreatic acinar imaging of exocytosis using syncollin-pHluorin. Am. J. Physiol. Cell Physiol. 2011;300:C1513–C1523. doi: 10.1152/ajpcell.00433.2010. [DOI] [PubMed] [Google Scholar]
- 12.Axelrod D. Cell-substrate contacts illuminated by total internal reflection fluorescence. J. Cell Biol. 1981;89:141–145. doi: 10.1083/jcb.89.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bai L., Wang Y., Xu T. Dissecting multiple steps of GLUT4 trafficking and identifying the sites of insulin action. Cell Metab. 2007;5:47–57. doi: 10.1016/j.cmet.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 14.Jaiswal J.K., Rivera V.M., Simon S.M. Exocytosis of post-Golgi vesicles is regulated by components of the endocytic machinery. Cell. 2009;137:1308–1319. doi: 10.1016/j.cell.2009.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mattheyses A.L., Simon S.M., Rappoport J.Z. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J. Cell Sci. 2010;123:3621–3628. doi: 10.1242/jcs.056218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Axelrod D., Omann G.M. Combinatorial microscopy. Nat. Rev. Mol. Cell Biol. 2006;7:944–952. doi: 10.1038/nrm2062. [DOI] [PubMed] [Google Scholar]
- 17.Huang S., Lifshitz L.M., Czech M.P. Insulin stimulates membrane fusion and GLUT4 accumulation in clathrin coats on adipocyte plasma membranes. Mol. Cell. Biol. 2007;27:3456–3469. doi: 10.1128/MCB.01719-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sebastian R., Diaz M.E., Toomre D. Spatio-temporal analysis of constitutive exocytosis in epithelial cells. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2006;3:17–32. doi: 10.1109/TCBB.2006.11. [DOI] [PubMed] [Google Scholar]
- 19.Vallotton P., James D.E., Hughes W.E. Towards fully automated identification of vesicle-membrane fusion events in TIRF Microscopy. AIP Conf. Proc. 2007;952:3–10. [Google Scholar]
- 20.Mele, K., A. Coster, …, P. Vallotton. 2009. Automatic identification of fusion events in TIRF microscopy image sequences. Computer Vision Workshops (ICCV Workshops). IEEE 12th International Conference. 578–584.
- 21.Südhof T.C. The presynaptic active zone. Neuron. 2012;75:11–25. doi: 10.1016/j.neuron.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohara-Imaizumi M., Ohtsuka T., Nagamatsu S. ELKS, a protein structurally related to the active zone-associated protein CAST, is expressed in pancreatic beta cells and functions in insulin exocytosis: interaction of ELKS with exocytotic machinery analyzed by total internal reflection fluorescence microscopy. Mol. Biol. Cell. 2005;16:3289–3300. doi: 10.1091/mbc.E04-09-0816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lang T., Wacker I., Almers W. Role of actin cortex in the subplasmalemmal transport of secretory granules in PC-12 cells. Biophys. J. 2000;78:2863–2877. doi: 10.1016/S0006-3495(00)76828-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J., Richards D.A. Spatial regulation of exocytic site and vesicle mobilization by the actin cytoskeleton. PLoS ONE. 2011;6:e29162. doi: 10.1371/journal.pone.0029162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmoranzer J., Simon S.M. Role of microtubules in fusion of post-Golgi vesicles to the plasma membrane. Mol. Biol. Cell. 2003;14:1558–1569. doi: 10.1091/mbc.E02-08-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beffert U., Dillon G.M., Ho A. Microtubule plus-end tracking protein CLASP2 regulates neuronal polarity and synaptic function. J. Neurosci. 2012;32:13906–13916. doi: 10.1523/JNEUROSCI.2108-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duman J.G., Chen L., Hille B. Contributions of intracellular compartments to calcium dynamics: implicating an acidic store. Traffic. 2006;7:859–872. doi: 10.1111/j.1600-0854.2006.00432.x. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y., Wang Y., Lippincott-Schwartz J. Rab10 and myosin-Va mediate insulin-stimulated GLUT4 storage vesicle translocation in adipocytes. J. Cell Biol. 2012;198:545–560. doi: 10.1083/jcb.201111091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allersma M.W., Wang L., Holz R.W. Visualization of regulated exocytosis with a granule-membrane probe using total internal reflection microscopy. Mol. Biol. Cell. 2004;15:4658–4668. doi: 10.1091/mbc.E04-02-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olivo-Marin J.C. Extraction of spots in biological images using multiscale products. Pattern Recognit. 2002;35:1989–1996. [Google Scholar]
- 31.Jaqaman K., Loerke D., Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kozka I.J., Clark A.E., Holman G.D. The effects of insulin on the level and activity of the GLUT4 present in human adipose cells. Diabetologia. 1995;38:661–666. doi: 10.1007/BF00401836. [DOI] [PubMed] [Google Scholar]
- 33.Jiang L., Fan J., Xu T. Direct quantification of fusion rate reveals a distal role for AS160 in insulin-stimulated fusion of GLUT4 storage vesicles. J. Biol. Chem. 2008;283:8508–8516. doi: 10.1074/jbc.M708688200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.An S.J., Grabner C.P., Zenisek D. Real-time visualization of complexin during single exocytic events. Nat. Neurosci. 2010;13:577–583. doi: 10.1038/nn.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nemoto T., Kimura R., Kasai H. Sequential-replenishment mechanism of exocytosis in pancreatic acini. Nat. Cell Biol. 2001;3:253–258. doi: 10.1038/35060042. [DOI] [PubMed] [Google Scholar]
- 36.Scepek S., Lindau M. Focal exocytosis by eosinophils—compound exocytosis and cumulative fusion. EMBO J. 1993;12:1811–1817. doi: 10.1002/j.1460-2075.1993.tb05829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cochilla A.J., Angleson J.K., Betz W.J. Differential regulation of granule-to-granule and granule-to-plasma membrane fusion during secretion from rat pituitary lactotrophs. J. Cell Biol. 2000;150:839–848. doi: 10.1083/jcb.150.4.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alvarez de Toledo G., Fernandez J.M. Compound versus multigranular exocytosis in peritoneal mast cells. J. Gen. Physiol. 1990;95:397–409. doi: 10.1085/jgp.95.3.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takahashi N., Hatakeyama H., Kasai H. Sequential exocytosis of insulin granules is associated with redistribution of SNAP25. J. Cell Biol. 2004;165:255–262. doi: 10.1083/jcb.200312033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ripley B.D. Modeling spatial patterns. J. R. Stat. Soc. Series B. State Methodol. 1977;39:172–212. [Google Scholar]
- 41.Zhu D., Zhang Y., Gaisano H.Y. Dual role of VAMP8 in regulating insulin exocytosis and islet β cell growth. Cell Metab. 2012;16:238–249. doi: 10.1016/j.cmet.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 42.Degtyar V.E., Allersma M.W., Holz R.W. Increased motion and travel, rather than stable docking, characterize the last moments before secretory granule fusion. Proc. Natl. Acad. Sci. USA. 2007;104:15929–15934. doi: 10.1073/pnas.0705406104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anantharam A., Onoa B., Axelrod D. Localized topological changes of the plasma membrane upon exocytosis visualized by polarized TIRFM. J. Cell Biol. 2010;188:415–428. doi: 10.1083/jcb.200908010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H., Foss S.M., Voglmaier S.M. Concurrent imaging of synaptic vesicle recycling and calcium dynamics. Front Mol. Neurosci. 2011;4:34. doi: 10.3389/fnmol.2011.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagamatsu S., Ohara-Imaizumi M. Imaging exocytosis of single insulin secretory granules with TIRF microscopy. Methods Mol. Biol. 2008;440:259–268. doi: 10.1007/978-1-59745-178-9_20. [DOI] [PubMed] [Google Scholar]
- 46.Zhu D., Zhou W., Xu T. Synaptotagmin I and IX function redundantly in controlling fusion pore of large dense core vesicles. Biochem. Biophys. Res. Commun. 2007;361:922–927. doi: 10.1016/j.bbrc.2007.07.083. [DOI] [PubMed] [Google Scholar]
- 47.Jaqaman K., Kuwata H., Grinstein S. Cytoskeletal control of CD36 diffusion promotes its receptor and signaling function. Cell. 2011;146:593–606. doi: 10.1016/j.cell.2011.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nemoto T., Kojima T., Kasai H. Stabilization of exocytosis by dynamic F-actin coating of zymogen granules in pancreatic acini. J. Biol. Chem. 2004;279:37544–37550. doi: 10.1074/jbc.M403976200. [DOI] [PubMed] [Google Scholar]
- 49.Lansbergen G., Grigoriev I., Akhmanova A. CLASPs attach microtubule plus ends to the cell cortex through a complex with LL5beta. Dev. Cell. 2006;11:21–32. doi: 10.1016/j.devcel.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 50.Stehbens S.J., Paszek M., Wittmann T. CLASPs link focal-adhesion-associated microtubule capture to localized exocytosis and adhesion site turnover. Nat. Cell Biol. 2014;16:561–573. doi: 10.1038/ncb2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Humeau Y., Doussau F., Poulain B. Synapsin controls both reserve and releasable synaptic vesicle pools during neuronal activity and short-term plasticity in Aplysia. J. Neurosci. 2001;21:4195–4206. doi: 10.1523/JNEUROSCI.21-12-04195.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen A., Leikina E., Chernomordik L.V. Fusion-pore expansion during syncytium formation is restricted by an actin network. J. Cell Sci. 2008;121:3619–3628. doi: 10.1242/jcs.032169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.