

Abstract
The major histocompatibility complex (MHC) class II protein can bind peptides of different lengths in the region outside the peptide-binding groove. Peptide-flanking residues (PFRs) contribute to the binding affinity of the peptide for MHC and change the immunogenicity of the peptide/MHC complex with regard to T cell receptor (TCR). The mechanisms underlying these phenomena are currently unknown. The molecular flexibility of the peptide/MHC complex may be an important determinant of the structures recognized by certain T cells. We used single-molecule x-ray analysis (diffracted x-ray tracking (DXT)) and fluorescence anisotropy to investigate these mechanisms. DXT enabled us to monitor the real-time Brownian motion of the peptide/MHC complex and revealed that peptides without PFRs undergo larger rotational motions than peptides with PFRs. Fluorescence anisotropy further revealed that peptides without PFRs exhibit slightly larger motions on the nanosecond timescale. These results demonstrate that peptides without PFRs undergo dynamic motions in the groove of MHC and consequently are able to assume diverse structures that can be recognized by T cells.
Introduction
T cell receptor (TCR) recognizes peptides that are derived from antigenic proteins and are bound to host major histocompatibility complex (MHC) molecules present on the surface of antigen-presenting cells (1). As a result of VDJ recombination, a large number of TCRs can be generated within the body. However, the possible number of peptide/MHC complexes is enormous and far larger than the number of potential recombined TCRs; therefore, a given TCR must be able to cross-react with thousands of antigens (2,3). The cross-reactivity of TCR is well documented, and crystal structures of free TCR in complex with MHC clearly reveal repositioning of the loops of the complementarity-determining region (4–8). On the other hand, class I-bound peptides in the MHC groove deform upon TCR binding; thus, longer peptides can bind class I MHC and still activate T cells, which flatten the bulged peptide loops upon binding (9,10). Furthermore, class I MHC exhibits a conformational variation in the α-helix that is dependent on bound peptide (11).
No such studies have been performed for class II MHC (MHC II), however, because peptides are believed to be a part of the MHC folding complex, and the Debye-Waller factor (B factor) indicates that peptides in the crystal adopt rigid conformations (12). However, the notion that MHC II binds peptides primarily via hydrogen bonds raises the possibility of flexible motion of peptides in the binding groove of MHC II (12–18). Furthermore, it has become clear that single peptides in complex with an MHC II molecule adopt multiple conformations (a minimum of two, called the type A and type B complexes) (19,20). Type A complexes acquire peptides in late endosomes through DM catalysis, whereas type B complexes acquire peptides in recycling endosomes or at the cell surface (19–21). DM translocates the peptide into the binding groove by an as-yet-unknown mechanism. Thus, we can assume that type A complexes adopt a rigid conformation, whereas type B complexes adopt a flexible conformation; however, beyond this rough assessment (22), the structural differences between type A and type B MHC complexes remain unknown.
In addition to DM catalysis in the late endosome, the length of the peptide affects the affinity of peptides for MHC II: specifically, the addition of a few residues to the region outside the groove is necessary for efficient peptide binding (23). Longer peptides that contain peptide-flanking residues (PFRs) have a greater tendency to form type A MHC complexes (22). PFRs also alter the antigenicity of the peptide in a way that corresponds to the ratio of the proportions of the type A and type B complexes (24,25). Hen egg lysozyme peptides (pHELs) of different lengths are a well-studied system for investigating the antigenicity of the type A and type B conformations (19,20), and we employed that system in this study. The HEL48-61 peptide contains four N-terminal PFRs and primarily forms a type A MHC II complex. By contrast, the HEL52-61 peptide lacks the N-terminal PFRs and primarily forms a type B MHC II complex. These differences were exploited in this study.
The structural isotypes of MHC II may arise from differences in the flexibilities of the peptide complexes. To determine the differences between the structural isotypes, we used the diffracted x-ray tracking (DXT) method (26,27), which utilizes gold nanocrystals (average size 20–25 nm) that are covalently attached to the protein of interest. Irradiation of the labeled protein with white x-rays results in single diffraction spots that can be monitored in real time, and the trajectories of the diffraction spots can be translated into movements in real space. Next, we analyzed an I-Ak/peptide complex by using fluorescence anisotropy, which revealed increased motion of the peptides on the nanosecond timescale. Comparative molecular-dynamics (MD) simulations exhibited large conformational fluctuations, particularly in the additional N-terminal amino acids that lie outside the groove, and a rather rigid structure throughout the rest of the peptide.
Materials and Methods
Peptides
Peptides were purchased from Peptide 2.0 (Chantilly, VA). Peptide sequences are; pHEL48-61 (DGSTDYGILQINSR); pHEL48-61(52A) (DGSTAYGILQINSR); pHEL52-61 (DYGILQINSR); pHEL48-61(56C) (DGSTDYGICQINSR); pHEL48-61 (52A, 56C) (DGSTAYGICQINSR); pHEL52-61(56C) (DYGCQINSR); and FITC-pHEL50-61 (STDGSTDYGILQINSR).
Production and purification of soluble MHC
The ectodomain of MHC with the class II-associated invariant chain peptide (CLIP) covalently bound to the N-terminus of the β chain was generated as previously described (28). The I-Ak/CLIP construct contained a thrombin cleavage site in the N-terminus of CLIP. Six-histidine tags were attached to the C-terminus of both the α and β chains. Constructs were integrated into pFastBac Dual (Thermo Fisher Scientific, Waltham, MA) and baculoviruses were generated according to the manufacturer’s protocol. SF+ cells were infected and cultured with SF-900 II serum-free medium (Thermo Fisher Scientific) at 18°C for 6–7 days. Culture supernatants containing the proteins were concentrated and exchanged with 10 mM sodium phosphate (pH 5.6) and 300 mM NaCl using a Vivaflow 50 (Sartorius, Goettingen, Germany). After adjustment to pH 7.0, the proteins were isolated with Co-Sepharose (TALON metal affinity resin; Clontech, Mountain View, CA) in 20 mM imidazole, 50 mM phosphate buffer (PB, pH 7.0), and 0.5 M NaCl, and eluted with 150 mM imidazole, 50 mM PB (pH 7.0), and 0.5 M NaCl. Aggregated proteins were removed by gel filtration chromatography using a Superdex-200 column (GE Healthcare, Pittsburgh, PA). The peptide was exchanged by digestion of I-Ak/CLIP with thrombin (Novagen, Madison, WI) in the presence of the desired peptides, followed by incubation at pH 5.2 overnight at room temperature.
Competition assay
Soluble I-Ak/CLIP (20 nM) was bound to 1 μM fluorescein isothiocyanate (FITC)-pHEL-50-61 and 0–400 μM of unlabeled competitor peptide with thrombin. After incubation for 48 h at room temperature, the I-Ak/ FITC-HEL50-61 complex was separated from the free peptide with a Micro Bio-Spin 30 column (Bio-Rad, Hercules, CA). FITC intensity was measured by injecting the sample into an LC-20AT high-performance liquid chromatography (HPLC) unit (Shimadzu, Tokyo, Japan) equipped with a BioSep-SEC-S 3000 column (300 × 7.8 mm; Phenomenex, Los Angeles, CA) connected to an in-line fluorescence detector (RF-10AXL fluorescence detector; Shimadzu, Tokyo, Japan). The strength of each peptide’s affinity was determined from the concentration of competitor peptide that yielded the half-maximum of the fluorescence intensity.
Production of gold nanocrystals
Gold nanocrystals were produced by epitaxial growth on an NaCl(100) substrate (29). The samples were prepared by thermal vacuum evaporation (ANELVA L-300EK; Canon Anelva, Kanagawa, Japan) of 99.95% gold from a tungsten basket in a vacuum of ∼2.0 × 10−4 Pa. Cleaved NaCl(100) (10 × 10 mm) was used as the substrate and maintained at ∼475°C during the deposition. The distance between the filament and the substrate was 150 mm. Gold was deposited at a rate of 0.1 nm/min onto the substrate, and a constant filament temperature of 1486.7 ± 4.3 K, as determined with a radiation thermometer (CHINO; IR-AH, Tokyo, Japan), was used in the experiments. To detach the gold nanocrystals from the NaCl(100) substrate, the substrates were dissolved in an-decyl-B-D maltoside solution (50 mM, pH 7.0). A dynamic light scattering device (DLS-8000DS; Otsuka Electronics, Osaka, Japan) with a 488 nm argon-ion laser was used to measure the particle size distribution and the stability of the gold nanocrystals in aqueous solution.
DXT
Complexes of I-Ak/CLIP (1–4 μM) were exchanged to either cysteine-substituted peptides or unsubstituted peptides by incubation with thrombin (Novagen, Madison, WI) and a 10-fold molar excess of the desired peptides for 2 h at room temperature, followed by exchange of the pH to 5.2 overnight and neutralization. Complexes of I-Ak/peptide were immobilized onto quartz basal plates via histidine tags. Immobilized I-Ak/peptide complexes were labeled with the gold nanocrystals via an electrical charge in the thiol groups of cysteine. Labeled complexes on the basal plates were locked in a PBS-filled chamber and irradiated with white x-rays (SPring-8, BL40XU; Sayo-gun, Hyogo, Japan) to record Laue diffraction from the gold nanocrystals. The diffraction spots were monitored with an x-ray image intensifier (V5445P; Hamamatsu Photonics, Shizuoka, Japan) and a CCD camera (C4880-82; Hamamatsu Photonics) at 656 × 494 pixels. The diffraction images were obtained at a video rate of 36 ms/frame (ms/f) and total exposure time of 3.2 s (90 frames). The effective size of the detector was 150 mm in diameter with a 100 mm sample-to-detector distance. Custom software written for IGOR Pro (Wavemetrics, Lake Oswego, OR) was used to track the diffraction spots’ trajectories and to analyze those motions statistically. The DXT experiments were performed with the approval of the Japan Synchrotron Radiation Research Institute (proposal numbers 2008A1857, 2009A1888, 2010B1153, 2011A1236, 2011B1317, 2012A1396, 2014A1030, and 2014B1246).
We analyzed the absolute angular displacement of the peptide and MHC in the θ and χ directions, and constructed histograms by recording the segments of angular displacement per 720 ms (20 frames). We then fitted the histograms to a normal Gaussian function and compared the fitting parameters of the distributions. To reduce noise in an experimental trajectory from the DXT data, we averaged the data points within a single trajectory, which yielded the mean-square angular displacement (MSD) for that trajectory from the DXT data. The data from the DXT analysis of the MHC-peptide complex were fit to the MSD. The measured spots represent ∼50 trajectories in all of the MSD curves. The values of the diffusion coefficient (D) and velocity (v) were determined from the plots of (Δθ2) versus the interval times (30).
Fluorescence anisotropy
pHEL48-61(L56C) and pHEL52-61(L56C) were labeled with fluorescein-5 maleimide (Thermo Fisher Scientific, Waltham, MA) after Tris(2-carboxyethyl)phosphine (Thermo Fisher Scientific) treatment and isolated by a peptide column using an HPLC system.
CLIP in complex with I-Ak was exchanged to fluorescein-labeled pHEL48-61(L56C) or pHEL52-61(L56C). Fluorescein-labeled I-Ak/pHEL48-61 and I-Ak/pHEL52-61 were isolated by gel filtration chromatography using a Superdex-200 (GE Healthcare, Pittsburg, PA) column equilibrated with 20 mM PB (pH 7.0), 150 mM NaCl, and 0.5 mM EDTA. Time-resolved measurements were performed with a FluoroCube 5000U time-correlated single-photon counting fluorometer (HORIBA, Kyoto, Japan) with a 457-nm NanoLED pulsed excitation source. The excitation wave length was 452 nm and the emission wave length was 520 nm. Experiments were performed at room temperature. The anisotropy decay was fitted with model function:
| (1) |
where γ (t) is anisotropy, B is the transition electric dipole moment angle constant, t is time, φ1 is the rotational correlation time for fluorescein, and φ2 is the rotational correlation time for peptide.
The data were processed and analyzed using Igor software (WaveMetrics, Lake Oswego, OR).
MD simulation
Initial structures of I-Ak/pHEL50-62, I-Ak/pHEL52-58, I-Ak/pHEL52-61, I-Ak/pHEL48-61, and I-Ak alone for MD simulations were built based on the x-ray crystal structure of I-Ak/HEL50-62 (31) using the molecular modeling software MOE (Chemical Computing Group, Montreal, Canada). All of the initial structures for the MD simulations were spherically surrounded by ∼30,000 TIP3P water molecules (32).
After the energy minimizations, all MD simulations were conducted for 6 ns at 300 K using the Amber 8.0 (33) modified for the MDGRAPE-3 system (34,35). The Amber ff03 force field (36) was adopted and the simulation time step was set at 1 fs. The bond lengths involving hydrogen atoms were constrained to equilibrium lengths using the SHAKE method (37). The temperature was maintained constant at 300 K using the method of Berendsen et al. (38).
To analyze the correlation rate for protein motion, we calculated a cross-correlation coefficient for the centers of mass of the residues (39). This value can vary from −1, which represents a completely anticorrelated motion, to +1, which corresponds to a completely correlated motion.
We calculated the binding free energies by the molecular-mechanics Poisson-Boltzmann/surface area (MM-PB/SA) method (40) using the last 1 ns of the MD trajectories.
Results
Peptides without PFRs and in complex with MHC move significantly within the binding groove of MHC
The affinities of peptides for I-Ak were measured by competition assay. The dissociation constants exhibited a 5-fold difference (1.4 μM for pHEL48-61 and 6.8 μM for pHEL52-61). Although the affinities of peptides seem relatively low, peptides are believed to be a part of folding complex with an MHC class II protein. Thus, these peptides should not be released in the neutral pH condition as we measured the motions in phosphate buffer.
To determine the active structure recognized by type A and type B T cells, we analyzed the motion of the peptide within the binding groove of MHC II using the DXT method. Fig. 1 A presents the geometry of the DXT experiment. Geometrical correlations between the movements of the vector in real space and those of the spots on the image frame are indicated. The diffraction spots from single nanocrystals were video-recorded at a rate of 36 ms/f and the trace locations were translated into movements in real space.
Figure 1.
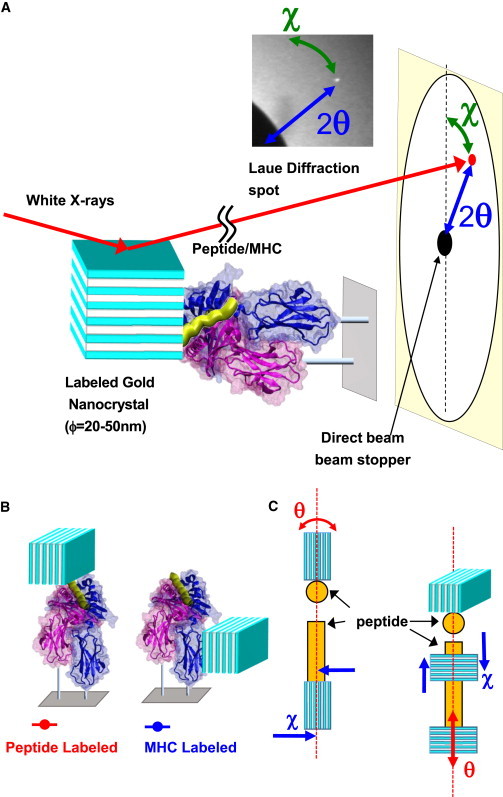
Principles of single-molecule measurement using DXT. (A) The relative positions of the gold nanocrystal-labeled peptide/MHC complex on the basal plate, the radiation beam, and the image plane are shown. Diffraction spots from single nanocrystals were video-recorded at a rate of 36 ms/f and the trace locations were translated into motion in real space. The large black circle on the left is the umbra of the beam stop for the direct x-ray beam. θ, tilting motion; χ, rotational motion. (B) Schematic model for the peptide (with the L56C mutation) and MHC (without mutation) labeled with gold nanocrystals. (C) The θ direction of the Laue pattern movement is assigned to the tilted motion of the peptide/I-Ak complex. The χ direction of the Laue pattern movement is assigned to the rotational motion of the peptide/I-Ak complex. To see this figure in color, go online.
The I-Ak/CLIP complex was expressed in baculovirus, and the cysteine-containing peptide was loaded after CLIP was cleaved from the groove. The complex was bound to a Ni2+-NTA-coated surface using histidine residues introduced into both the α and β chains of MHC, and then labeled with gold nanocrystals (Fig. 1 B). The gold nanocrystals preferentially bound to the solvent-exposed cysteine residues of the peptide. However, in the absence of a cysteine in the peptide, the gold nanocrystals preferentially bound to methionine 160 of the MHC II β chain because no cysteine residues are exposed in the MHC II molecule (Fig. 1 B). The motion of gold nanocrystals on the MHC complex were defined as the polar coordinate of the vector normal to the x-ray diffraction plane of the nonocrystal. The polar coordinates in the diffraction plane were converted to the orthogonal axes (χ-θ coordinates). The quartz surface was irradiated with primary white x-rays and irradiation was carried out in parallel to the longitudinal axis of the adsorbed MHC molecule. The orientation of the MHC molecule was controlled by two six-histidine tags on the quartz surface. If the dynamic movement of an adsorbed MHC molecule was equal to that of the labeled nanocrystal, the x-ray diffraction spots were read as the movement of the MHC molecule. When the MHC molecule tilted, the diffraction spot moved radially, as defined by the angle θ (Fig. 1 C). However, when the adsorbed MHC molecule rotated, the spot also rotated on the image plane, with angle χ. Thus, χ represents the rotational angle and θ represents the tilt angle (27).
To quantify these movements, we introduced the diffusion coefficients for both peptides and MHC from the MSD analysis (Fig. 2, left panel). The diffusion coefficients for the labeled peptide versus the labeled MHC of I-Ak/pHEL48-61, I-Ak/pHEL52-61, and I-Ak/pHEL48-61(52A) were 33.6 mrad2/s vs. 20.1 mrad2/s, 48.1 mrad2/s vs. 32.9 mrad2/s, and 74 mrad2/s vs. 41 mrad2/s, respectively. This result suggests that the motion of the peptide is always slightly larger than the MHC itself. The diffusion constants of both the peptide and MHC are larger with pHEL52-61 than with pHEL48-61, indicating that the PFRs affect the motion of the peptide and even the MHC. The anchorless peptide pHEL48-61(52A) shows greater motion than pHEL52-61, although I-Ak/pHEL48-61(52A) has similar specificity in terms of the T cell activation profile.
Figure 2.
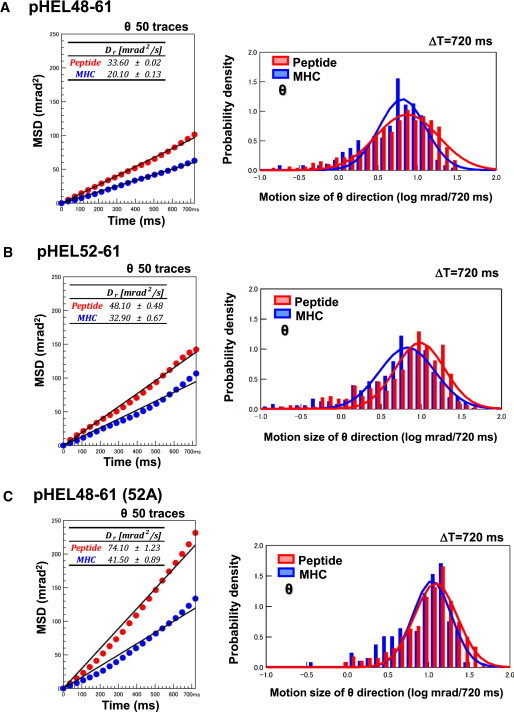
The peptide-labeled I-Ak/pHEL52-61 complex exhibited larger motion for the θ direction than the MHC-labeled complex. (A–C) CLIP in complex with I-Ak was exchanged with pHEL48-61 (A), pHEL52-61 (B), or pHEL48-61(52A) (C) that contained the L56C mutation (red) or did not contain the mutation (blue). The complexes were labeled with gold nanocrystals (peptide labeled and MHC labeled) and analyzed by DXT. Left panels: MSD curves for the tilted angle Δθ2. Right panels: Gaussian plots for the tilted angle at 720 ms. To see this figure in color, go online.
To examine the properties of each motion, we utilized a Gaussian plot of 100 sampled trajectories, which simultaneously visualized the motions of individual spots at each time point. Fig. 2 (right panel) shows an overlay histogram of the probability density plot for the absolute displacement of θ (tilt angle) at 720 ms. Both I-Ak/pHEL48-61 and I-Ak/pHEL48-61(52A) exhibited similar peaks in the histograms for the labeled MHC and the labeled peptide. Conversely, I-Ak/pHEL52-61 exhibited a larger peak for the labeled peptide than for the labeled MHC. The histograms of the labeled MHC for the pHEL48-61- and pHEL52-61-bound complexes were similar, suggesting that the peptides in the I-Ak/pHEL52-61 complex moved more dynamically than in the other complexes. We compared the Gaussian plots as single peaks representing single motions. However, pHEL48-61(52A)-bound I-Ak seems to have composite motion, as the small peak appeared at 100.5 (= 3.2) mrad/720 ms, suggesting that replacement of the anchor residue of bound peptide caused a more dynamic structural transition than the PFRs. Subsequently, therefore, we concentrated on studying the role of PFRs and compared pHEL48-61 and pHEL52-61.
We next analyzed the movements of the rotational angle χ as measured by DXT. Fig. 3 A shows the two-dimensional probability density plots for the absolute displacement of θ and χ for two peptides. Both I-Ak/pHEL52-61 at 360 ms and I-Ak/pHEL52-61 at 720 ms exhibited larger probability densities in the χ direction than I-Ak/pHEL48-61 at 360 ms and I-Ak/pHEL48-61 at 720 ms. Fig. 3 B presents the overlay histogram of the absolute displacement for the χ movements of I-Ak/pHEL52-61 and I-Ak/pHEL48-61 at 720 ms. I-Ak/pHEL48-61 exhibited a peak at 1.5 mrad, whereas I-Ak/pHEL52-61 exhibited a peak at 2.2 mrad, reflecting the degree of peptide movement in the groove. Although the difference in the peak positions between IAk/pHEL48-61 and IAk/pHEL52-61 in Fig. 3 B was subtle, the full width half maximum for the movement distribution in the peptide without PFRs (pHEL52-61) was larger. This motion width observed rotationally was converted to the translational one. This estimated value was ∼1 Å which might be the same size as the width of activating conformations for certain T cells.
Figure 3.
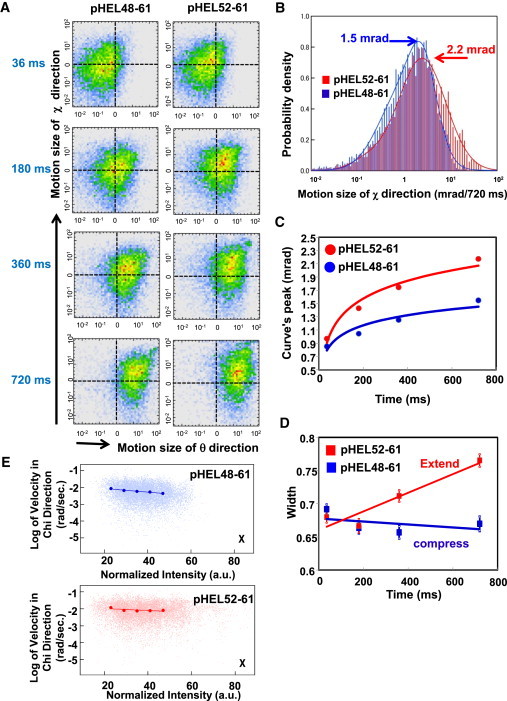
The complex of a peptide without PFRs, I-Ak/pHEL52-61, demonstrates larger χ-directed Brownian motion in DXT. CLIP in complex with I-Ak was exchanged to either pHEL48-61 or pHEL52-61 containing the L56C mutation. The complexes were labeled with gold nanocrystals and analyzed by DXT. (A) Two-dimensional probability density plots of the absolute displacement for the tilted angle θ (horizontal) and absolute displacement for the rotational angle χ (vertical) at 36 ms, 180 ms, 360 ms, and 720 ms. (B) Histograms of the absolute displacement, indicating the Gaussian distribution for the χ movement at 720 ms for I-Ak/pHEL48-61 and I-Ak/pHEL52-61, were obtained in the same experiment shown in A. (C) Analysis of the peaks of the Gaussian plots for the χ movement. (D) Analysis of the width of the Gaussian plots for the χ movement. (E) The observed angular velocity as a function of normalized intensity of the diffraction spot for I-Ak/pHEL48-61 and I-Ak/pHEL52-61. To see this figure in color, go online.
In analysis of the x direction of the Gaussian plots, these time-dependent peak positon's curves had saturation values (Fig. 3 C). This means that detected movements were assigned to restricted ones. Additionally, it was indicated that the movements of the peptide without PFRs (pHEL52-61) was clearly bigger than that of the peptide with PFRs (pHEL48-61) (Fig. 3 C). In addition, the peptide without PFRs exhibited a broader Gaussian distribution, whereas the peptide containing PFRs (pHEL48-61) exhibited a Gaussian distribution that narrowed over time (Fig. 3 D).
Because the size of the gold nanocrystal is comparable to that of MHC proteins, one should consider the effect of the gold nanocrystal on the protein’s dynamics when using the DXT method. We estimated the degree of disturbance of the inherent motion of the target protein from the labeled gold nanocrystal by plotting the angular velocity of the χ direction versus normalized intensity, which we inferred to be the size of the gold nanocrystal (41). The size of gold nanocrystal bound to the target protein was between 20 and 80 nm, and within this range we could not find much difference in velocity, which suggests that the size effects of gold nanocrystals are minimal in such measurements (Fig. 3 E). Taken together, these findings indicate that pHEL52-61 is highly flexible, resulting in variations in the structure of the peptide/MHC II complex (i.e., the TCR ligand).
A peptide without PFRs in complex with MHC exhibits faster motion as determined by fluorescence anisotropy
To investigate the motion of peptides on the nanosecond timescale, we examined the time decay of anisotropy using single-photon-counting time-resolved fluorescence anisotropy. Both pHEL48-61 and pHEL52-61 were labeled with fluorescein at residue 56. In this experiment, we followed the decay of anisotropy after a rapid pulse. We calculated the decay rates using an equation with two variables: the rotation of fluorescein itself and the motion of the peptide. As shown in Fig. 4, the anisotropy decay in fluorescein-labeled I-Ak/pHEL52-61 was slightly faster (φ2 = 0.96 ns) than that in fluorescein-labeled I-Ak/pHEL48-61 (φ2 = 1.51 ns), indicating that I-Ak/pHEL52-61 was more flexible than I-Ak/pHEL48-61 on the nanosecond timescale. The tumbling motion of the peptide/MHC complex should be almost the same between two complexes because the total mass and overall structures are the same (42); therefore, we conclude that the difference is due to the peptide motion within the binding groove.
Figure 4.
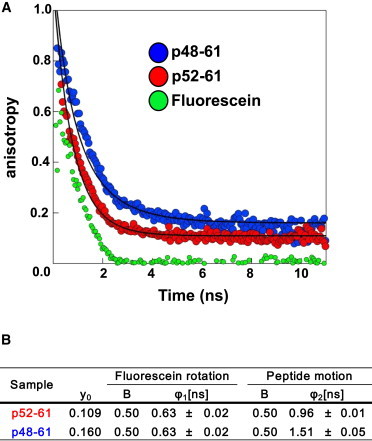
The I-Ak/pHEL52-61 complex exhibited larger motion than the I-Ak/pHEL48-61 complex in bulk solvent. (A) Anisotropies of I-Ak/pHEL48-61 and IAk/pHEL52-61 complexes and fluorescein alone. Since the components are peptide motion and fluorescence rotation, the data were analyzed by secondary fitting using Eq. 1. (B) The differential dynamics of the IAk/pHEL52-61 (φ2 = 0.96 ns) revealed by steady-state anisotropy can be attributed to increased fluctuations occurring over the nanosecond timescale compared with I-Ak/pHEL48-61 (φ2 = 1.51 ns). To see this figure in color, go online.
Comparative MD simulations
To confirm that pHEL52-61 undergoes larger motions than pHEL48-61 in the groove of I-Ak, we conducted comparative MD simulations. The structures of the I-Ak/peptide complexes used for the MD simulations were built based on the crystal structure of I-Ak molecules in complex with pHEL50-62 (31). As shown in Fig. 5 A, the atomic fluctuation values of residues of pHEL48-61, pHEL52-61, and pHEL52-58 within the groove of I-Ak tended to be similar to those of the crystal structure, suggesting that these simulations did not contain obvious artifacts, whereas the N-terminal PFRs of pHEL48-61, located outside the I-Ak groove, exhibited larger atomic fluctuations. The final simulated structure of I-Ak/pHEL48-61 exhibited larger conformational differences in the N-terminal PFRs and mild displacement of the C-terminal residues, which are stabilized by peptide-MHC interactions inside the groove. Taken together, these data indicate that the large fluctuation of the N-terminus of pHEL48-61 stabilizes the I-Ak complex, whereas without the fluctuation in the PFRs, a shorter peptide may not be able to maintain a protein complex that is stable enough to have biological function.
Figure 5.

Comparative MD simulations confirmed that the PFR-peptide MHC complex is stabilized by a large fluctuation of the peptide N-terminus that lies outside the binding groove. (A) Atomic fluctuations from MD simulations for I-Ak/pHEL48-61, I-Ak/pHEL52-61, and I-Ak/pHEL52-58, including the B factors from the crystal structure, are shown. (B and C) The correlation rates of the protein dynamics are presented for I-Ak/pHEL48-61 (B) and I-Ak/pHEL52-61 (C). Positive correlation rates are colored red (correlation rate: 0.75–1.0) or pink (correlation rate: 0.5–0.75). Negative correlation rates are colored blue (correlation rate: 0.75–1.0) or light blue (correlation rate: 0.5–0.75). Positive correlations are shown in the lower-right triangle, and negative correlations are shown in the upper-left triangle. The intensity of the shading is proportional to the magnitude of the correlation rate. The horizontal and vertical axes indicate the residue number (I-Akα chain: 1-182; I-Akβ chain: 183-367; pHEL48-61: 368-381; and pHEL52-61: 368-377). (D) The binding free energy was calculated using the MM-PB/SA method. Blue bars indicate the binding free energy, red bars indicate the enthalpy, and yellow bars indicate the entropy. To see this figure in color, go online.
Subsequently, we analyzed the correlation rate for protein dynamics. The collective motion of I-Ak/pHEL48-61 exhibited a highly positive correlation between the C-terminus of the peptide and the binding sites in both the I-Akα and I-Akβ chains (Fig. 5 B). However, there was no correlation between the N-terminus of the peptide and the binding site of I-Ak. Within I-Ak/pHEL48-61, there were weak and negative correlation sites between the I-Akα and I-Akβ chains. Conversely, the C-terminus of the peptide correlated weakly with the binding groove of the I-Akα chain in the I-Ak/pHEL52-61 complex (Fig. 5 C). Overall, the pHEL48-61 complex had a tighter conformation than pHEL52-61, suggesting that the latter peptide engages in a greater degree of independent motion relative to MHC.
Next, we calculated the binding free energy of I-Ak/HEL52-58, I-Ak/HEL52-61, and I-Ak/pHEL48-61 using the MM-PB/SA method (Fig. 5 D). The binding free energy increased as the size of the peptide increased, and this tendency was dominated by the value of the binding enthalpy. Conversely, the calculated entropy did not linearly increase with the size of the peptide. Thus, the large fluctuations of the PFRs in I-Ak/pHEL48-61 contribute to entropic stabilization of the complex.
Discussion
Our results demonstrate that peptides bound in the MHC groove exhibit distinct motions, based on differences in the conformations adopted by peptides of varying length within these complexes. Some T cells may be biased toward interaction with a peptide that undergoes conformational fluctuations in the groove of the MHC molecule, likely due to recognition of a transitional conformation generated by the rapidly fluctuating peptide. Borbulevych et al. (6) proposed that the dynamic properties (i.e., the molecular motions of the peptide and MHC molecule) of HLA-A2 are differentially tuned by the Tax and Tel1p peptides (8,11,43). This antigen-dependent tuning of peptide/MHC molecular flexibility may be similar to the phenomenon we observed in our DXT experiments. In fact, we initiated this study based on the observation that I-Ak/HEL52-61 can engage with multiple isotypes more easily than I-Ak/pHEL48-61 because the former peptide activates both type A and type B T cells to comparable extents, whereas the latter mainly activates type A T cells (19).
It is worth noting that both DXT and anisotropy showed a similar tendency in terms of the difference between the peptides, even though the timescales were different. The nanosecond timescale of anisotropy may represent the real motion of the peptide itself, and the slow millisecond motion detected by DXT may represent the coherent motion of the peptide with the MHC α-helix, which is slower than the peptide itself. This may impact TCR recognition, since the on-rates of TCR for MHC/peptides are usually very slow (44).
We found that in addition to creating diversity in the TCR ligand, PFRs played an essential role in the MHC II complex. Because most bound peptides are longer than the groove, the additional increase in the stability of the peptide/MHC II complex may be a general feature of MHC II-binding peptides (22). Here, we demonstrate the rationale for this increased stability: N-terminal PFRs undergo massive structural fluctuations, which increase the entropy. In normal protein interactions, the changes in enthalpy and entropy are compensated for, resulting in a marginal gain in free energy (45). In the case of MHC II, it is logical for PFR regions to be free to move so that they can provide strong stability to the complexes.
Conclusions
This study, in which we measured tiny motions related to immune functions, may be the first successful effort of its kind. Previous studies examined the peptide/MHC-TCR interaction using structural data obtained by x-ray crystallography (4). Although those studies provided new insights that furthered our understanding of antigen recognition by T cells, their results were obtained from the most compressed and stable forms of the molecules involved in the interactions. However, proteins in solution are flexible and undergo their own unique movements, determined by intra- and intermolecular interactions. Thus, the extra flexibility of the peptide/MHC complex could play a critical role in the interaction with TCR.
DXT was developed with the goal of acquiring structural information about the internal MD of individual molecules (30). Currently, single-molecule fluorescence resonance energy transfer (FRET) (46) is used to measure nanometer-scale distances and changes in distances or intermolecular orientations between the two fluorophores, both in vitro and in vivo. However, it is difficult to measure intramolecular structural changes of single protein molecules using single-molecule FRET due to the instability of the signal intensity and the lack of monitoring precision under physiological conditions (47). The DXT method has been used to observe individual DNA molecules (26), denatured proteins (48), and functional membrane proteins (49) such as KcsA (27), as well as for quantitative single-molecule analysis of antigen-antibody interactions (50), monitoring of super-weak forces (on the order of piconewtons) (26), and studies of ATP-dependent group II chaperonin (51).
The idea that molecular motion plays an important role in protein-protein interactions is an emerging concept in biology. Here, we used peptide-MHC interactions to demonstrate, to our knowledge, a novel protein-protein interaction; however, it is possible that a wide variety of biological systems may also depend on such interactions. Using DXT in combination with biological, biochemical, and structural analyses in a wide variety of systems should provide information regarding the dynamic roles of molecular motion in protein-protein interactions.
Acknowledgments
We thank Dr. N. Ohta and Dr. N. Yagi (JASRI/SPring-8) for their technical assistance during the DXT experiments at SPring-8. Fluorescence anisotropy was performed in collaboration with Horiba Ltd. Kyoto.
This work was supported by the CREST, Japan Science and Technology Agency.
Contributor Information
Haruo Kozono, Email: kozonoh@rs.noda.tus.ac.jp.
Yuji C. Sasaki, Email: ycsasaki@k.u-tokyo.ac.jp.
References
- 1.Blum J.S., Wearsch P.A., Cresswell P. Pathways of antigen processing. Annu. Rev. Immunol. 2013;31:443–473. doi: 10.1146/annurev-immunol-032712-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today. 1998;19:395–404. doi: 10.1016/s0167-5699(98)01299-7. [DOI] [PubMed] [Google Scholar]
- 3.Sewell A.K. Why must T cells be cross-reactive? Nat. Rev. Immunol. 2012;12:669–677. doi: 10.1038/nri3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rudolph M.G., Stanfield R.L., Wilson I.A. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 5.Macdonald W.A., Chen Z., McCluskey J. T cell allorecognition via molecular mimicry. Immunity. 2009;31:897–908. doi: 10.1016/j.immuni.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 6.Borbulevych O.Y., Piepenbrink K.H., Baker B.M. T cell receptor cross-reactivity directed by antigen-dependent tuning of peptide-MHC molecular flexibility. Immunity. 2009;31:885–896. doi: 10.1016/j.immuni.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin Y., Mariuzza R.A. The multiple mechanisms of T cell receptor cross-reactivity. Immunity. 2009;31:849–851. doi: 10.1016/j.immuni.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Baker B.M., Scott D.R., Hawse W.F. Structural and dynamic control of T-cell receptor specificity, cross-reactivity, and binding mechanism. Immunol. Rev. 2012;250:10–31. doi: 10.1111/j.1600-065X.2012.01165.x. [DOI] [PubMed] [Google Scholar]
- 9.Burrows S.R., Chen Z., Rossjohn J. Hard wiring of T cell receptor specificity for the major histocompatibility complex is underpinned by TCR adaptability. Proc. Natl. Acad. Sci. USA. 2010;107:10608–10613. doi: 10.1073/pnas.1004926107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y.C., Chen Z., Gras S. The energetic basis underpinning T-cell receptor recognition of a super-bulged peptide bound to a major histocompatibility complex class I molecule. J. Biol. Chem. 2012;287:12267–12276. doi: 10.1074/jbc.M112.344689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hawse W.F., Gloor B.E., Baker B.M. Peptide modulation of class I major histocompatibility complex protein molecular flexibility and the implications for immune recognition. J. Biol. Chem. 2013;288:24372–24381. doi: 10.1074/jbc.M113.490664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Painter C.A., Stern L.J. Conformational variation in structures of classical and non-classical MHCII proteins and functional implications. Immunol. Rev. 2012;250:144–157. doi: 10.1111/imr.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fremont D.H., Crawford F., Kappler J. Crystal structure of mouse H2-M. Immunity. 1998;9:385–393. doi: 10.1016/s1074-7613(00)80621-4. [DOI] [PubMed] [Google Scholar]
- 14.McFarland B.J., Beeson C., Sant A.J. Cutting edge: a single, essential hydrogen bond controls the stability of peptide-MHC class II complexes. J. Immunol. 1999;163:3567–3571. [PubMed] [Google Scholar]
- 15.McFarland B.J., Katz J.F., Sant A.J. Energetic asymmetry among hydrogen bonds in MHC class II∗peptide complexes. Proc. Natl. Acad. Sci. USA. 2001;98:9231–9236. doi: 10.1073/pnas.151131498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anders A.K., Call M.J., Wucherpfennig K.W. HLA-DM captures partially empty HLA-DR molecules for catalyzed removal of peptide. Nat. Immunol. 2011;12:54–61. doi: 10.1038/ni.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Painter C.A., Negroni M.P., Stern L.J. Conformational lability in the class II MHC 310 helix and adjacent extended strand dictate HLA-DM susceptibility and peptide exchange. Proc. Natl. Acad. Sci. USA. 2011;108:19329–19334. doi: 10.1073/pnas.1108074108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pos W., Sethi D.K., Wucherpfennig K.W. Crystal structure of the HLA-DM-HLA-DR1 complex defines mechanisms for rapid peptide selection. Cell. 2012;151:1557–1568. doi: 10.1016/j.cell.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lovitch S.B., Pu Z., Unanue E.R. Amino-terminal flanking residues determine the conformation of a peptide-class II MHC complex. J. Immunol. 2006;176:2958–2968. doi: 10.4049/jimmunol.176.5.2958. [DOI] [PubMed] [Google Scholar]
- 20.Mohan J.F., Unanue E.R. Unconventional recognition of peptides by T cells and the implications for autoimmunity. Nat. Rev. Immunol. 2012;12:721–728. doi: 10.1038/nri3294. [DOI] [PubMed] [Google Scholar]
- 21.Pu Z., Lovitch S.B., Unanue E.R. T cells distinguish MHC-peptide complexes formed in separate vesicles and edited by H2-DM. Immunity. 2004;20:467–476. doi: 10.1016/s1074-7613(04)00073-1. [DOI] [PubMed] [Google Scholar]
- 22.Nelson C.A., Petzold S.J., Unanue E.R. Identification of two distinct properties of class II major histocompatibility complex-associated peptides. Proc. Natl. Acad. Sci. USA. 1993;90:1227–1231. doi: 10.1073/pnas.90.4.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Brien C., Flower D.R., Feighery C. Peptide length significantly influences in vitro affinity for MHC class II molecules. Immunome Res. 2008;4:6. doi: 10.1186/1745-7580-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carson R.T., Vignali K.M., Vignali D.A. T cell receptor recognition of MHC class II-bound peptide flanking residues enhances immunogenicity and results in altered TCR V region usage. Immunity. 1997;7:387–399. doi: 10.1016/s1074-7613(00)80360-x. [DOI] [PubMed] [Google Scholar]
- 25.Arnold P.Y., La Gruta N.L., Vignali D.A. The majority of immunogenic epitopes generate CD4+ T cells that are dependent on MHC class II-bound peptide-flanking residues. J. Immunol. 2002;169:739–749. doi: 10.4049/jimmunol.169.2.739. [DOI] [PubMed] [Google Scholar]
- 26.Sasaki Y.C., Okumura Y., Yagi N. Picometer-scale dynamical x-ray imaging of single DNA molecules. Phys. Rev. Lett. 2001;87:248102. doi: 10.1103/PhysRevLett.87.248102. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu H., Iwamoto M., Oiki S. Global twisting motion of single molecular KcsA potassium channel upon gating. Cell. 2008;132:67–78. doi: 10.1016/j.cell.2007.11.040. [DOI] [PubMed] [Google Scholar]
- 28.Kozono H., White J., Kappler J. Production of soluble MHC class II proteins with covalently bound single peptides. Nature. 1994;369:151–154. doi: 10.1038/369151a0. [DOI] [PubMed] [Google Scholar]
- 29.Okumura Y., Miyazaki T., Sasaki Y.C. Fabrications of dispersive gold one-dimensional nanocrystals using vacuum evaporation. Thin Solid Films. 2005;471:91–95. [Google Scholar]
- 30.Sasaki Y.C., Suzuki Y., Yanagihara M. Tracking of individual nanocrystals using diffracted x rays. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 2000;62(3 Pt B):3843–3847. doi: 10.1103/physreve.62.3843. [DOI] [PubMed] [Google Scholar]
- 31.Fremont D.H., Monnaie D., Unanue E.R. Crystal structure of I-Ak in complex with a dominant epitope of lysozyme. Immunity. 1998;8:305–317. doi: 10.1016/s1074-7613(00)80536-1. [DOI] [PubMed] [Google Scholar]
- 32.Jorgensen W.L., Chandrasekhar J., Madura J.D. Computer simulations of organic reactions in solution. Ann. N. Y. Acad. Sci. 1986;482:198–209. doi: 10.1111/j.1749-6632.1986.tb20951.x. [DOI] [PubMed] [Google Scholar]
- 33.Case D.A., Pearlman D.A., Kollman P.A. University of California; San Francisco: 2004. AMBER 8. [Google Scholar]
- 34.Narumi, T., Y. Ohno, …, M. Taiji. 2006. A 185 Tflops simulation of amyloid-forming peptides from yeast prion Sup35 with the special-purpose computer system MD-GRAPE3. Proc. Supercomputing 2006 (CD-ROM).
- 35.Tajii, M. 2004. A 165 Gflops application specific LSI for molecular dynamics simulations. IEEE Comput. Soc. (CD-ROM).
- 36.Duan Y., Wu C., Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 37.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 38.Berendsen H.J.C., Vangunsteren P.J., Haak A., Jr. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 39.McCammon J.A. Protein dynamics. Rep. Prog. Phys. 1984;47:1–46. [Google Scholar]
- 40.Kollman P.A., Massova I., Cheatham T.E., 3rd Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 41.Sekiguchi H., Suzuki Y., Sasaki Y.C. Real time ligand-induced motion mappings of AChBP and nAChR using X-ray single molecule tracking. Sci. Rep. 2014;4:6384. doi: 10.1038/srep06384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keskin O., Jernigan R.L., Bahar I. Proteins with similar architecture exhibit similar large-scale dynamic behavior. Biophys. J. 2000;78:2093–2106. doi: 10.1016/S0006-3495(00)76756-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pöhlmann T., Böckmann R.A., Alexiev U. Differential peptide dynamics is linked to major histocompatibility complex polymorphism. J. Biol. Chem. 2004;279:28197–28201. doi: 10.1074/jbc.C400128200. [DOI] [PubMed] [Google Scholar]
- 44.Aleksic M., Dushek O., van der Merwe P.A. Dependence of T cell antigen recognition on T cell receptor-peptide MHC confinement time. Immunity. 2010;32:163–174. doi: 10.1016/j.immuni.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dunitz J.D. Win some, lose some: enthalpy-entropy compensation in weak intermolecular interactions. Chem. Biol. 1995;2:709–712. doi: 10.1016/1074-5521(95)90097-7. [DOI] [PubMed] [Google Scholar]
- 46.Hohng S., Joo C., Ha T. Single-molecule three-color FRET. Biophys. J. 2004;87:1328–1337. doi: 10.1529/biophysj.104.043935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roy R., Hohng S., Ha T. A practical guide to single-molecule FRET. Nat. Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sasaki Y.C., Okumura Y., Higurashi T. Observations of x-ray radiation pressure force on individual gold nanocrystals. Appl. Phys. Lett. 2006;89:053121. [Google Scholar]
- 49.Okumura Y., Oka T., Sasaki Y.C. Picometer-scale dynamical observations of individual membrane proteins: the case of bacteriorhodopsin. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2004;70:021917. doi: 10.1103/PhysRevE.70.021917. [DOI] [PubMed] [Google Scholar]
- 50.Sagawa T., Azuma T., Sasaki Y.C. Dynamical regulations of protein-ligand bindings at single molecular level. Biochem. Biophys. Res. Commun. 2007;355:770–775. doi: 10.1016/j.bbrc.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 51.Sekiguchi H., Nakagawa A., Sasaki Y.C. ATP dependent rotational motion of group II chaperonin observed by X-ray single molecule tracking. PLoS ONE. 2013;8:e64176. doi: 10.1371/journal.pone.0064176. [DOI] [PMC free article] [PubMed] [Google Scholar]