

Summary
Most B cell lymphomas arise in the germinal center (GC), where humoral immune responses evolve from potentially oncogenic cycles of mutation, proliferation, and clonal selection. Although lymphoma gene expression diverges significantly from GC-B cells, underlying mechanisms that alter the activities of corresponding regulatory elements (REs) remain elusive. Here we define the complete pathogenic circuitry of human follicular lymphoma (FL), which activates or decommissions REs from normal GC-B cells and commandeers enhancers from other lineages. Moreover, independent sets of transcription factors, whose expression was deregulated in FL, targeted commandeered versus decommissioned REs. Our approach revealed two distinct subtypes of low-grade FL, whose pathogenic circuitries resembled GC-B or activated B cells. FL-altered enhancers also were enriched for sequence variants, including somatic mutations, which disrupt transcription factor binding and expression of circuit-linked genes. Thus, the pathogenic regulatory circuitry of FL reveals distinct genetic and epigenetic etiologies for GC-B transformation.
Introduction
B cell lymphoma is one of the most common human cancers. Its prevalence is a byproduct of the complex cellular and molecular processes that tailor humoral immune responses to antigens in the germinal center (GC). In secondary lymphoid organs, GC-B cells undergo clonal expansion while activation-induced cytidine deaminase (AICDA) targets their genomes for DNA damage-associated alterations. In the GC dark zone, proliferative centroblasts (CBs) perform somatic hypermutation (SHM) to fine-tune the affinity of immunoglobulin (Ig) variable regions for their cognate antigens. Some of the CB then migrate to the light zone, where they become non-cycling centrocytes (CCs), are selected for antigen affinity, and undergo class-switch recombination (Allen et al., 2007). Recent studies indicate that, rather than being two distinct stages of differentiation, CC and CB are alternate activation states of GC-B cells (Victora et al., 2012).
Although essential for optimization of humoral responses, coincident genome damage and rapid proliferation during the GC reaction increases the risk of oncogenic lesions that underlie most types of B cell lymphoma (Rui et al., 2011). For example, follicular lymphoma (FL), a common form of non-Hodgkin lymphoma (NHL), is thought to arise from CC (Victora et al., 2012). The pathologic hallmark of FL is an AICDA-mediated t(14;18) [IgH-BCL2] translocation, which causes over-expression of the anti-apoptotic BCL2 protein (Leich et al., 2011). However, this primary genetic lesion is insufficient to initiate FL. Indeed, BCL2 transgenic mice infrequently develop FL, and t(14;18) bearing B cells are found in healthy individuals who do not develop lymphoma (Cheung et al., 2009; Dölken et al., 1996). A fundamental focus of lymphoma biology remains the identification of cooperative mechanisms that promote oncogenesis.
One clear outcome of these cooperative mechanisms is a substantial revision of normal GC expression programs during transformation to NHL (Klein and Dalla-Favera, 2008), which culminates from a combination of genetic and epigenetic changes. First, compared with GC-B cells, NHL have altered expression of at least some transcription factors (TFs) that are essential to the GC reaction (Alizadeh et al., 2000). Second, NHL have mutations in histone modifiers, leading to altered chromatin landscapes (Morin et al., 2011; Yap et al., 2011). These epigenomic perturbations may coordinately silence large cohorts of genes (e.g., tumor suppressors), while activating genes involved in proliferation (Baylin and Jones, 2011). Third, many diseases are commonly linked to genetic or epigenetic differences in non-coding sequences that encompass transcriptional regulatory elements (REs) (Akhtar-Zaidi et al., 2012; Maurano et al., 2012). This shared genetic–epigenetic etiology may be especially relevant to NHL, due to its inherent genome instability and dysregulation of histone modifiers, both of which could impact RE function.
To understand the oncogenic processes that sculpt NHL transcriptomes, it is critical to identify altered distal REs (DREs) and assign them to target genes, thereby defining pathogenic circuitry that drives cellular transformation. Analyses of chromatin modification patterns have linked some DREs with their target promoters in normal cells, revealing aspects of lineage-specific regulatory circuits (Gerstein et al., 2012; Maurano et al., 2012). To decipher oncogenic changes to regulatory circuits, purified malignant cells from primary tumors should be compared with normal counterparts rather than using unsorted biopsy samples containing a mixture of cell types, or cancer lines, which accumulate widespread revisions to the genome and epigenome during long-term culture (Masters, 2000; Victora et al., 2012). To circumvent these obstacles, we focused on FL, an indolent B cell malignancy that is incurable and often transforms to a more aggressive lymphoma (Lenz and Staudt, 2010).
We report an integrative analysis of –omics data from purified human FL-B cells and their normal GC-B counterparts, exposing the oncogenic regulatory circuitry of this cancer. The FL circuitry is a composite of enhancers that are engaged normally in CC or CB subsets, but are chronically activated or attenuated in FL, as well as DREs usurped from a wide range of other, non-B lineages. Importantly, augmented or attenuated DREs are each targeted by a distinct set of TFs that exhibit altered expression in FL. Our approach also revealed two previously unappreciated subtypes of low-grade FL, with distinct expression profiles and corresponding sets of aberrant DREs. The FL-altered DREs are also enriched for disease-associated single nucleotide polymorphisms (SNPs) and, remarkably, for somatic mutations. Indeed, several of the variants, located in attenuated DREs, impair binding of TFs that play key roles in GC reactions and correspond to reductions in target gene expression. These discoveries establish the epigenetic and genetic etiologies that mediate transformation of normal GC-B cells into a common cancer and pave the way for personalized epigenetic therapies that target subtype-specific control elements.
Results
The FL Regulome Reflects its Centrocyte Origins
Rewiring of gene expression in cancer evolves from a series of genetic and epigenetic changes that impinge on the activities of transcriptional promoters and their DREs. Deciphering how these processes drive cancer requires characterization of regulomes in purified malignant cells to compare with normal counterparts. Thus, we purified malignant B cells from lymph node biopsies of 18 FL patients (Table S1). Control B cells were purified from the peripheral blood of healthy individuals (PBB), FL patients, and excised tonsils (TsB). Chromatin, RNA, and genomic DNA harvested from these samples were analyzed by ChIP-seq, RNA-seq, expression arrays, and SNP arrays, respectively. We used FAIRE-seq to identify putative cis-elements in each sample (Giresi and Lieb, 2009) and analyzed hallmark chromatin features (Bernstein et al., 2012), including H3K27ac and H3ac, to assess relative RE activities among B cell populations. To standardize comparisons between samples, the data were normalized for differences in read depth, and overlapping peaks were merged, generating a consolidated list of putative REs.
Based on gene expression profiles, FL and Diffuse Large B Cell Lymphoma (DLBCL) most closely resemble GC-B cells (Victora et al., 2012). We purified CC and CB from human tonsils by flow cytometry using the following surface markers: CD19+CD10+CD44loCXCR4+ (CB) and CD19+CD10+CD44loCXCR4− (CC). Successful separation was confirmed by comparing the expression of genes known to discriminate CC from CB (Figure S1A). Importantly, the close relationship between CC and CB, compared with other hematopoietic lineages, is evident from clustering analyses of open chromatin regions (Figure S1B), underscoring the strong functional links between regulomes and transcriptomes.
To determine the most appropriate control cell type for defining pathogenic FL circuitry, we first compared expression of genes that distinguish CC from CB (Victora et al., 2012). Consistent with a previous study, expression of these genes in FL is most similar to CC, diverging almost completely from CB (Figure 1A). In contrast, cultured NHL cell lines resemble the highly proliferative CB subset. Epigenomic analyses complement these transcriptome comparisons; the majority of REs in FL are shared with GC-B, though a greater proportion overlap with CC than CB (Figure 1B). Together, these data support the notion that FL arises from CC (Victora et al., 2012), and that comparison to CC should be used for identification of REs with altered function in FL.
Figure 1. The Centrocyte Origins and Alterations to the FL Regulome.
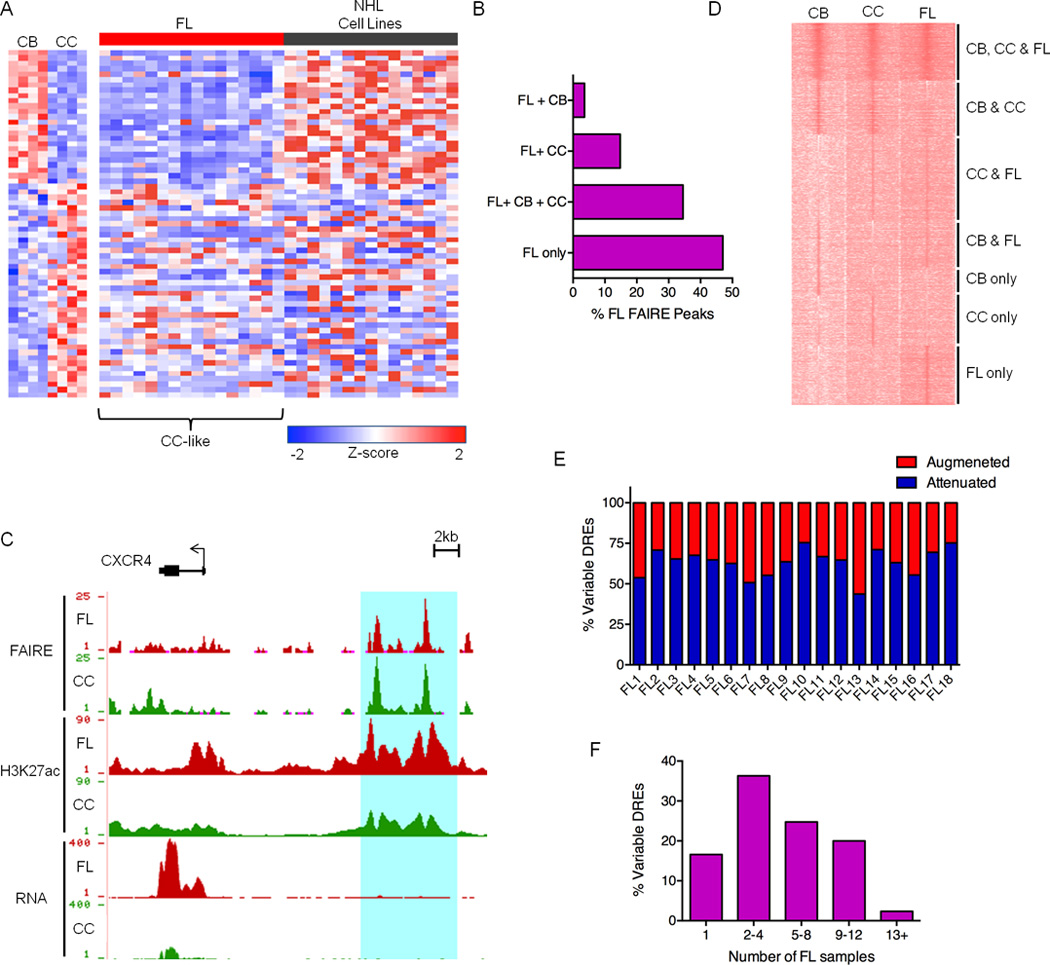
A) Expression profiles of FL and NHL cell lines for a panel of genes differentially expressed in CB versus CC. B) Bar graph showing unique and shared FAIRE peaks in FL and GC-B cell populations. C) UCSC Genome Browser views of FAIRE-Seq, H3K27ac ChIP-Seq and RNA-Seq data from FL and CC samples, illustrating a collection of DREs located near the CXCR4 gene. FAIRE and ChIP data are presented as number of reads per million mapped reads and plotted on an axis of 1–25 (FAIRE) and 1–90 (H3K27ac). RNA data are presented as number of aligned, in silico extended reads per 10 bp, on a scale of 1–400 reads. D) H3K27ac ChIP-Seq intensities for DREs in representative CB, CC, and FL samples. Data are presented as k-means clustering of tag densities per 200 bp within a window of 10 kb around the DREs. E) Percent of variable DREs with ≥ 2-fold increase (augmented) or decrease (attenuated) in FAIRE-seq, H3ac ChIP-seq or H3K27ac ChIP-seq signal for FL samples relative to CC. F) Recurrence rates of variable DREs depicted by proportion detected in number of FL samples.
Defining the FL-altered Regulome
Pathogenic gene expression in many diseases, including cancer, is caused by changes in DRE activity, which in turn, alter promoter function (Akhtar-Zaidi et al., 2012). Accordingly, we set out to identify DREs with augmented or attenuated activities in FL compared to normal CC. Although FAIRE-seq data mark DRE locations in the genome, both poised and active cis-elements emerge as peaks. To measure the relative activity of DREs, we incorporated ChIP-seq data for H3K27ac and H3ac, which mark active DREs. For example, nearly identical FAIRE peaks are detected in both CC and FL samples for a cis-element located near the CXCR4 gene (Figure 1C), which encodes a chemokine receptor required for light (CC) and dark (CB) zone organization (Allen et al., 2007). This region is substantially enriched for H3K27ac in FL, indicating augmented activity. Indeed, FL also exhibits higher CXCR4 expression when compared with CC controls. A global view of H3K27ac levels at DREs in FL- and GC-B cells segregates these regions into multiple categories based on relative activity, including FL hyper-activation or attenuation of DREs characteristic of CC, CB, or neither of these closely related subsets (Figure 1D).
To define the FL dysregulome, we assigned variable DREs as regions that differ more than two-fold in FAIRE, H3K27ac, or H3ac intensity for an individual lymphoma versus averaged values for the same regions in CC samples. As expected from similarities between FL and CC, the majority of REs remain unchanged in lymphoma B cells, while the remainder exhibit altered signal intensity (15–30K per FL). In nearly all FL, more DREs are attenuated, though a substantial proportion exhibit enhanced activity (25–55%) (Figure 1E). Epigenetic alteration of DREs cannot be attributed exclusively to mutations in chromatin modifiers, which occurred in only a subset of samples (Table S2). As shown in Figure S1C, H3K27ac peak intensity in CC control samples is highly reproducible; less than 5% of DREs are variable in CC samples, whereas 45–50% of these elements are variable in FL (Figure S1D). Augmented or attenuated DRE intensities were also similar when comparing FL to patient-matched PBB or any of the CC controls (Figure S1E). Underscoring their relevance to common pathways of lymphomagenesis, the majority of variable DREs are recurrent, with greater than 80% identified in at least 2 FL (Figure 1F).
The genomic location of variable DREs – distal, non-promoter regions – coupled with their epigenetic profiles (FAIRE+H3K27ac+) indicates enhancer function. Indeed, luciferase reporter assays revealed enhancer activity for 7/10 DREs in cultured lymphoma cells (Figure S1F). A subtype of DRE, termed super-enhancers (SEs), are composed of large RE clusters and drive the expression of lineage-specifying genes (Hnisz et al., 2013; Whyte et al., 2013). In addition, SEs may contribute to pathologic gene expression in DLBCL (Chapuy et al., 2013). To evaluate the variability of SEs in FL versus CC, we identified these elements by their unusually high H3K27ac density (Figure S1G and Table S3) (Whyte et al., 2013). Consistent with previous reports, genes within 500 kb of SEs are expressed more robustly than those near conventional enhancers (Figure S1H). Using our criteria, a subset (~10%) of SEs can be categorized as variable DREs in FL (Figure S1I). However, the proportion of variable SEs is substantially lower than that of conventional enhancers with altered activity in FL (45–50%). The relative lack of variability in SEs likely reflects their role in maintaining B cell identity. Indeed, genes involved in immune responses and lymphocyte activation are enriched in the SE circuits (Table S3). We conclude that a majority of variable DREs are conventional enhancers whose altered function resets the transcriptional circuitry of B cells to drive transformation.
Pathogenic FL Regulatory Circuits
To decipher the pathogenic regulatory circuitry of FL, variable DREs must be connected to their target genes. Chromatin accessibility at enhancers and nearby promoters often correlate, providing a pattern-based strategy to assign regulatory circuits (Thurman et al., 2012). Using this approach, we connected variable DREs to genes within 500 kb that exhibit concordant changes in chromatin at their transcription start sites (TSS) (Figure S2A). Approximately one-third of the variable DREs were linked to a single TSS, whereas the majority had potential connections to two or more target genes (Figure 2A). Moreover, the average expression of genes with concordant TSS chromatin is significantly elevated or attenuated compared to that of genes near unchanged DREs (Figure 2B) or to all neighboring genes (Figure S2B).
Figure 2. Pathogenic circuitry of FL.
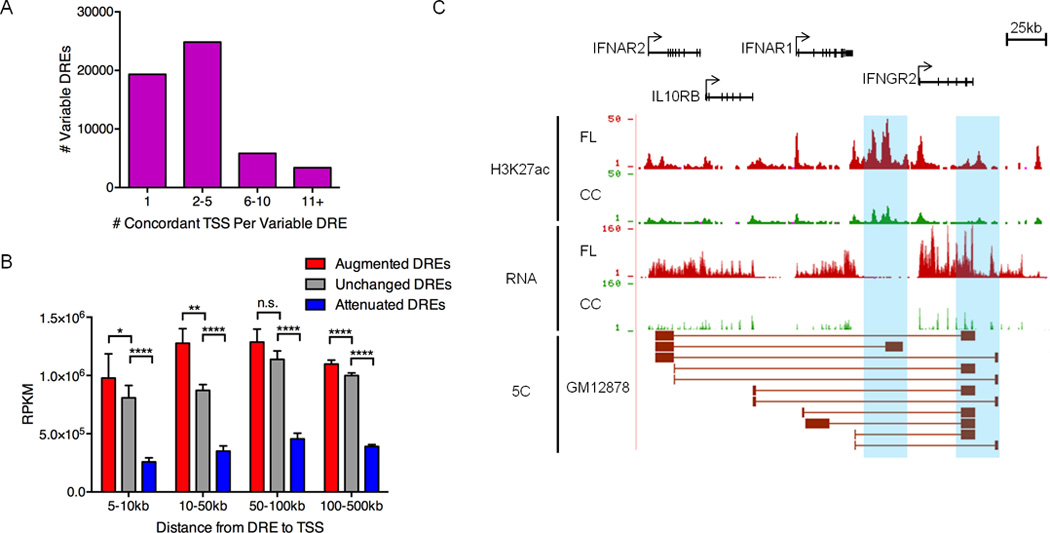
A) Number of TSSs with chromatin fold-change that is concordant with nearby variable DREs (within 500kb). B) Mean transcript abundance as determined by RNA-Seq for genes linked to augmented, unchanged or attenuated DREs and located within the distances shown from the DREs. Statistical significance (Mann-Whitney test): *p≤ 0.05 ** p≤ 0.01, **** p≤ 0.0001. C) UCSC Genome Browser views of H3K27ac ChIP-seq data, showing augmented DREs (highlighted in blue). RNA-seq data depict the corresponding up-regulation of mRNA from DRE target genes. The bottom track shows significant spatial interactions in GM12878 B cells (ENCODE, 5C data) between restriction fragments encompassing the DREs and restriction fragments near the target gene TSSs (Sanyal et al., 2012).
Nevertheless, chromatin-based correlations do not evaluate the functional output of putative DRE-gene connections, i.e., expression levels. Therefore, we applied a new level of stringency: concordant changes in the expression of genes predicted to be targets of DREs by chromatin profiling (Figure S2A). Only 53% of putative target genes were verified by this expression filter (Table S4), indicating a 47% false-positive rate for predicting DRE-promoter connections using only chromatin patterns. Approximately 40% of the circuits with altered activity in FL are normally employed in the GC reaction, including CC- or CB-specific connections, as well as those used by both subsets (Figure S2C). However, the majority of altered FL circuits are not engaged in either subset of GC-B lymphocytes, indicating a widespread change in the B cell regulome.
The validity of connections identified by this strategy is highlighted by the type 2 cytokine receptor locus that includes the IFNAR1, IFNAR2, IL10RB, and IFNGR2 genes (Figure 2C). Activation of these receptors alters B cell responses, including enhanced viability and proliferation. However, to our knowledge, overexpression of these genes has not been linked to FL. The pathogenic circuits for each of the two augmented DREs at the 3’ end of this cluster include predicted regulatory interactions with multiple TSSs of the receptor genes. Published 5C data for the B lymphoblastoid cell line GM12878 confirms interactions between these DREs and regions proximal to many of the TSSs (Sanyal et al., 2012). Thus, integrative analysis of transcriptome and epigenome data reveals a core pathogenic circuitry for FL that incorporates regulome components from both GC subsets. The newly defined circuitry also identifies variable DRE connections to genes whose roles in FL pathogenesis were previously unknown.
Epigenome-Centric Analysis Segregates FL into Two Distinct Subtypes
Despite significant heterogeneity in clinical behavior and therapeutic response, low-grade FL is uniformly classified for prognosis and treatment (Izutsu, 2014). One source of FL heterogeneity may be utilization of distinct B cell regulatory circuits during oncogenesis. To test this, we performed unbiased hierarchical clustering of chromatin features for FL and normal B cell populations, including CC, CB, resting PBB, and in vitro activated PBB, which resemble plasmablasts (Tarte et al., 2002). Unexpectedly, this epigenomic analysis revealed two distinct FL subtypes with DREs that differ significantly (p <0.01). While the subtype 1 enhancer profile parallels that of CC, the subtype 2 profile includes DREs with activities similar to in vitro activated PBB (Figure 3A). Thus, enhancer profiling identifies two distinct subtypes of FL that resemble different stages of B cell activation and differentiation, much like previous expression profiling studies have segregated subtypes of DLBCL (Alizadeh et al., 2000).
Figure 3. Epigenome-centric analyses reveal distinct FL subtypes.
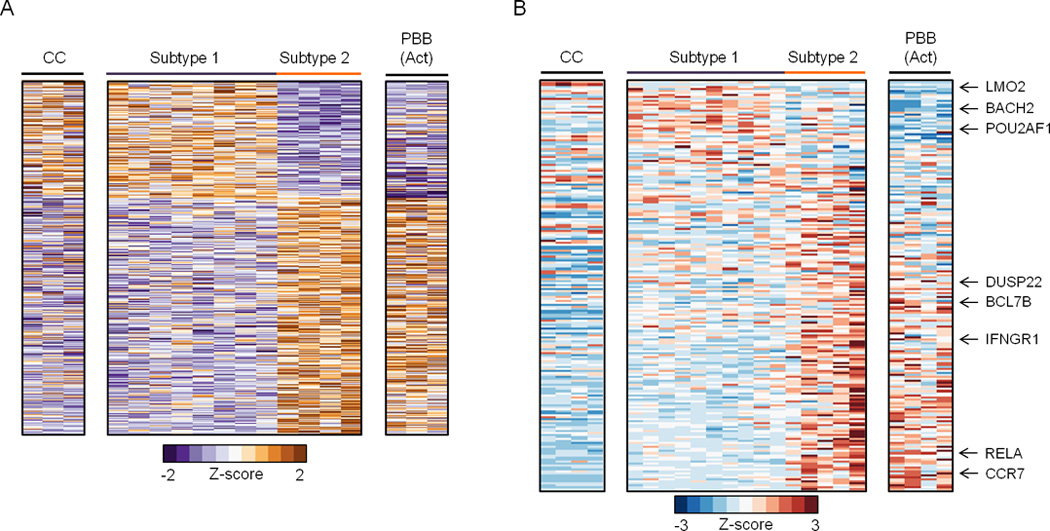
A) Heatmap for variable DREs with significantly different levels of open chromatin (FAIRE, normalized reads), which separate FL into two subtypes. B) Heatmap for genes that are differentially expressed in distinct stages of B cell activation or differentiation (Longo et al., 2009) as determined by Gene Set Enrichment Analysis of microarray data (FDR q<0.0001, GSEA).
To gain insight into the genes regulated by subtype 1 and 2 DREs, we focused on a cohort shown previously to distinguish GC- from other mature B cell populations (Longo et al., 2009). Strikingly, subtype-specific DREs form regulatory circuits with >95% of these genes. Figure 3B shows expression data for genes linked to subtype-specific DREs, confirming that components of CC and plasmablast transcriptional programs are differentially engaged by enhancers specific to subtypes 1 and 2, respectively. Importantly, when we focus on these genes defined by our epigenome-centric approach, the divergent expression patterns are also evident in microarray data from larger FL sample sets (Figure S3A) (Compagno et al., 2009; Dave et al., 2004). For example, a subtype 2-specific circuit connects a variable DRE with enhanced expression of the CCR7 gene. CCR7 encodes the CCL19 and CCL21 chemokine receptor (Figure S3B), which has been linked to NHL retention within the lymph node, thereby providing transformed cells with a survival advantage (Rehm et al., 2011). The two FL subtypes also have remarkably different expression patterns for genes involved in a range of relevant biological processes, including DNA repair and NF-κB signaling pathways (Figure S3C).
Though sample numbers are limited, we observe no association of either subtype with patient age, gender, tumor purity, grade, or presence of BCL2 translocations (Tables S1 and S5). However, patients in subtype 2 were more likely to have received chemotherapy for lymphoma compared to subtype 1 (3/6 vs 2/10). Our epigenome-centric approach uncovered two subtypes of low-grade FL characterized by distinct enhancer profiles and linked transcriptional circuitries, suggesting divergent modes of pathogenesis.
FL Discards Non-Essential Components of GC Circuitry
Humoral immune responses in the GC are evolutionary processes that incorporate molecular and cellular mechanisms unique to B lymphocytes, including Ig isotype switching, somatic mutation, and B cell receptor signaling (Victora and Nussenzweig, 2012). Many of these antigen-induced responses may be dispensable or deleterious for FL survival. Indeed, more than half of the DREs identified in CC are attenuated in at least one FL sample (Figure 1E). When genes connected to attenuated DREs were subjected to ontology analysis, we found enriched pathways that likely are detrimental to lymphomagenesis, including cell cycle checkpoints and apoptosis (Figure 4A). Many pathways critical for the GC reaction, but presumably dispensable for generating FL, including somatic hypermutation of Ig genes and B cell-mediated immunity, also were down-regulated.
Figure 4. FL suppresses REs associated with GC identity.
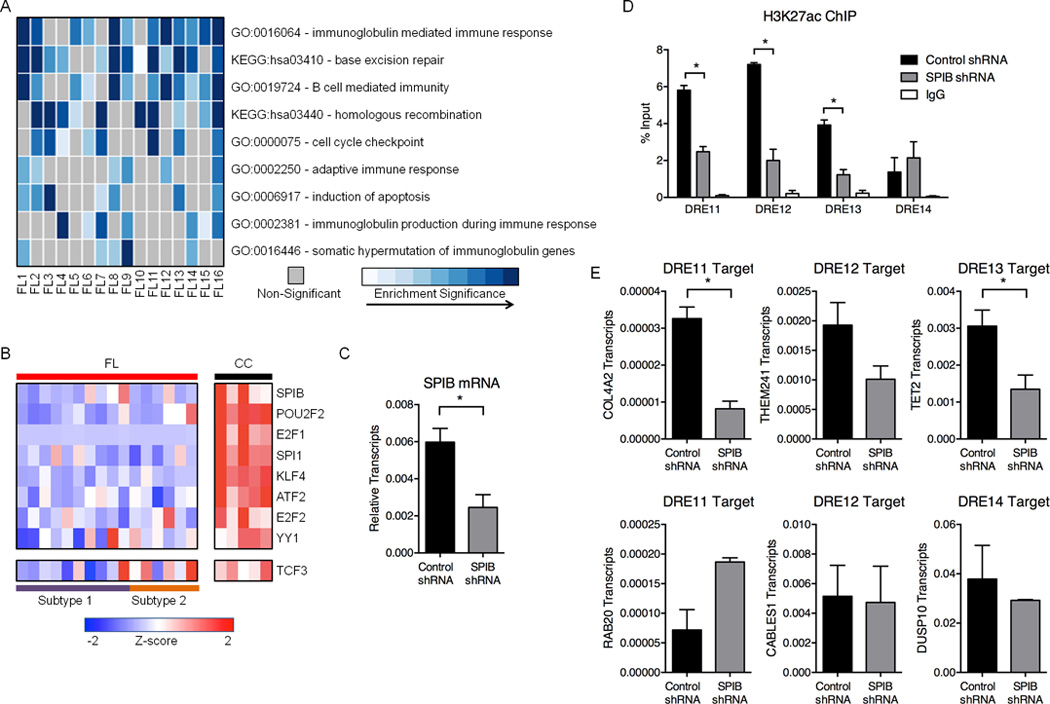
A) Heatmap representation of enriched Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways identified from down-regulated genes within 500kb of attenuated DREs. B) Expression profiles (microarray) for TFs predicted to bind motifs in attenuated DREs. Refer to Table S6 for expression values. C) SPIB transcripts, as measured by qPCR, in GM12878 cells transfected with either control or SPIB shRNA. Results represent the mean ± SEM of three independent experiments. D) H3K27ac ChIP assays in GM12878 cells transfected with control or SPIB-specific shRNA. Associated DNA was analyzed via qPCR using primers spanning DREs harboring SPIB binding sites. Results represent the mean ± SEM of three independent experiments. E) Transcript levels for DRE target genes, as measured by qPCR, in GM12878 cells transfected with either control or SPIB-specific shRNA. Results represent the mean ± SEM of three independent experiments. Statistical significance (unpaired t-test): * p≤ 0.05.
One strategy for decommissioning large blocks of DREs could involve suppression of their cognate TFs. To test this possibility, we identified TF binding motifs enriched in attenuated DREs, and found that most serve key functions in lymphocyte development and the GC reaction, including POU2F2, SPIB, and TCF3 (Figure 4B) (Hagman and Lukin, 2006). Indeed, mRNA expression of these TFs is considerably decreased in all FL samples compared with CC, with relatively greater suppression of TCF3 in subtype 1 compared to subtype 2 (Figure 4B). To test whether diminished levels of one such TF contribute to the decommissioning of DREs, we expressed a validated shRNA specific for SPIB in GM12878 B cells (Figure 4C). We selected for focused analysis four DREs that are known to bind the related ETS family factor PU.1 in these cells (Neph et al., 2012) and are attenuated in FL that exhibit low levels of SPIB expression. Levels of H3K27ac were reduced significantly at three of the four DREs in cells that express the SPIB-specific, but not the control shRNA (Figure 4D). The loss of H3K27ac at these three DREs coincided with attenuated expression of at least one putative target gene, whereas the putative target for the fourth DRE was unaffected (Figure 4E). Our findings indicate that coordinated suppression of B cell TFs and their target REs is a key component of the pathologic circuitry for FL transformation and survival.
FL Usurps Regulatory Circuits from Other Cell Lineages
In addition to discarding non-essential GC circuitry, FL may evolve by co-opting circuits from other cell lineages that promote growth and survival. Indeed, ~20% (11,318) of the variable DREs identified in FL are completely absent in all control B cell populations tested, including CC, CB, TsB, resting or activated PBB, and the B cell line GM12878. Nearly one-third of these “de novo” DREs overlap a region of open chromatin found in at least one of 27 primary cell types (Figure S4A) (Bernstein et al., 2012; Thurman et al., 2012). Many of the remaining de novo DREs are shared with one or more non-B cell cancers (Figure 5A). The pathologic relevance of these elements is highlighted by a de novo DRE overlapping the promoter of an anti-sense non-coding RNA in the HOXA10 locus. This de novo DRE coincides with enhanced expression of HOXA10 in FL compared to CC (Figure S4B). HOXA genes are master TFs of embryonic development and are aberrantly expressed in many cancers (Shah and Sukumar, 2010). Indeed, overexpression of HOXA10 in BM is associated with a block in B cell differentiation and induction of leukemia in mice (Argiropoulos and Humphries, 2007). Thus, FL commandeers DREs that are irrelevant for normal B cell identity or activation, suggesting that they control the expression of genes involved in cellular transformation.
Figure 5. Novel DREs in pathogenic FL regulatory circuits.
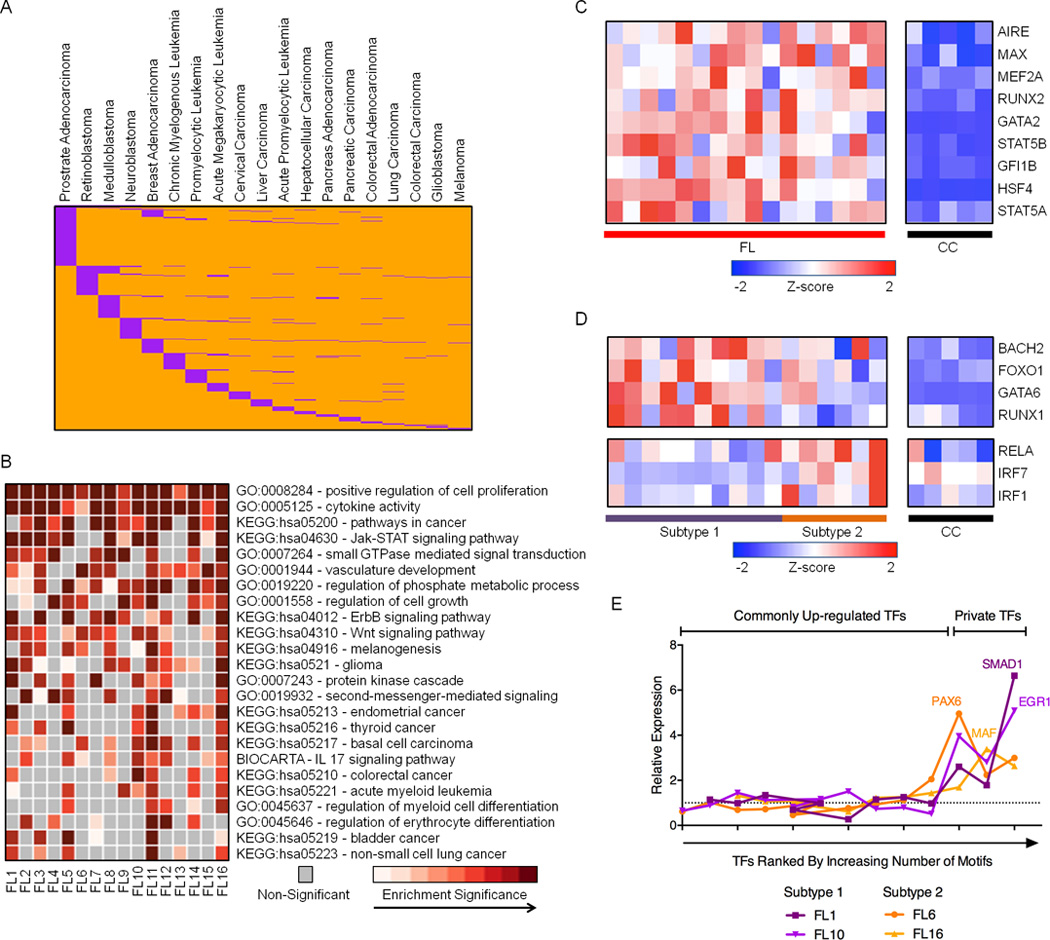
A) Distribution of 706 de novo DREs (orange) that overlap REs in other cancer types (purple). B) Heatmap representation of enriched GO terms and KEGG pathways identified from up-regulated genes within 500kb of de novo DREs. C) RNA expression profiles (microarray) for TFs predicted to bind de novo DREs that are consistently up-regulated in FL samples. Refer to Table S6 for expression values. D) RNA expression profiles (microarray data) for TFs predicted to bind de novo DREs that are differentially up-regulated in either subtype 1 or subtype 2 FL. Refer to Table S6 for expression values. E) Relative expression of TFs, ranked by number of corresponding TF motifs within private DREs, in individual FL samples compared to the average expression in all FL. Additional data are shown in Figure S4C.
To investigate this possibility, we performed ontology analysis on genes connected to de novo REs in our FL circuitry. Consistent with cell type distribution of the de novo DREs, many of the enriched pathways govern biological or oncogenic processes in other lineages, such as vasculature development and gliomagenesis (Figure 5B). Other pathways correspond to more general mechanisms of oncogenesis, including regulation of cell signaling and growth. Similar to attenuated DREs, the de novo DREs are enriched in motifs for TFs that exhibit higher expression levels across FL samples (Figure 5C). Dysregulated TFs include those involved in general cellular transformation (MAX, GFI1B) and the development of non-B cells (MEF2A, RUNX2). A subset of the enriched motifs corresponds to TFs whose augmented expression distinguishes subtype 1 from subtype 2 FL (Figure 5D). TFs with higher expression in subtype 1 FL, which resemble CC, include BACH2, a transcriptional repressor that cooperates with BCL6 to regulate gene expression in GC B cells (Huang et al., 2014), as well as TFs with diverse roles in hematopoiesis (FOXO1, GATA6). TFs with relatively higher expression in subtype 2 FL, which resemble activated B cells, include the RELA subunit of NF-κB, and IRF1, a component of the MYD88 signaling cascade (Ikushima et al., 2013). We next considered whether variable DREs unique to individual FLs arise from deregulation of additional TFs, which create these “private” DREs. We find that when an FL expresses substantially elevated levels of a unique TF compared with other FL samples, its private DREs are enriched for that TF binding site (Figures 5E and S4C). We conclude that FL commandeers oncogenic regulatory circuits, in part, by activating expression of general or lineage-inappropriate TFs.
Variable DREs Are Enriched for Inherited and Acquired Sequence Alterations
Prior studies have shown that sequence variants co-localize with regulatory regions and disrupt TF binding motifs, providing a potential genetic source for perturbations in DRE activity and, consequently, target gene expression (Corradin et al., 2014; Huang et al., 2014; Khurana et al., 2013; Maurano et al., 2012). To explore whether this mechanism is active in FL, we selected high quality sequence variants in DREs that were present in multiple RNA-, FAIRE- or ChIP-Seq datasets in an individual FL sample. We assigned each variant as a SNP based on its annotation in the 1000 Genomes or dbSNP-All SNPs databases, or as a putative somatic single nucleotide variant (SNV). To validate this variant calling method, Sanger sequencing was performed using FL samples and patient-matched peripheral blood mononuclear cells (PBMCs) when available. Of 28 identified variants, 24 were confirmed (86%). Twelve were present in FL and matched PBMC, suggesting that they are private SNPs; nine were present in the FL sample (no matched PBMC), and three were confirmed to be of somatic origin (in FL but not PBMC, Table S7). Thus, our informatics approach for identification of DRE sequence variants is highly accurate.
Consistent with a potential function in altering DRE activity, we found that both SNPs and SNVs are significantly enriched (p< 0.0001) in variable DREs (Figure 6A). Analysis of disease- and trait-associated index SNPs from the GWAS catalog revealed a significant enrichment for B cell cancer SNPs in variable DREs relative to the fraction of total SNPs in these regions (Figure 6B). In contrast, variable DREs are depleted for SNPs associated with other cancers and several unrelated traits. Although somatic mutation levels vary in subtypes of some cancers (Kandoth et al., 2013; Pasqualucci et al., 2014), there were no significant differences in SNV load for the variable DREs present in FL subtype 1 versus subtype 2 (Figure 6C). Similarly, we did not identify DRE sequence variants significantly associated with either subtype. Together, these findings suggest that the activity of some DREs is altered in FL via sequence variation, and that a subset of these pathologic elements may arise from somatic mutation during lymphomagenesis.
Figure 6. Variable DREs are enriched for inherited and acquired sequence variants.
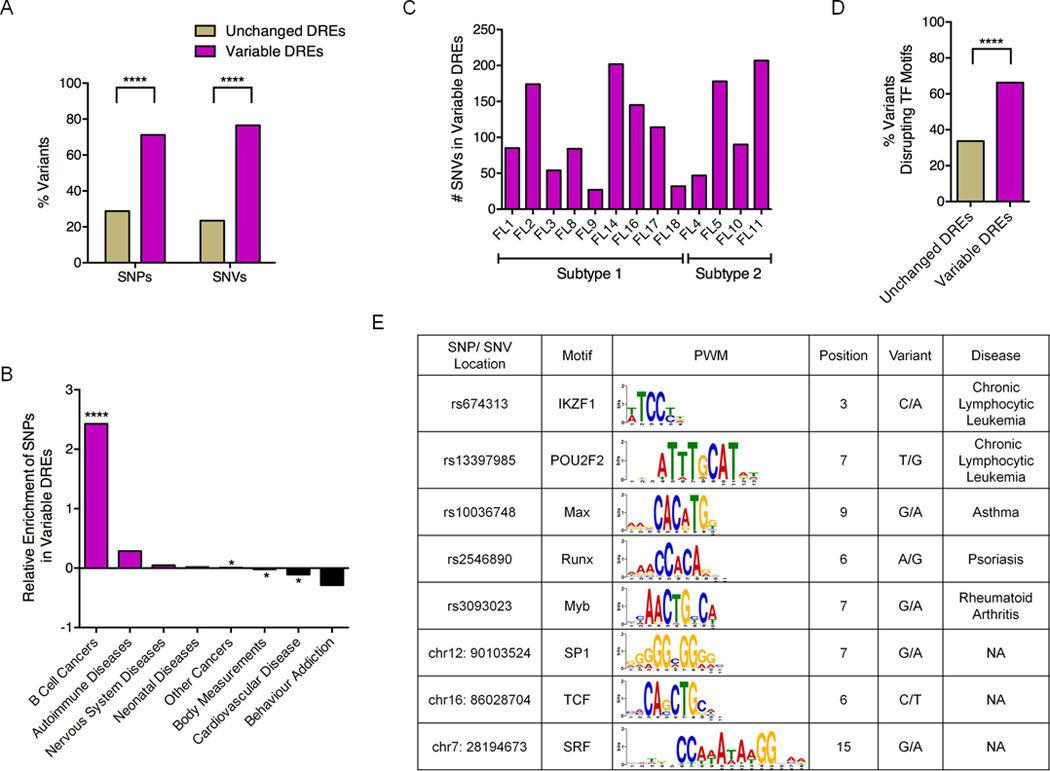
A) Percentage of variable and unchanged DREs with either SNPs and SNVs. B) Enrichment or depletion of disease- or trait-associated SNPs in variable DREs. C) Number of SNVs within variable DREs in each FL sample. D) Fraction of sequence variants that disrupt TF motifs in unchanged versus variable DREs. Statistical significance for panels A, B, and D (χ2 test): **** p≤ 0.0001. E) Representative disease-associated SNPs and SNVs predicted to disrupt TF motifs.
To explore the impact of sequence variants on DRE function, we identified SNPs or mutations predicted to disrupt TF motifs using TRANSFAC and FunSEQ (Khurana et al., 2013; Matys et al., 2006). A significantly greater fraction of motif-disrupting variants occurred in variable compared to unchanged DREs, suggesting a functional role in altering enhancer activity (Figure 6D). Moreover, a subset of these variants overlap binding motifs for TFs important in GC-B cell biology, including POU2F2, IKZF1, and TCF3 (Figure 6E). We selected three sequence variants, all located in attenuated DREs, for more in-depth analysis (Figure 7). Although not previously linked to FL, a SNP associated with familial Chronic Lymphocytic Leukemia (rs674313) (Slager et al., 2011) is located in a binding motif for IKZF1, a TF that regulates cell-fate decisions during lymphopoiesis. The second variant likely corresponds to a private SNP, since it is also present in the patient’s PBMCs, but is not found in available SNP databases (Table S5). The private SNP overlaps a binding site for SP1, a ubiquitous TF that governs many biological processes, including cell cycle regulation and apoptosis (Archer, 2011). The third variant is a somatic mutation that was present in the FL sample but not the matched PBMCs (Table S5). The mutation is located in a predicted binding site for TCF3, a TF important for many aspects of B cell development and activation (Hagman and Lukin, 2006).
Figure 7. Polymorphisms and mutations in FL-altered DREs disrupt enhancer function.
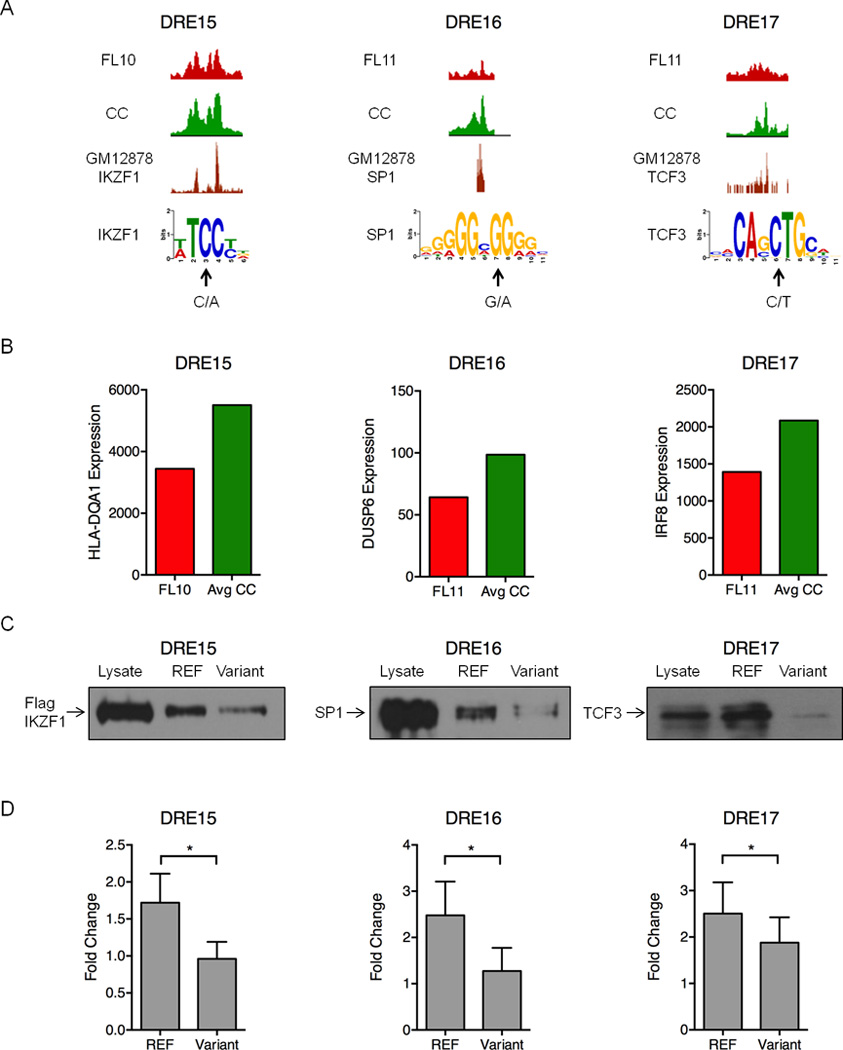
A) UCSC Genome Browser views of H3K27ac ChIP-seq data illustrating attenuated DREs harboring the indicated sequence variants in FL and the reference sequence in CC (upper 2 tracks). The bottom track shows ChIP-seq data for the indicated TFs performed in the GM12878 B cell line (Neph et al., 2012). Arrows indicate the variant sequence and location in PWMs for each TF. B) Expression of the DRE target genes quantified by microarray analysis. C) Oligonucleotide precipitation assays demonstrate reduced TF binding in variant-containing compared to reference sequences. Western blots were probed with antibodies specific to the TF of interest (representative of three experimental replicates). D) Luciferase reporter assays performed in lymphoma cell lines demonstrate significantly reduced activity for the variant-containing compared to reference sequences in the attenuated DREs. Luciferase activity is presented as a fold change for the enhancer vector relative to a reporter containing only the SV40 promoter (set to a value of 1.0). Results represent the mean ± SEM of three independent experiments. Statistical significance (paired t-test): * p≤ 0.05.
Binding of each TF to its predicted site in the attenuated DRE is supported by IKZF1, SP1, and TCF3 ChIP-seq data from GM12878 B cells (Figure 7A) (Neph et al., 2012). Importantly, expression of target genes predicted by FL pathogenic circuitry is also reduced in FLs harboring the altered DREs (Figure 7B). Each of the target genes has a demonstrated role in B cell cancer: allelic variants of HLA-DQA1 have been associated with an increased risk of childhood acute lymphocytic leukemia (Urayama et al., 2013); DUSP6, a MAP kinase phosphatase specific for ERK1/2, is deregulated in multiple cancers (Bermudez et al., 2010); and IRF8, a TF that regulates BCL6 and AICDA in GC reactions, is mutated in several types of NHL (Morin et al., 2011; Pasqualucci et al., 2014; Wang and Morse, 2009). A functional impact for the variants is demonstrated by oligonucleotide precipitation assays. Each TF binds robustly to a sequence corresponding to the reference allele. In contrast, binding is substantially diminished when oligonucleotides contain the identified variants (Figure 7C). Finally, enhancer activity of each DRE, as measured by luciferase reporter assays, is significantly attenuated when the reference enhancer is mutated to its corresponding sequence variant (Figure 7D). To our knowledge, this is the first reported example of an acquired mutation in an enhancer that attenuates TF binding and target gene expression in cancer. Together, our data indicate that DRE sequence variants, whether inherited or acquired, contribute to altered gene expression programs that drive lymphomagenesis.
Discussion
A key question in lymphomagenensis is how the normal gene expression programs of GC-B cells are dramatically altered during transformation. We now provide a comprehensive wiring scheme for pathogenic gene expression circuits in a common B cell cancer, which is a composite of changes to normal GC-B circuitry and regulatory circuits commandeered from other cell lineages. Importantly, the collection of regulatory elements linked into FL pathogenic circuits is significantly enriched for sequence variants, either SNPs or somatic mutations, some of which disrupt TF binding and attenuate the expression of their target genes.
Our epigenome-centric approach revealed two distinct subtypes of low-grade FL, which is considered a single diagnostic entity despite clinical heterogeneity. Each subtype exhibits a characteristic pathogenic enhancer profile. The variable DREs and linked genes specific to subtype 1 most resemble the regulatory circuits seen in CC, whereas subtype 2 FLs acquire components of normal plasmablast circuitry. Notably, the expression of genes linked to subtype-specific DREs largely recapitulates patterns observed for subtypes of DLBCL, termed GC and activated B cell (ABC). Similar to DLBCL, the distinct variable DREs and target genes likely reflect divergent modes of FL pathogenesis, with subtype 1 maintaining survival pathways downstream of tonic B cell receptor (BCR) signaling (GC). In contrast, subtype 2 variable DREs govern genes that are responsive to chronic BCR signaling (ABC) (Rui et al., 2011). Our definition of FL regulatory circuits should inform mechanistic studies into common and distinct modes of lymphoma pathogenesis. Variable DREs that distinguish subtype-specific circuits may be leveraged for new precision medicine strategies by directly targeting these elements with sequence-specific chromatin modifiers to reverse their pathogenic function.
A key insight from our integrative analysis of FL regulatory circuits is the central role for TFs in driving pathogenic changes to DRE function. Specifically, we identified distinct cohorts of TFs associated with attenuated versus enhanced activity of DREs in FL. Compared with normal CC counterparts, FL decommissions some circuitry by attenuating the expression of TFs that either regulate GC processes dispensable for lymphomagenesis, such as Ig class-switch recombination, or are prohibitive for transformation, such as pro-apoptotic pathways. Our study provides experimental support for this general mechanism, demonstrating that depletion of one such factor, SPIB, reduces the activity of predicted enhancer targets and genes linked in the pathogenic FL circuitry. In contrast, a second set of TFs overexpressed in FL activate DREs that are normally silent in all subsets of mature B cells. The circuits targeted by these TFs include genes involved in general mechanisms of oncogenesis, such as cell signaling, survival, and proliferation.
The central role of TFs in rewiring regulatory circuits was manifested at a genetic level by enrichment of sequence variants in DREs that impact TF binding. The inherited sequence variants in FL are associated with other types of B cell cancer and autoimmune disorders, suggesting that disruption of these regulatory circuits is a shared mechanism for a spectrum of lymphocyte-mediated diseases. In addition to SNPs, we provide the first evidence, to our knowledge, of a somatic mutation in a DRE that attenuates TF binding, enhancer function, and expression of its target gene in cancer. The precise source of somatic mutations remains to be established. However, an intriguing possibility is that some acquired SNVs arise during B cell activation, perhaps as off-target consequences of somatic hypermutation (Khodabakhshi et al., 2012).
In summary, our epigenome studies provide a rich resource for deciphering aberrations in the transcriptional circuitry that fosters pathogenesis of B cell lymphoma. In particular, we find that FL co-opts beneficial regulatory circuits and prunes potentially deleterious connections to construct pathogenic cistromes via diverse mechanisms, including inappropriate TF expression and the acquisition of somatic mutations in DREs.
Experimental Procedures
Detailed methods for sample collection, ChIP-seq, FAIRE-seq, and informatics analyses can be found in Supplemental Experimental Procedures.
B cell Isolation
Single cell suspensions of CD19+ B lymphocytes from each FL biopsy were isolated by physical disruption and magnetic-assisted cell sorting (MACS, Human CD19 Microbeads, Miltenyi, Bergisch Gladbach, Germany). PBB were isolated from blood samples by negative sorting (Human B cell Isolation Kit II, Miltenyi) and immediately processed (resting PBB) or subjected to in vitro activation using IL4, anti-CD40, IgM, and IgD (activated PBB). Tonsillar tissues were mechanically disrupted and digested with collagenase for 1h. GC-B cells were isolated by MACS (CD19+ sorting) and flow cytometry to sort CD10+CD44loCXCR4+ (CB) and CD10+CD44loCXCR4− (CC) populations from tonsil (Caron et al., 2009).
ChIP- and FAIRE-seq
ChIP and FAIRE assays were performed as described (Giresi and Lieb, 2009; Koues et al., 2008). At least 3 ng of FAIRE, ChIP, or input DNA was used for indexed library preparation. Samples were pooled (9 samples), and subjected to 42 bp single-end sequencing.
Knockdown experiments
Knockdown of SPIB was achieved by electroporation of control (target sequence: AAGCTGGAGTACAACTACA) or SPIB shRNA (target sequence: TACAGCTGAAGTGTGGCCCGTC) plasmid containing a selectable marker (CD4). Transfected cells were purified 48 hours post transfection using magnetic bead isolation for CD4 (Stem Cell Technologies, Vancouver, Canada).
Gene Expression Analysis
RNA was purified (Qiagen RNeasy), amplified (Nugen Ovation Pico SL or Ovation Pico), labeled (Nugen Encore Biotin), and hybridized on Affymetrix Human Gene 1.0ST arrays. Expression was quantified using Expression Console software (v1.2.0.20) with probe-level RMA and default settings. rRNA-depleted (Ribo-Zero, Epicentre) libraries were prepared using TruSeq RNA Sample kits with indexed adaptors (Illumina), pooled (3 libraries), and subjected to 100 bp paired-end sequencing. RNA-Seq data were aligned to the reference genome (build GRCh37/hg19) with TopHat (Trapnell et al., 2012). FPKM values were obtained using Cufflinks with default parameters.
Mutation Analysis
SNVs were identified from RNA- and ChIP-seq files using samtools (Li et al., 2009). Variants were filtered to identify non-coding SNVs common to multiple sequencing formats and known SNPs were removed by comparison to all SNPs in dbSNP, build 138. SNVs predicted to disrupt TF binding motifs were identified using TRANSFAC or FunSEQ (Khurana et al., 2013; Matys et al., 2006). For 28 randomly selected SNVs, the regions flanking the SNV positions were PCR-amplified from tumor and PBMC genomic DNA and the products were sequenced.
Luciferase Assays
DREs were PCR-amplified and inserted downstream of the luciferase coding sequence in the SV40 promoter-driven pGL3 plasmid (Promega, France). Reporter vectors were transfected by electroporation into lymphoma cell lines OCI-LY7 (DREs 1–10), Raji (DREs 15–16) and RL (DRE17). Dual luciferase assays were performed in duplicate according to the manufacturer’s protocol (Promega).
Oligonucleotide Precipitation Analysis
Assays were performed as described (Basu et al., 2009). 293T cells (ATCC, Manassass, VA, USA) transfected with TCF3, IKZF1, or SP1 expression plasmid were lysed in HKMG buffer (10 mM Hepes (pH 7.9), 100 mM KCl, 5 mM MgCl2, 10% glycerol, 1 mM DTT and 1% Nonidet P-40). Lysates were incubated overnight at 4°C with biotinylated double-stranded oligonucleotides spanning the reference or variant TF sequence in the presence of 10-fold excess poly(dI–dC). DNA-bound proteins were collected with NeutrAvidin UltraLink Resin (Thermo Scientific, Waltham, MA) and assayed by Western blot.
Supplementary Material
Acknowledgements
This work was supported by NIH grants CA156690 (J.E.P. and E.M.O.), CA090547 (O.I.K.), TR000448 (WU-ICTS), CA91842 (Siteman Cancer Center). We thank B. Sleckman, T. Fehniger, N. Mosammaparast, M. Artyomov, and T. Wang for comments; the WUSM Lymphoma Banking Program, Dept. of Medicine, Division of Medical Oncology, N. Bartlett, N. Wagner-Johnston, K. Carson, and T. Fehniger for lymphoma biopsies and patient histories; M. Colonna for tonsil samples, and the Genome Technology Access Center for assistance with – omics analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession Numbers
Data have been deposited in NCBI's Gene Expression Omnibus (GSE62246).
Author Contributions
E.M.O., J.E.P. and O.I.K. conceptualized the study and designed experiments. E.M.O. and J.E.P. supervised all aspects of the project. Experiments and data analyses were performed by O.I.K., J.E.P., R.A.K., S.C.P., L-W.C., J.A.S, H.L., L.E.S., J.J.B. and T.B.H. Specimens were processed by O.I.K., R.A.K., J.A.S. and A.F.C. The manuscript was written by E.M.O., J.E.P. and O.I.K. with input from A.F.C.
Competing Financial Interests
The authors declare no competing financial interests.
References
- Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, Myeroff L, Lutterbaugh J, Jarrar A, Kalady MF, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science. 2012;336:736–739. doi: 10.1126/science.1217277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Allen CDC, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity. 2007;27:190–202. doi: 10.1016/j.immuni.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer MC. Role of sp transcription factors in the regulation of cancer cell metabolism. Genes Cancer. 2011;2:712–719. doi: 10.1177/1947601911423029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26:6766–6776. doi: 10.1038/sj.onc.1210760. [DOI] [PubMed] [Google Scholar]
- Basu S, Liu Q, Qiu Y, Dong F. Gfi-1 represses CDKN2B encoding p15INK4B through interaction with Miz-1. Proc. Natl. Acad. Sci. U. S. A. 2009;106:1433–1438. doi: 10.1073/pnas.0804863106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat. Rev. Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermudez O, Pagès G, Gimond C. The dual-specificity MAP kinase phosphatases: critical roles in development and cancer. Am. J. Physiol. Cell Physiol. 2010;299:C189–C202. doi: 10.1152/ajpcell.00347.2009. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron G, Le Gallou S, Lamy T, Tarte K, Fest T. CXCR4 expression functionally discriminates centroblasts versus centrocytes within human germinal center B cells. J. Immunol. 2009;182:7595–7602. doi: 10.4049/jimmunol.0804272. [DOI] [PubMed] [Google Scholar]
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MGM, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung K-JJ, Shah SP, Steidl C, Johnson N, Relander T, Telenius A, Lai B, Murphy KP, Lam W, Al-Tourah AJ, et al. Genome-wide profiling of follicular lymphoma by array comparative genomic hybridization reveals prognostically significant DNA copy number imbalances. Blood. 2009;113:137–148. doi: 10.1182/blood-2008-02-140616. [DOI] [PubMed] [Google Scholar]
- Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–721. doi: 10.1038/nature07968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradin O, Saiakhova A, Akhtar-Zaidi B, Myeroff L, Willis J, Cowper-Sal lari R, Lupien M, Markowitz S, Scacheri PC. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res. 2014;24:1–13. doi: 10.1101/gr.164079.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, Fisher RI, Braziel RM, Rimsza LM, Grogan TM, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N. Engl. J. Med. 2004;351:2159–2169. doi: 10.1056/NEJMoa041869. [DOI] [PubMed] [Google Scholar]
- Dölken G, Illerhaus G, Hirt C, Mertelsmann R. BCL-2/JH rearrangements in circulating B cells of healthy blood donors and patients with nonmalignant diseases. J. Clin. Oncol. 1996;14:1333–1344. doi: 10.1200/JCO.1996.14.4.1333. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan K-K, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giresi PG, Lieb JD. Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements) Methods. 2009;48:233–239. doi: 10.1016/j.ymeth.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagman J, Lukin K. Transcription factors drive B cell development. Curr. Opin. Immunol. 2006;18:127–134. doi: 10.1016/j.coi.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Geng H, Boss I, Wang L, Melnick A. Cooperative transcriptional repression by BCL6 and BACH2 in germinal center B-cell differentiation. Blood. 2014;123:1012–1020. doi: 10.1182/blood-2013-07-518605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikushima H, Negishi H, Taniguchi T. The IRF Family Transcription Factors at the Interface of Innate and Adaptive Immune Responses. Cold Spring Harb. Symp. Quant. Biol. 2013;78:105–116. doi: 10.1101/sqb.2013.78.020321. [DOI] [PubMed] [Google Scholar]
- Izutsu K. Treatment of follicular lymphoma. J. Clin. Exp. Hematop. 2014;54:31–37. doi: 10.3960/jslrt.54.31. [DOI] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodabakhshi AH, Morin RD, Fejes AP, Mungall AJ, Mungall KL, Bolger-Munro M, Johnson NA, Connors JM, Gascoyne RD, Marra MA, et al. Recurrent targets of aberrant somatic hypermutation in lymphoma. Oncotarget. 2012;3:1308–1319. doi: 10.18632/oncotarget.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana E, Fu Y, Colonna V, Mu XJ, Kang HM, Lappalainen T, Sboner A, Lochovsky L, Chen J, Harmanci A, et al. Integrative Annotation of Variants from 1092 Humans: Application to Cancer Genomics. Science (80-.) 2013;342:1235587–1235587. doi: 10.1126/science.1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat. Rev. Immunol. 2008;8:22–33. doi: 10.1038/nri2217. [DOI] [PubMed] [Google Scholar]
- Koues OI, Dudley RK, Truax AD, Gerhardt D, Bhat KP, McNeal S, Greer SF. Regulation of acetylation at the major histocompatibility complex class II proximal promoter by the 19S proteasomal ATPase Sug1. Mol. Cell. Biol. 2008;28:5837–5850. doi: 10.1128/MCB.00535-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leich E, Ott G, Rosenwald A. Pathology, pathogenesis and molecular genetics of follicular NHL. Best Pract. Res. Clin. Haematol. 2011;24:95–109. doi: 10.1016/j.beha.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Lenz G, Staudt LM. Aggressive lymphomas. N. Engl. J. Med. 2010;362:1417–1429. doi: 10.1056/NEJMra0807082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo NS, Lugar PL, Yavuz S, Zhang W, Krijger PHL, Russ DE, Jima DD, Dave SS, Grammer AC, Lipsky PE. Analysis of somatic hypermutation in X-linked hyper-IgM syndrome shows specific deficiencies in mutational targeting. Blood. 2009;113:3706–3715. doi: 10.1182/blood-2008-10-183632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters JR. Human cancer cell lines: fact and fantasy. Nat. Rev. Mol. Cell Biol. 2000;1:233–236. doi: 10.1038/35043102. [DOI] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489:83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, Ouillette P, Trifonov V, Rossi D, Tabbò F, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6:130–140. doi: 10.1016/j.celrep.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm A, Mensen A, Schradi K, Gerlach K, Wittstock S, Winter S, Büchner G, Dörken B, Lipp M, Höpken UE. Cooperative function of CCR7 and lymphotoxin in the formation of a lymphoma-permissive niche within murine secondary lymphoid organs. Blood. 2011;118:1020–1033. doi: 10.1182/blood-2010-11-321265. [DOI] [PubMed] [Google Scholar]
- Rui L, Schmitz R, Ceribelli M, Staudt LM. Malignant pirates of the immune system. Nat. Immunol. 2011;12:933–940. doi: 10.1038/ni.2094. [DOI] [PubMed] [Google Scholar]
- Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer. 2010;10:361–371. doi: 10.1038/nrc2826. [DOI] [PubMed] [Google Scholar]
- Slager SL, Rabe KG, Achenbach SJ, Vachon CM, Goldin LR, Strom SS, Lanasa MC, Spector LG, Rassenti LZ, Leis JF, et al. Genome-wide association study identifies a novel susceptibility locus at 6p21.3 among familial CLL. Blood. 2011;117:1911–1916. doi: 10.1182/blood-2010-09-308205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarte K, De Vos J, Thykjaer T, Zhan F, Fiol G, Costes V, Rème T, Legouffe E, Rossi J-F, Shaughnessy J, et al. Generation of polyclonal plasmablasts from peripheral blood B cells: a normal counterpart of malignant plasmablasts. Blood. 2002;100:1113–1122. [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urayama KY, Thompson PD, Taylor M, Trachtenberg EA, Chokkalingam AP. Genetic variation in the extended major histocompatibility complex and susceptibility to childhood acute lymphoblastic leukemia: a review of the evidence. Front. Oncol. 2013;3:300. doi: 10.3389/fonc.2013.00300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victora GD, Nussenzweig MC. Germinal centers. Annu. Rev. Immunol. 2012;30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, Nussenzweig MC. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood. 2012;120:2240–2248. doi: 10.1182/blood-2012-03-415380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Morse HC. IRF8 regulates myeloid and B lymphoid lineage diversification. Immunol. Res. 2009;43:109–117. doi: 10.1007/s12026-008-8055-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap DB, Chu J, Berg T, Schapira M, Cheng S-WG, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117:2451–2459. doi: 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.