

Abstract
Alkali halides MX, have been viewed as typical ionic compounds, characterized by 1:1 ratio necessary for charge balance between M+ and X−. It was proposed that group I elements like Cs can be oxidized further under high pressure. Here we perform a comprehensive study for the CsF-F system at pressures up to 100 GPa, and find extremely versatile chemistry. A series of CsFn (n ≥ 1) compounds are predicted to be stable already at ambient pressure. Under pressure, 5p electrons of Cs atoms become active, with growing tendency to form Cs (III) and (V) valence states at fluorine-rich conditions. Although Cs (II) and (IV) are not energetically favoured, the interplay between two mechanisms (polyfluoride anions and polyvalent Cs cations) allows CsF2 and CsF4 compounds to be stable under pressure. The estimated defluorination temperatures of CsFn (n = 2,3,5) compounds at atmospheric pressure (218°C, 150°C, -15°C, respectively), are attractive for fluorine storage applications.
In general, for a given ionic compound AmBn, the stoichiometry reflects the ratio of valences (or the ratio of cationic and anionic formal charges). Yet, some ionic compounds do not strictly obey this rule. For instance, MgO2 can be prepared at very high oxygen fugacities, in which anions form the peroxide group [O2]2− 1. The variation of stoichiometry comes from the formation of polyatomic anions (such as those in peroxides, superoxides2, polyiodides3, etc), without changing cation valences. It was found that peroxides (usually unstable or metastable) become thermodynamically stable under higher pressures1.
It appears that increasing pressure promotes the formation of increasing oxidation states. Our recent work has discovered that Xe will form stable oxides under high pressure, in which Xe exhibits the oxidation states of +2, +4 and +64. In this case, the new stoichiometry is no longer from the anion-anion bonds (O-O), but from the higher valence of Xe. Elements around Xe in the Periodic Table are expected to undergo similar transitions. In particular, Cs, in the electronic configuration [Xe]6s1, is a natural choice to study this possibility. Indeed, Miao5 has recently reported that Cs, under pressure, can adopt oxidation states higher than +1 to form a series of stable CsFn compounds. According to Miao's calculation, CsF2 becomes stable at 5 GPa, CsF3 at 15 GPa, and CsF5 at 50 GPa. Considering [CsF2]− and [CsF5] are isoelectronic to the well-known molecular XeF2 (Ref. 6) and [XeF5]− (Ref. 7), this picture makes sense. However, Miao suggested that CsF2 and CsF4 adopt structures similar to those of XeF2 and XeF4, with the oxidation states of Cs being +2 and +4. In particular, an I4/mmm CsF2 was proposed to be thermodynamically stable at 10 GPa, which appears to break the isoelectronic analogy and involve Cs2+ ions isoelectronic to unknown and unstable Xe+ ion. To resolve this, we performed a comprehensive investigation of CsFn system under pressures up to 100 GPa. Our calculation uncovers quite a different scenario from Miao's report. We further explain that interplay between two mechanisms (polyfluoride anions and increase of Cs oxidation state) results in an unexpected variety of stable CsFn compounds under moderate pressure.
Results and Discussion
We have performed variable-composition structure searches using the USPEX code8,9,10,11 with up to 24 atoms in the unit cell at pressures of 0 and 100 GPa for the Cs-F system, in which we found only CsFn (n ≥ 1) to be stable, with CsF being stable at all pressures. Thus we focused our search on the CsF-F system at pressures 0, 30, 50, 75, 100 GPa. As shown in Fig. 1, these searches yielded the correct crystal structures for CsF and F, and a series of polyfluoride compounds as stable states, including CsF2, CsF3, CsF5, which are thermodynamically stable already at ambient pressure. As pressure increases, CsF2 becomes unstable above 19 GPa, while CsF4 appears on the convex hull (i.e. is thermodynamically stable) at pressures in the range between 17–80 GPa. CsF3 and CsF5 are stable in the entire pressure range up to 100 GPa. However, all of the stable compounds undergo a series of phase transitions, with dramatic changes of the electronic structure.
Figure 1. Convex hull diagrams of CsFn at different pressures.

Let us first look at CsF3 phases. At ambient pressure, we find CsF3 adopts a rhombohedral structure (space group 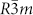 ), which is made of Cs+ and linear symmetric [F3]− species. The F-F distance in the [F3] is 1.736 Å, indicating, as expected, weaker bonding than in the F2 molecule (F-F bond length 1.442 Å). Bader analysis also supports this conclusion: there is a charge transfer of 0.950 e from Cs to F3 species, very close to the value in CsF (0.928 e), but the charge distribution within F3 is not even: two end F atoms have the charge of -0.406 e, while the central atom only has -0.138 e. Trihalide anions [X3]− (X = Br, I) are well known, but the [F3]− species has only been experimentally found as CsF3 complexes in argon matrix12,13. Here, we for the first time report its existence in a thermodynamically stable crystalline phase. According to DFT calculation,
), which is made of Cs+ and linear symmetric [F3]− species. The F-F distance in the [F3] is 1.736 Å, indicating, as expected, weaker bonding than in the F2 molecule (F-F bond length 1.442 Å). Bader analysis also supports this conclusion: there is a charge transfer of 0.950 e from Cs to F3 species, very close to the value in CsF (0.928 e), but the charge distribution within F3 is not even: two end F atoms have the charge of -0.406 e, while the central atom only has -0.138 e. Trihalide anions [X3]− (X = Br, I) are well known, but the [F3]− species has only been experimentally found as CsF3 complexes in argon matrix12,13. Here, we for the first time report its existence in a thermodynamically stable crystalline phase. According to DFT calculation, 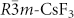 is stable against decomposition to CsF and F2 (the formation energy is about -0.189 eV/atom at T = 0 K, P = 1 atm). At 27 GPa, CsF3 undergoes a phase transition to a monoclinic phase C2/c. More interestingly, this structural transition coincides with a striking increase in Cs's Bader charge, as shown in Fig. 2. At 30 GPa, Cs has a charge of +1.7 e, far beyond the +1 formal charge of alkali elements under ambient conditions, suggesting Cs+ has been further oxidized. Our previous study has shown that Xe under high pressure can be oxidized to +2, +4 and +6 states. Cs3+ is isoelectronic to Xe2+. This phase transition can be interpreted as a transition from Cs+[F3]− to [CsF2]+[F]−. This is also evidenced by the dramatic change of Cs-F distance. In the C2/c phase, there are two types of F atoms (F1 and F2), each Cs is surrounded by 2 F1 and 4 F2. At 50 GPa, the calculated Cs-F1 distance is 2.01 Å, while the Cs-F2 distance is 2.58 Å, appearing to be consistent with a Jahn-Teller distortion related to the open-shell Cs3+ configuration. However, compared with Cs-F bond length (2.69 Å) in ionic CsF, we conclude that Cs-F2 interaction is very close to a typical ionic Cs-F bond, while each individual Cs-F1 bond has much stronger interaction (more covalent bonding). Therefore, it can be viewed as [CsF2]+[F]− complex. A similar discussion can be also found in Miao's work5. But his previously proposed C2/m structure is less stable than the C2/c structure found here.
is stable against decomposition to CsF and F2 (the formation energy is about -0.189 eV/atom at T = 0 K, P = 1 atm). At 27 GPa, CsF3 undergoes a phase transition to a monoclinic phase C2/c. More interestingly, this structural transition coincides with a striking increase in Cs's Bader charge, as shown in Fig. 2. At 30 GPa, Cs has a charge of +1.7 e, far beyond the +1 formal charge of alkali elements under ambient conditions, suggesting Cs+ has been further oxidized. Our previous study has shown that Xe under high pressure can be oxidized to +2, +4 and +6 states. Cs3+ is isoelectronic to Xe2+. This phase transition can be interpreted as a transition from Cs+[F3]− to [CsF2]+[F]−. This is also evidenced by the dramatic change of Cs-F distance. In the C2/c phase, there are two types of F atoms (F1 and F2), each Cs is surrounded by 2 F1 and 4 F2. At 50 GPa, the calculated Cs-F1 distance is 2.01 Å, while the Cs-F2 distance is 2.58 Å, appearing to be consistent with a Jahn-Teller distortion related to the open-shell Cs3+ configuration. However, compared with Cs-F bond length (2.69 Å) in ionic CsF, we conclude that Cs-F2 interaction is very close to a typical ionic Cs-F bond, while each individual Cs-F1 bond has much stronger interaction (more covalent bonding). Therefore, it can be viewed as [CsF2]+[F]− complex. A similar discussion can be also found in Miao's work5. But his previously proposed C2/m structure is less stable than the C2/c structure found here.
Figure 2. Bader charge of Cs in stable CsF3 compounds as a function of pressure.

Insets (a), 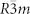 CsF3 structure at 0 GPa; (b) C2/c CsF3 structure at 70 GPa.
CsF3 structure at 0 GPa; (b) C2/c CsF3 structure at 70 GPa.
Similar to CsF3, CsF5 is also stable in the entire investigated pressure range between 0–100 GPa. At 0 GPa, we found a monoclinic P21 phase is stable against decomposition to any other stable compositions (Cs, CsF, CsF3, F). P21-CsF5 can be described as packing of Cs+ and [F5]− species. [F5]− ion has a V-shape and F-F bond lengths are 1.617, 1.953, 1.858, 1.617 Å, and F-F-F bond angle at central F atom is 98.592°. Bader analysis in Fig. 3 shows that the entire F5 group has charge -0.958 e. The hypothetical pentafluoride anion [F5]− has also been proposed by Riedel12, and we confirm it can exist at ambient pressure in a stable compound. At 4 GPa, a new phase with [F5]− groups and C2/c symmetry becomes stable. Around 21 GPa, C2/c phase transforms to another monoclinic C2/m phase, which can be represented as [CsF2] [F3]. The [CsF2] unit is very similar to the one in C2/c-CsF3 (Bader charge is 0.820e), indicating that Cs achieves the +3 oxidation state. At the same time, [F3] is a typical polyfluoride anion with Bader charge -0.820e; thus the whole structure can be viewed as [CsF2]+[F3]−. At 47 GPa, consistent with Miao's results5, we found a structure based on the packing of CsF5 molecules. We again plot the variation of Cs's Bader charge in stable CsF5 compounds with pressure. Indeed, analysis indicates a two-step oxidation of Cs +1 → +3 → +5, coinciding with the transition sequence (from C2/c to C2/m at 21 GPa, and from C2/m to Fddd at 47 GPa).
Figure 3. Bader charge of Cs in stable CsF5 compounds as a function of pressure.

Insets (a), P21-CsF5 structure at 0 GPa; (b) C2/m-CsF5 structure at 30 GPa; (c) Fddd-CsF5 structure at 70 GPa.
Our results show CsF3 and CsF5 are stable alongside the known compound CsF in the whole investigated pressure range (0–100 GPa). Unlike the recently discovered exotic sodium chlorides14, most of which are metallic, all of the predicted caesium fluorides are insulators. There are two factors determining the stoichiometry of these insulating compounds: (1) Cs's valence state transition (I → III → V); (2) formation of polyfluoride anions ([F3]−, [F5]−). Note that this is different from the previous study5, in which the latter factor was overlooked, along with a large number of stable phases. Due to these two competing mechanisms, one can expect other stoichiometries can be stabilized as well. Indeed, we found CsF2 and CsF4 can be stable at intermediate pressure ranges.
Previously, a tetragonal (I4/mmm) XeF2-like molecular structure was proposed to be stable at 5-20 GPa5. Our search found molecular CsF2 crystal to be unstable against decomposition to CsF3 and CsF at all pressures. A class of CsF2 compounds, however, has been found to be stable at low pressures in our prediction. At 0 GPa, another I4/mmm CsF2 phase is found to be stable. As shown in Fig. 4c, it consists of [Cs]+ and [F4]2− ions. The calculated Bader charges are 0.924 e for [Cs], and -1.848 e for [F4], suggesting the formation of [Cs]+ and [F4]2−. Therefore, [Cs]2+ is not favoured by energy, but CsF2 can be stabilized due to the formation of the [F4]2− anion. [F4]2− has not been observed by chemists so far, except that Riedel et al12 theoretically investigated the possibility of [F4]−. Yet our comprehensive structural search suggests that [F4]2− based CsF2 should be stable. Tetraiodide anion [I4]2− is known3. Our results suggest that fluorine follows that same trend under high pressure. I4/mmm-CsF2 would undergo a phase transition to an orthorhombic phase (Pbam) at 2.8 GPa, which also contains Cs+ and [F4]2− ions. Above 19 GPa, CsF2 is no longer stable as there is a dramatic change in the valence of Cs from (I) to (III). At around 20 GPa, CsF4 becomes stable in a monoclinic form (C2/m) (Fig. 5a). One can clearly see from the electron localization function (ELF) that half of Cs atoms have strong bonding with two neighbouring F atoms, and the other half of Cs atoms are simple Cs+ cations. The remaining F atoms form [F3]− ions, as we already saw above in both CsF3 and CsF5. Thus, it can be viewed as [CsF2]+[Cs]+2[F3]−. Bader analysis also supports this interpretation. Half of Cs atoms have Bader charge of 1.049e, half of Cs have 1.773e. This suggests that Cs firstly achieves III valence state in CsF4. At 31 GPa, all Cs atoms are oxidized to +3 state (Bader charge 1.829e), and the structure has space group P-1. As shown in Fig. 5b, the whole structure can be represented as 2[CsF2]+[F4]2− ([F4]2− anions appear again!). Miao investigated the possibility of CsF4 molecule structurally similar to XeF4, which is contradictory to chemical intuition (CsF4 can be neither isostructural nor isoelectronic to XeF4). Indeed, our results suggest that Cs4+ based compound is energetically unfavored. But we found that the most stable CsF4 structure above 57 GPa shows the oxidation state higher than +3. As shown in Fig. 5c, it can be viewed as [CsF2]+[F]− · [CsF5]0. Therefore, half of Cs atoms (in [CsF2]) have III valence, while the other half of Cs (in [CsF5]) have V valence state. The resulting structure, crystallizing in P-1 symmetry, is stable up to 79 GPa between C2/c-CsF3 and Fddd-CsF5. We note that there also exists a stable phase of XeF3 that can be represented as [XeF2]· [XeF4]15.
Figure 4.

(a) Enthalpy of formation relative to Pbam-CsF2 as a function of pressure; (b) unstable molecular I4/mmm-CsF2 at 0 GPa; (c) stable I4/mmm-CsF2 structure (stable between 0-2.8 GPa); (d) Pbam-CsF2 structure (stable between 2.8-19 GPa).
Figure 5.

The stable crystal structures of CsF4 and their corresponding (sliced or isosurfaced) ELF pictures at pressures of (a) 20 GPa, (b) 50 GPa, (c) 80 GPa.
Light halogens, fluorine (F) and chlorine (Cl), at normal conditions are highly reactive and toxic gases. For chemical industry and laboratory use, this presents great inconvenience. Their storage in the gaseous form (even as liquefied gases) is very inefficient, and compressed gas tanks may explode, presenting great dangers. At normal conditions, the volume of 22.4 litres (L) of pure fluorine gas weighs just 36 grams (g), illustrating the dismal inefficiency of storage in this form. To the best of our knowledge, no effective and safe fluorine storage materials are known. Both F and Cl have a huge range of industrial applications, which would benefit from such storage materials, especially if they can achieve high storage capacity, stability and reversibility.
In this work, we found that a series of CsFn (n = 1, 2, 3, 5) compounds can be stable at zero temperature and ambient pressure. One mole of CsF5 (227.9 g, occupying the volume of 0.07 L) contains 2 moles of F2 gas (which in the free state would occupy the volume of 44.8 L - hence, storage in the form of CsF5 is three orders of magnitude more efficient, and much safer, than in the form of pure F2 gas). The reaction CsF5 = CsF + 2F2(gas), is thermodynamically unfavourable at zero temperature (the enthalpy of this reaction is 88.41 kJ/mol), but will be favourable on increasing temperatures, due to the higher entropy of the F2 gas (202.8 J/(mol·K) at standard conditions)16. The calculated thermodynamic properties of these defluorination reactions are given in Table I. It can be seen that such compounds as CsF3 can be thermally decomposed, and then again be synthesized at lower temperatures at nearly room temperature window. CsF5, having the highest F content, can be used for fluorine storage at low temperature conditions. Thermodynamic stability of the predicted polyfluorides at atmospheric pressure means that there will be ways to “recharge” them with fluorine after defluorination, and such reversibility is a strong advantage of the proposed fluorine storage materials.
Table 1. Investigated reactions of the CsF-F system at ambient pressure conditions. wt% gives the weight content of released F2 gas. ΔH0K and ΔH300K are the calculated enthalpies at T = 0 K and 300 K, including the vibrational energies in (kJ/mol). ΔS300K is the corresponding formation entropy in J/(K·mol). Tc is the predicted decomposition temperature at standard atmosphere (1 bar). Note that F2 is treated as crystalline solid at 0 K.
| Reactions | wt % | ΔH0K | ΔH300K | ΔS300K | Tc(°C) |
|---|---|---|---|---|---|
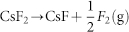 |
11.1 | 44.30 | 37.59 | 78.25 | 218 |
| CsF3 → CsF + F2(g) | 20.0 | 72.24 | 63.41 | 152.29 | 150 |
| CsF5 → CsF + 2F2(g) | 33.3 | 88.41 | 76.73 | 284.96 | -15 |
We have presented a comprehensive study of possible stable compounds in the CsF-F binary system under pressure. CsFn phases show extremely rich chemistry. At ambient pressure, novel and unexpected compounds CsF2, CsF3, CsF5 are thermodynamically stable because of the formation of polyfluoride anions of [F3]−, [F4]2−, [F5]−. Our results confirm the previously proposed polyfluoride anions (F3−, F5−), and suggest a new ion (F42−). Under high pressure, 5p electrons of Cs atoms can become chemically active, making Cs3+ and Cs5+ energetically favourable. Although our prediction found Cs2+ and Cs4+ states are far from being stable, stochiometric compounds CsF2 and CsF4 can become stable at 0–20 and 15–80 GPa, respectively, but these contain Cs+, Cs3+, Cs5+, [F4]2−, [F3]−. As shown in Fig. 6, crystal structures of caesium polyfluorides can be summarized as packings of Cs-containing cations (Cs+, [CsF2]+), polyfluoride anions ([F3]−, [F4]2−, [F5]−), and neutral molecular species (CsF5). Our hope is that this report will stimulate further experimental studies and serve as a guide for the design of fluorine storage materials.
Figure 6. Comparison of CsFn (n = 2,3,4,5) stability phase diagram with respect to pressure.

(a) revised results from this study; (b) results from previous study5. Note that that each colour represents distinct Cs's valence state in the given compounds (grey: Cs+; green: Cs2+; blue: Cs3+; red: Cs5+), while the gradient colour indicate mixed valence states in between. The I4/mmm-CsF2 structure in (a) and (b) are very different. The only common phase between (a) and (b) is Fddd-CsF5.
Methods
Searches for the stable compounds and structures were performed using an evolutionary algorithm, as implemented in the USPEX code8,9,10,11. The most significant feature of USPEX we used in this work is the capability of optimizing the composition and crystal structures simultaneously - as opposed to the more usual structure predictions at fixed chemical composition1,14. The compositional search space is described via building blocks (for example, search for all compositions in a form of [xCsF + yF]). During the initialization, USPEX samples the whole range of compositions of interest randomly and sparsely. Chemistry-preserving constraints in the variation operators are lifted and replaced by the block correction scheme which ensures that a child structure is within the desired area of compositional space, and a special “chemical transmutation” operator is introduced. Stable compositions are determined using the convex hull construction: a compound is thermodynamically stable if the enthalpy of its decomposition into any other compounds is positive. Structure prediction was done in conjunction with ab initio structure relaxations based on density functional theory (DFT) within the Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation (GGA)17 as implemented in the VASP code18. For structural relaxation, we used the all-electron projector-augmented wave (PAW) method and the plane wave basis set with the 600 eV kinetic energy cutoff; the Brillouin zone was sampled by uniform Gamma-centered meshes with the resolution 2π × 0.06 Å−1. For post-processing, the selected low-enthalpy structures were treated by using hard PAW potential of F (F_h), using a energy cut off of 1000 eV. Such calculations provide an excellent description of the known structures (CsF and F2) and their energetics. To ensure that the obtained structures are dynamically stable, we calculated phonon frequencies throughout the Brillouin zone using the finite-displacement approach as implemented in the Phonopy code19. The vibrational entropies and enthalpies are obtained by directly summing over the calculated phonon frequencies, in order to calculate the free energy (see online supporting information, similar methods have been widely used for simulation of dehydration reactions for hydrogen storage materials20,21). Charge transfer was investigated on the basis of the electron density using Bader's analysis22 as implemented in a grid-based algorithm without lattice bias23. Electron localization function (ELF)24 is also calculated in order to analyze chemical bonding for the selected compounds.
Author Contributions
Q.Z. designed the project and carried out structure prediction and electronic structure calculations. Q.-F.Z performed phonon calculations. Q.Z. and A.R.O analysed the data and wrote the paper.
Supplementary Material
Supplementary Online Materials: Formation of Stoichiometric CsFn Compounds
Acknowledgments
This work is funded by DARPA (Grants No. W31P4Q1210008 and No. W31P4Q1310005), by the Government (No. 14.A12.31.0003) of Russian Federation, Foreign Talents Introduction and Academic Exchange Program (No. B08040), the Basic Research Foundation of NWPU (Grant No. JCY20130114), and the Natural Science Foundation of China (Grants No. 51372203 and No. 51332004). Calculations were performed on the supercomputer of Center for Functional Nanomaterials, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under contract No. DE-AC02 98CH10086. The authors also acknowledge the High Performance Computing Center of NWPU for the allocation of computing time on their machines. ARO thanks V. Mukhanov for the idea of fluorine storage applications.
References
- Zhu Q., Oganov A. R. & Lykhov A. O. Novel Stable Compounds in the Mg-O System under High Pressure. Phys. Chem. Chem. Phys 35, 1654 (2013). [DOI] [PubMed] [Google Scholar]
- Vannerberg N. G. Peroxides, Superoxides, and Ozonides of the Metals of Groups Ia, IIa, and IIb. in Progress in Inorganic Chemistry, Volume 4 Cotton, F. A. (ed.) (John Wiley Sons, Inc.HobokenNJ. USA, 1962). [Google Scholar]
- Svensson P. H. & Kloo L. Synthesis, Structure, and Bonding in Polyiodide and Metal Iodide-Iodine Systems. Chem. Rev., 103, 1649 (2003). [DOI] [PubMed] [Google Scholar]
- Zhu Q. et al. Stability of xenon oxides at high pressures. Nature Chem. 5, 61–65 (2013). [DOI] [PubMed] [Google Scholar]
- Miao M.-S. Caesium in high oxidation states and as a p-block element. Nature Chem. 5, 846–852 (2013). [DOI] [PubMed] [Google Scholar]
- Levy H. A. & Agron P. A. The Crystal and Molecular Structure of Xenon Difluoride by Neutron Diffraction. J. Am. Chem. Soc. 85, 241–242 (1963). [Google Scholar]
- Christe K. O. et al. The pentafluoroxenate(IV) anion, XeF5-: the first example of a pentagonal planar AX5 species. J. Am. Chem. Soc. 113, 3351–3361 (1991). [Google Scholar]
- Oganov A. R. & Glass C. W. Crystal structure prediction using ab initio evolutionary techniques: principles and applications. J. Chem. Phys. 124, 244704 (2006). [DOI] [PubMed] [Google Scholar]
- Lyakhov A. O., Oganov A. R., Stokes H. T. & Zhu Q. New developments in evolutionary structure prediction algorithm USPEX. Comp. Phys. Comm. 184, 1172–1182 (2013). [Google Scholar]
- Zhu Q., Oganov A. R., Glass C. W. & Stokes H. T. New developments in evolutionary structure prediction algorithm USPEX. Acta Cryst. B68, 215–226 (2012). [DOI] [PubMed] [Google Scholar]
- Zhu Q., Li L., Oganov A. R. & Allen P. B. Evolutionary method for predicting surface reconstructions with variable stoichiometry. Phys. Rev. B 87, 195317 (2013). [Google Scholar]
- Riedel S., Kochner T., Wang X. & Andrews L. Polyfluoride Anions, a Matrix-Isolation and Quantum-Chemical Investigation. Inorganic Chemistry 49, 7156–7164 (2010). [DOI] [PubMed] [Google Scholar]
- Ault B. S. & Andrews L. Matrix reactions of alkali metal fluoride molecules with fluorine. Infrared and Raman spectra of the trifluoride ion in the M+F3- species. J. Am. Chem. Soc. 98, 1591–1593 (1976). [Google Scholar]
- Zhang W. W. et al. Unexpected Stable Stoichiometries of Sodium Chlorides. Science 342, 1502–1505 (2013). [DOI] [PubMed] [Google Scholar]
- Burns J. H., Ellison R. D. & Levy H. A. The crystal structure of the molecular addition compound xenon diluoride - xenon tetrafluoride. J. Phys. Chem. 67, 1569–1570 (1963). [Google Scholar]
- Chase M. W. J. NIST-JANAF Themochmical Tables. 4th Edition. (AIP Publishing, MelvilleNY, 1998).
- Perdew J. P., Burke K. & Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- Kresse G. & Furthmuller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996). [DOI] [PubMed] [Google Scholar]
- Togo A., Oba F. & Tanaka I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 78, 134106 (2008). [Google Scholar]
- Wolverton C., Ozolins V. & Asta M. Hydrogen in aluminum: First-principles calculations of structure and thermodynamics. Phys. Rev. B 69, 144109 (2004). [Google Scholar]
- Ozolins V., Majzoub E. H. & Wolverton C. First-Principles Prediction of Thermodynamically Reversible Hydrogen Storage Reactions in the Li-Mg-Ca-B-H System, J. Am. Chem. Soc. 131,230–237 (2009). [DOI] [PubMed] [Google Scholar]
- Bader R. F. W. Atoms in Molecules - A Quantum Theory (Oxford University Press, 1990). [Google Scholar]
- Tang W., Sanville E. & Henkelman G. A grid-based Bader analysis algorithm without lattice bias. J. Phys: Condensed Matter 21, 084204 (2009). [DOI] [PubMed] [Google Scholar]
- Becke A. D. & Edgecombe K. E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 92, 5397–5403 (1990). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Online Materials: Formation of Stoichiometric CsFn Compounds