

Abstract
ATP-binding cassette transporter A1 (ABCA1) mediates cholesterol efflux to lipid-free apolipoprotein A-I (apoA-I) and apolipoprotein E (apoE). ABCA1 is an essential regulator of High Density Lipoproteins (HDL) and reverse cholesterol transport – a role that determines its importance for atherosclerosis. Over the last 10 years studies have provided convincing evidence that ABCA1, via its control of apoE lipidation, also has a role in Alzheimer’s disease (AD). A series of reports have revealed a significant impact of ABCA1 on Aβ deposition and clearance in AD model mice, as well as an association of common and rare ABCA1 gene variants with the risk for AD. Since APOE is the major genetic risk factor for late onset AD, the regulation of apoE level or its functionality by ABCA1 may prove significant for AD pathogenesis. ABCA1 is transcriptionally regulated by Liver X receptors (LXR) and Retinoic X Receptors (RXR) which provides a starting point for drug discovery and development of synthetic LXR and RXR agonists for treatment of metabolic and neurodegenerative disorders. This review summarizes the recent results of research on ABCA1, particularly relevant to atherosclerosis and AD.
Keywords: Abca1, HDL, apoE, apoA-I, Alzheimer’s disease, amyloid beta, cardiovascular disease, LXR, RXR
Introduction
Alzheimer’s disease (AD) is a senile dementia characterized by the presence of senile plaques made of amyloid β (Aβ), neurofibrillary tangles, and cognitive decline. Although the inheritance of ε4 allele of APOE is the major genetic risk factor for late-onset sporadic form of AD (LOAD), the mechanisms underlying this association remain elusive. It is conceivable that additional genetic factors influence the risk, precipitating the development of dementia.
There is overwhelming data suggesting a link between lipid metabolism and AD (Hanson et al., 2013; Hughes et al., 2014; Reed et al., 2014; Simons et al., 1998). Genetic linkage and association studies have identified genes involved in cholesterol metabolism or transport as AD susceptibility genes (Harold et al., 2009; Jones et al., 2010). Dyslipidemia is a shared risk factor for cardiovascular disease and AD [reviewed in (Reitz, 2013)].
ATP binding cassette transporter A1 (ABCA1) belongs to the large superfamily of ABC transmembrane transporters (Koldamova et al., 2010; Oram and Vaughan, 2006). An important step towards understanding ABCA1 function was the discovery that mutations in its sequence cause Tangier disease (TD) characterized by impaired cellular cholesterol efflux, low levels of HDL particles and inefficient reverse cholesterol transport (RCT). Transcription of ABCA1 is regulated by Liver X Receptors (LXR), Retinoic X Receptors (RXR) and Peroxisome Proliferator-activated Receptors (PPARs). ABCA1 regulates cholesterol efflux to cholesterol acceptors, primarily lipid-free apoA-I and apoE but not to large HDL particles. ABCA1 is an essential mediator of HDL generation and loss of its function results in almost complete absence of HDL and apoA-I and a decrease of apoE. The role of ABCA1 as a regulator of HDL level determines its significance for atherosclerosis and cardiovascular disease.
The significance of ABCA1 for AD originates from its effect on apoE lipidation and stability. Experimental and clinical data suggest that apoE is involved in Aβ aggregation, toxicity and clearance [reviewed in Tai et al. (Tai et al., 2014)], therefore it is conceivable to expect that ABCA1 as a modulator of apoE metabolism will have a role in AD pathogenesis. Data from experimental animals demonstrated that Abca1 deficiency abolishes the lipidation of apoE and increases amyloid plaques in AD model mice (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005a; Koldamova et al., 2005b; Wahrle et al., 2005). In contrast treatment of AD model mice with LXR, RXR or PPAR agonists ameliorates AD phenotype (Cramer et al., 2012; Donkin et al., 2010; Fitz et al., 2010; Koldamova et al., 2005b; Terwel et al., 2011; Yamanaka et al., 2012). Additional topic of interest for AD is the effect of ABCA1 on HDL in plasma and HDL-like lipoproteins in brain. Association studies have shown that lower concentration of HDL (Reed et al., 2014) and apoA-I (Merched et al., 2000) correlate with increased risk for AD. The results from Genome Wide Association Studies (GWAS) designed to reveal genetic association of ABCA1 with AD are controversial, however.
In this review, we summarize the results of research exploring the role of ABCA1 in metabolic diseases, mainly atherosclerosis and diabetes, and pathogenesis of LOAD. We will focus on the mechanism of cholesterol efflux and generation of HDL, and how they affect cardiovascular and neurodegenerative disease.
ABCA1 mediated regulation of cholesterol efflux and HDL generation
ABCA1 is a transmembrane protein that transfers phospholipids and cholesterol to lipid free apoA-I or other apolipoproteins for generation of discoidal HDL particles (Oram and Vaughan, 2006). Discoidal HDL particles are composed of 100–200 lipid molecules and are surrounded by two apoA-I molecules (Lund-Katz and Phillips, 2010). A major function of HDLs is to participate in reverse cholesterol transport, a process by which excess cholesterol is removed from the cells and transported to the liver where it is metabolized for excretion (Oram and Vaughan, 2006). ABCA1 is comprised of two halves each having 6 transmembrane domains, and two large extracellular domains connected by intramolecular disulfide bonds that are important for the direct binding to apoA-I (Nagata et al., 2013; Oram and Vaughan, 2006). The mechanism by which ABCA1 translocates cholesterol and phospholipids is not fully agreed upon but it is established that binding of ABCA1 to lipid-free apolipoproteins is critical for initiating the process of lipid efflux (Westerterp et al., 2014). In contrast to ABCA1, another transporter - ABCG1 mediates cholesterol efflux to HDL particles but not to lipid-free apolipoproteins (Gelissen et al., 2006; Wang et al., 2004).
According to an earlier model ABCA1 initiates translocation of phospholipids and cholesterol from the cytoplasmic to exofacial leaflet of the plasma membrane which leads to membrane bending and formation of exovesiculated domain to which apoA-I binds with high affinity (Vaughan and Oram, 2003; Vedhachalam et al., 2007). The next step is a spontaneous solubilization of membrane phospholipids and cholesterol in the exovesiculated domain by the bound apoA-I followed by formation of discoidal HDL particles (Vedhachalam et al., 2007; Westerterp et al., 2014). A different model for the formation of discoidal HDL particles was recently proposed by Nagata et al. (Nagata et al., 2013) (Figure 1). The authors used a single-molecule fluorescence tracking technique to demonstrate that ABCA1 monomer diffuses freely and translocates lipids on the plasma membrane by an ATP-dependent mechanism (Figure 1, step 1). Upon reserving sufficient cholesterol and phospholipids ABCA1 undergoes conformational changes and forms dimers. The lipidated ABCA1 dimers interact with the actin filaments in the plasma membrane and are immobilized until lipid-free apoA-I directly binds to the extracellular domains of the ABCA1 dimer (Figure 1, step 2). In the last step apoA-I or another apolipoprotein accepts the lipids translocated by ABCA1 leading to the formation of discoidal HDL (Figure 1, step 3). After transferring cholesterol and phospholipids to apoA-I, ABCA1-dimer dissociates into monomers and resumes its function to translocate lipids in an ATP-dependent manner.
Figure 1. Mechanism of ABCA1-dependant cholesterol efflux.
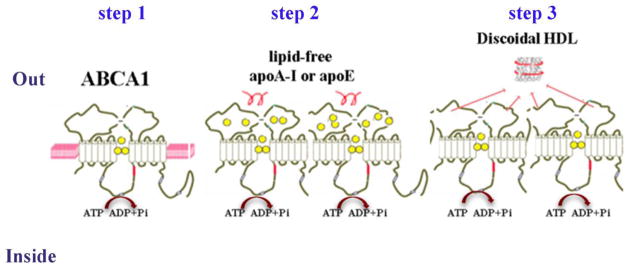
Step 1: ABCA1 monomer diffuses freely and translocates lipids on the plasma membrane by ATP-dependent mechanism. Step 2: Upon reserving sufficient cholesterol and phospholipids ABCA1 undergoes conformational changes and forms dimers. The lipidated ABCA1 dimers interact with the actin filaments in the plasma membrane and are immobilized until lipid-free apoA-I directly binds to the extracellular domains of ABCA1 dimer. Step 3: ApoA-I or another apolipoprotein accepts the lipids transclocated by ABCA1 leading to the formation of discoidal HDL. After transferring cholesterol and phospholipids to apoA-I, ABCA1-dimer dissociates into monomers and resumes its function to translocate lipids. (according to the model proposed by Nagata et al.)
Apolipoproteins that serve as lipid acceptor in ABCA1-mediated cholesterol efflux are mainly apoA-I which comprises 60–70% of the total HDL protein mass and to a lesser extent apoA-II and apoE (Tang and Oram, 2009). Liver is the main peripheral organ for the synthesis of HDL (approximately 70% of plasma HDL) and apolipoproteins, but intestine and other extrahepatic organs may also participate. Discoidal HDL formed only with apoE are slightly different than HDL containing only apoA-I, in that they are rather quasi-spheroidal (Lund-Katz and Phillips, 2010). When functional ABCA1 is lacking, the apolipoproteins which participate in HDL formation are left un-lipidated and are subjected to degradation in the kidney. Consequently, in patients without functional ABCA1 the levels of apoA-I and apoA-II fall to 1% and 7% of normal values respectively. Since apoE also participates in the formation of other lipoproteins such as very low density lipoproteins (VLDL), intermediate density lipoproteins (IDL) and chylomicrons, in patients with nonfunctional ABCA1 this apolipoprotein is partially protected from degradation and its level is decreased only by ~50% (Alaupovic et al., 1981; Mahley et al., 1991).
Transcriptionally, ABCA1 is regulated by nuclear receptors LXRs, RXRs and PPARs. Potential binding sites for other transcription factors in the proximal promoter of ABCA1 do exist (Santamarina-Fojo et al., 2001), but they have not been interrogated systematically so far. According to a widely accepted model, ligand activated LXR/RXR and PPAR/RXR heterodimers bind to response elements upstream of ABCA1 transcription start site and drive the transcription in response to specific extra- or intracellular signals. The understanding of the interplay between the nuclear receptors and co-regulators involved in the transcriptional control of ABCA1 and APOE is critical for successful development and testing of efficient ligand based therapies for AD (the topic is reviewed by Landreth et al., in this SI: Nuclear Receptors in Neurodegenerative Diseases, Neurobiol. Dis. (2014), http://dx.doi.org/10.1016/j.nbd.2014.04.001)
Role of ABCA1 in periphery
Cardiovascular disease
The decrease of cholesterol efflux, HDL, apoA-I and apoE as a result of loss-of-function mutations in ABCA1 affects different metabolic pathways in the periphery and central nervous system (CNS). In atherosclerosis and cardiovascular disease the effects of ABCA1 reflect its role in HDL formation and RCT. TD is a rare monogenic disorder caused by loss-of-function mutations in ABCA1 and characterized by extremely low HDL level (less than 2% of normal)(Hobbs and Rader, 1999; Rader and deGoma, 2012). Typical TD patients in addition to very low HDL often present with hepatosplenomegaly, peripheral neuropathy and enlarged yellow tonsils (Hobbs and Rader, 1999; Rader and deGoma, 2012). TD is inherited as an autosomal recessive trait and almost all of the patients with clinical phenotype are compound heterozygotes. At present, more than 180 ABCA1 mutations have been listed in Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ABCA1) and more than 100 of missense, nonsense and frameshift mutations had been identified in patients with TD and in subjects with a phenotype of HDL deficiency (Puntoni et al., 2012). While there is a significantly increased risk of cardiovascular disease in TD patients, the phenotypic presentation is not always exactly the same. Some TD patients, for example have a severe form of coronary heart disease whereas in other, typical symptoms including atherosclerotic lesions are missing (Hovingh et al., 2004). In addition to the reduced penetrance, variable expressivity of the mutant alleles and allelic interactions are possible explanations for the phenotypic variability observed in a sub-set of TD patients, where concomitant with HDL, there is a decrease in LDL level (Hobbs and Rader, 1999). Thus, in cases of ABCA1 functional impairment, not well understood environmental and other genetic factors could alleviate the effect of the otherwise significant functional impairment of cholesterol efflux and HDL generation.
Whereas TD patients are very rare (less than 100 diagnosed in the world; http://ghr.nlm.nih.gov/condition/tangier-disease), ABCA1 heterozygosity in the general population is present with a frequency of 3:1000 and also associates with decreased levels of HDL cholesterol (Frikke-Schmidt et al., 2008). Most of the published studies report an increase of atherosclerosis in heterozygous patients with missense mutations in ABCA1 (Bochem et al., 2013; Clee et al., 2000; Frikke-Schmidt et al., 2005; van Dam et al., 2002). However, not all of the functional mutations in ABCA1 are associated with an increased risk of cardiovascular disease (Clee et al., 2000; Frikke-Schmidt et al., 2008). The explanation of these seemingly controversial results is that the biochemical phenotype inherited as an autosomal co-dominant trait in heterozygotes, and revealed by plasma HDL-C and apoA-I levels, is highly variable - the values can be within the entire range of those measured in normal individuals and patients with TD. A differential contribution of missense mutations to the functional defect in cholesterol efflux determining the biochemical, cellular and clinical phenotypes was recognized early after the cloning of ABCA1. For example, some of the ABCA1 missense mutations ((P1065S, G1216V, N1800H, R2144X) cause only a mild decrease of cholesterol efflux which in heterozygous state results in a relatively small reduction of HDL (less than 30% decrease compared to the normal values) explaining the lack of atherosclerosis (Frikke-Schmidt et al., 2008). In contrast ABCA1 mutations which cause more than 50% decrease of HDL were associated with an increased risk for ischemic heart disease (Bochem et al., 2013; Clee et al., 2000; Frikke-Schmidt et al., 2005).
Recent meta-analysis of GWAS demonstrated that SNPs near ABCA1 associate with lower HDL and total cholesterol levels (Teslovich et al., 2010), but not with cardiovascular risk (Willer et al., 2008). However, as was discussed in many recent reports, whereas GWAS studies have a power to detect common variants of ABCA1, most of these variants in heterozygous patients result only in a small reduction of HDL and are less likely to affect the risk of cardiovascular disease (Frikke-Schmidt, 2011; Westerterp et al., 2014). In contrast using next generation sequencing and selecting a population at the extreme end of HDL (or other quantitative metabolic traits) could help identifying rare variants of ABCA1 with large effects on phenotype (cardio vascular disease or atherosclerosis) (Frikke-Schmidt et al., 2004; Service et al., 2014).
Studies with mice lacking ABCA1 in the whole body demonstrated dramatic reduction of HDL and apoA-I, a decrease of LDL and foam cell accumulation in the lungs (McNeish et al., 2000). Recent studies using targeted approach to delete Abca1 only in liver showed that expression of ABCA1 in this organ causes profound hypo-alphalipoproteinemia and kidney hypercatabolism of apoA-I (Timmins et al., 2005), and modulates the susceptibility to atherosclerosis (Brunham et al., 2009).
Type 2 diabetes mellitus
It was reported that mice with selective ablation of ABCA1 in pancreatic β-cells had markedly impaired glucose tolerance and defective insulin secretion but normal insulin sensitivity (Brunham et al., 2007). The results of the study suggested that dysfunctional ABCA1 contributes to the development of type 2 diabetes through increased cholesterol levels in pancreatic β-cells.
Studies in a limited number of TD patients demonstrated impaired insulin secretion from pancreatic β-cells suggesting, in support to the studies in genetically modified mice, that ABCA1 may be involved in insulin secretion (Koseki et al., 2009; Vergeer et al., 2010). A highly frequent non synonymous variant of ABCA1 (R230C) was identified in a Mexican population and shown to associate with obesity and type 2 diabetes (Villarreal-Molina et al., 2008). Also in a Mexican population it was reported that diabetic patients harboring this mutation needed a higher dose of glyburide to control glucose level (Aguilar-Salinas et al., 2013). However, a recent study examining several genetic variants in ABCA1 and ABCG1 did not find an association with an increased risk of type 2 diabetes (Schou et al., 2012).
ABCA1 and Alzheimer’s disease
The role of ABCA1 in amyloid deposition, clearance and memory deficits in experimental animals has been demonstrated and confirmed by the results of studies conducted in different laboratories. First, studies from our and other groups have demonstrated that lack of ABCA1 increases amyloid deposition and cognitive decline in different APP transgenic mice accompanied by significant decrease in the levels of soluble apoE (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005a; Lefterov et al., 2009; Wahrle et al., 2005). Second, it was shown that treatment with LXR and RXR ligands which increases global Abca1 expression in mice significantly ameliorates amyloid pathology (Cramer et al., 2012; Fitz et al., 2010; Jiang et al., 2008; Koldamova et al., 2005b). Third, transgenic mice overexpressing Abca1 in brain have less amyloid plaques (Wahrle et al., 2008a). Although similar data in humans are missing, the results of a recent case control genetic study with the application of advanced sequencing technologies clearly indicated that rare ABCA1 gene variants, some of them associated with higher levels of HDL, may have protective effect against AD (Lupton et al., 2014).
Physiological function of ABCA1 in central nervous system (CNS)
ABCA1 is expressed in all brain cell types and regulates cholesterol efflux to lipid-free apolipoproteins (Koldamova et al., 2003). ABCA1 mediates cholesterol efflux from astrocytes and microglia, brain capillary endothelial cells and pericytes to lipid-free apolipoproteins (Koldamova et al., 2003), (Kim et al., 2007; Panzenboeck et al., 2002; Saint-Pol et al., 2012). Since the blood brain barrier (BBB) is not penetrable for cholesterol and lipoproteins from periphery (Dietschy and Turley, 2001) brain HDL and ABCA1 do not contribute to RCT. In brain apoE is the main apolipoprotein that serves as an acceptor of the lipids translocated by ABCA1. ApoE-containing HDL lipoproteins were identified in astrocytes conditioned media, brain interstitial fluid (ISF) and cerebrospinal fluid (CSF) (Fitz et al., 2010; Pitas et al., 1987; Yu et al., 2010). ApoE-containing HDL lipoproteins isolated from astrocytes conditioned media are discoidal and do not contain cholesterol esters. In CSF apoE-containing HDL are spherical in shape and have the size of plasma HDL containing a core of cholesterol esters (Koch et al., 2001; Yu et al., 2010). ApoA-I-containing lipoproteins are also detected in CSF and are smaller than apoE-HDL (Pitas et al., 1987). ApoA-I passes BBB from the circulation in lipid-free form but there are some reports suggesting apoA-I expression in brain capillary endothelial cells (Panzenboeck et al., 2002). However, using a RNA-seq approach we were not able to detect an appreciable mRNA expression of Apoa-I in mouse brain or to detect apoA-I protein in ISF (Koldamova & Lefterov unpublished data).
The normal physiological role of ABCA1 in the brain is currently unknown however one can presume that it is to maintain cholesterol transport from glial cells (mainly astrocytes) to neurons. Previous studies have reported that cholesterol is needed for neurites outgrowth and repair, and synaptic vesicle formation and regeneration (Pfrieger and Barres, 1997). Recently, we examined the neurite architecture of pyramidal hippocampal neurons of Abca1ko mice (Fitz and Koldamova, unpublished data). Our data revealed a significant decrease in neurite length and number of neurite segments in CA1 region of hippocampus of Abca1ko mice when compared to WT mice suggesting importance of disruption of Abca1 for neurite degeneration in the brain.
In a similar way to periphery, lack of functional ABCA1 affects apoE lipidation and stability leading to a significant decrease of apoE in the brain of Abca1ko mice (Hirsch-Reinshagen et al., 2004; Wahrle et al., 2004). The effect of ABCA1 is apoE selective, since the levels of apoJ, another lipoprotein secreted by astrocytes, do not change regardless of the absence of functional ABCA1. Mice with a targeted disruption of brain Abca1 had a significant reduction of apoE in CNS and a very low level of apoE-HDL in CSF (Karasinska et al., 2009).
Genetic variation in ABCA1 and Alzheimer’s disease
Common genetic variants and risk for AD
The very first indication that there might be a link between AD and lipid metabolism came with the discovery of APOE as a major risk factor for LOAD (Corder et al., 1993; Poirier et al., 1993; Saunders and Roses, 1993). Recent GWAS studies have identified genes involved in cholesterol metabolism or transport as AD susceptibility genes, providing strong support for the association (Harold et al., 2009; Jones et al., 2010).
The identification of ABCA1 as a principal/master regulator of RCT and possibly a major risk factor for atherosclerosis in case of mutations or polymorphic variants, known to influence intracellular cholesterol efflux and HDL levels in periphery, reinvigorated the search for association of disturbed cholesterol metabolism in the CNS and the development of AD.
The establishment of association of non-synonymous common genetic variation in ABCA1 with altered lipoprotein levels and a modified risk for Coronary Artery Disease (CAD) (Clee et al., 2000) was followed by more than a dozen of targeted Genetic Association Studies (GAS) to test the hypothesis if common SNPs within the sequence of ABCA1 gene are related to the risk for AD (Cascorbi et al., 2013; Chu et al., 2007; Jiang et al., 2012; Katzov et al., 2004; Khorram Khorshid et al., 2011; Reynolds et al., 2009; Rodriguez-Rodriguez et al., 2007; Shibata et al., 2006; Sun et al., 2012; Sundar et al., 2007; Wahrle et al., 2007; Wang and Jia, 2007; Wavrant-De Vrieze et al., 2007; Wollmer et al., 2003). The association of higher levels of HDL-C in elderly individuals and lower risk of LOAD further rationalized studies to test the combination of ABCA1 variants and other cholesterol-related genes (Cascorbi et al., 2013; Xiao et al., 2012). R219K (rs2230806), I883M (rs4149313), and R1587K (rs2230808) are the non-synonymous variants most extensively investigated since they translate into amino acid changes and have been shown to associate with the risk for CAD. Altogether, the results of those studies were inconsistent with lack of reproducibility even in same ethnic groups and contradicting conclusions ranged from no effect, increasing or decreasing the risk for AD (detailed comments on earlier studies published before 2010 are available in (Koldamova et al., 2010). A meta-analysis based on those studies - 6,214 patients and 6,034 controls, concluded that the data so far does not support a model for association between ABCA1 and AD suggesting among other reasons, insufficient or low number of patients and controls investigated so far (Wang et al., 2013; Xiao et al., 2012). There have been two recent reports not included in the meta-analysis. Cascorbi et al. (Cascorbi et al., 2013) used cerebellar brain tissue for genotyping from 71 histopathologically verified AD cases and 81 non-demented controls and found no connections between rs2230806 or rs1800977 and amyloid deposition in brain. Xiao et al. (Xiao et al., 2012) investigated possible associations of common non-synonymous polymorphic markers in several lipid related genes including ABCA1R219K, with susceptibility to AD and plasma lipid levels (104 AD patients and 104 controls, Han Chinese). The results demonstrated association of significantly higher levels of HDL cholesterol and apoA-I, in the carriers of KK genotype and K allele, and a decreased AD risk.
Like in many other association studies the reasons for the controversial results in the studies discussed above are not clear: size of the sample, differences in genetic background, ethnicity and race, as well as significant differences in minor allele frequencies in many of those studies obviously exist. However, the results of all major GWAS studies on LOAD risk associations published during the last 5 years, and numerous meta-analysis reports based on those, demonstrate that it is very difficult or even impossible to overcome the problems listed above and the discovery of new significant associations of variants within ABCA1 (or perhaps any other gene) is unlikely (Ridge et al., 2013). Insufficient, or complete lack of understanding of the functional relationship of the majority of genes identified by GWAS and LOAD (APOE is perhaps example #1) add strongly to this rather disappointing conclusion. However, resent pathway analyses (Jones et al., 2010) based on association signals identified in 2 large GWAS studies (Harold et al., 2009; Lambert et al., 2009) identify sets of genes and physiological processes related to cholesterol metabolism and innate immune response and demonstrate that these processes are etiologically relevant and suitable targets for novel and existing therapeutic approaches. It is quite remarkable that while ABCA1 had not been identified in either of the GWAS, the gene is listed in the lipid related processes as a gene with a SNP significantly enriched at p<0.0001. There are two subsets of genes (Table 5 in (Jones et al., 2010)), that deserve a particular attention and provide further insight into ABCA1 role in LOAD: a) the first subset is composed of 3 genes: ABCA1, APOE and APOA-I. The significance of the proteins in the pathogenesis of LOAD has been underlined numerous times in this review; b) the second subset is represented by the following 6 genes: APOE, APOC1, APOC1, APOC2, APOC4, and ABCG1. All of those are transcriptionally regulated by LXR/RXR heterodimers and experimental therapeutic approaches have been successfully applied numerous times in in vitro and in vivo systems. Regardless of some controversies, the application of synthetic RXR agonists remains promising and is being evaluated in preclinical and Phase I clinical trials (Cramer et al., 2012; Fitz et al., 2013; Price et al., 2013; Tesseur et al., 2013; Veeraraghavalu et al., 2013). There is no such other group of genes within the lipid-related processes identified by Jones et al. (Jones et al., 2010). Overall, the representation of identified genes involved in cholesterol/lipid metabolism is impressive, lending further credibility to and suggesting a role for ABCA1, as well as other cholesterol transporters and APOE gene cluster in AD pathogenesis. These results also provide solid foundation for the development of relevant targeted therapeutic approaches.
Rare functional variants and AD pathology
A differential contribution of missense mutation to the biochemical, cellular and clinical phenotypes was recognized early after the cloning of ABCA1. There are two notable examples of ABCA1 mutations highly relevant to our understanding of its association to AD risk and AD pathogenesis.
N935S mutation was identified in a patient with extremely low levels of HDL, but without accelerated development of premature atherosclerosis and with signs of severe dementia and amyloid depositions in the brain at age of 60 (Walter et al., 1994a; Walter et al., 1994b). In vitro experiments with an immortalized cell line derived from the patient’s fibroblasts demonstrated the dominant negative effect of the mutant protein and the influence of ABCA1 mediated disturbance of cholesterol efflux on APP processing: the cells responded to the LXR ligand (T0901317) treatment with increased Aβ secretion (Koldamova et al., 2005b).
The second example is a compound heterozygous mutation (D1099Y and F2009S) identified in a subject with severe HDL cholesterol deficiency (Ho Hong et al., 2002). The patient had no history or clinical manifestation of CAD and no other cardiovascular disease risk factors, except for low HDL cholesterol. There were no clinical signs of TD either. The patient developed and died of complications related to cerebral amyloid angiopathy (CAA). These two examples point to the significance of rare functional variants of ABCA1 which can be associated with AD risk, most probably operating through HDL cholesterol levels, although other mechanisms, influencing APP processing cannot be excluded.
Recently, the contribution of rare non-synonymous gene variants to the risk of common diseases, including TREM2, APP, ABCA1, and AD has been demonstrated by deep re-sequencing. Lupton et al. (Lupton et al., 2014) used pooled DNA samples from 311 cases and 367 controls for next generation sequencing to identify low frequency, non-synonymous ABCA1 variation. They found a significantly higher proportion of rare ABCA1 variants in control individuals, compared to AD cases. The interpretation of the results, according to the authors was suggestive of a protective effect. Importantly, the number of non-synonymous alleles of previously identified rare variant E1172D, known to be associated with very high HDL-C levels, was more than twice higher in the control compared to the case samples. While the number of these studies is small and the protocol is still relatively expensive, the potential of those studies to reveal new gene-level associations that further explain the phenotypic variance in AD beyond the variants identified in GWAS studies, is obvious.
ABCA1 and the concept of pleomorphic risk loci
In 2011 A. Singleton and J. Hardy suggested that common disease, common variant (CDCV) and multiple rare variant (MRV) hypotheses are not mutually exclusive and these two ideas can be brought together as a general hypothesis for disease susceptibility (Singleton and Hardy, 2011). They coined the term “pleomorphic risk loci” (PRL) and outlined the steps necessary to conduct, including targeted resequencing, as to understand and delineate, in the context of the hypothesis, benign, risk and protective rare variants. The technological advent of NGS platforms, relatively easy to work out protocols to generate sequencing libraries and the availability of free, open source bioinformatics software now makes it possible to test the hypothesis. A candidate PRL would influence the disease through different biological effects, accomplished by several, distinct disease-related mechanisms that coexist at the same locus and on a single gene. The total number of mutations in ABCA1 is more than 180, and the majority of those in heterozygous form (excluding compound heterozygotes with Tangier disease) determine a biochemical phenotype distinguished by low, or even very low HDL levels. Epidemiological studies provide enough evidence that in a subset of patients those levels may underlie an increased risk for AD. The non-synonymous rare variants already identified in patients with amyloid depositions in brain, amyloid angiopathy and early cognitive decline provide additional support. On the other side, the application of newest sequencing technologies with samples from patients with AD and atherosclerosis in case-control studies demonstrate that there are rare, yet protective ABCA1 gene variants. It is possible to predict, that the majority of those 180 mutations within the sequence of the entire ABCA1 will be benign with no detectable effect relevant to any disease. We are predicting soon we will have a better idea if ABCA1 is a PRL.
ABCA1 and AD model mice
In APP transgenic mice ABCA1 deficiency increases parenchymal amyloid plaques and CAA (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005a; Wahrle et al., 2005). In these mice the level of soluble apoE and apoA-I in brain was significantly reduced while insoluble apoE was not changed. In contrast, transgenic mice overexpressing Abca1 in brain have less amyloid plaques. It was also demonstrated that overexpression of ABCA1 resulted in apoE-containing particles with a larger size than normal, while the total level of apoE was decreased (Wahrle et al., 2008b)
Haplodeficiency of Abca1 was examined in APP mice expressing mouse apoE and human apoE3 or E4 isoforms. In old APP23 mice expressing mouse apoE absence of one copy of Abca1 deteriorates cognitive deficits in correlation with an increased level of soluble oligomers but not with amyloid plaques (Lefterov et al., 2009). In a recent study Fitz et al. demonstrated that Abca1 haplodeficiency had a differential effect on the phenotype of mice expressing apoE3 or apoE4 (Fitz et al., 2012). The lack of one copy of Abca1 significantly aggravates memory deficits, Aβ plaques and clearance in APP/E4 but not in APP/E3 mice (Fitz et al., 2012) suggesting that apoE4 confers less resistance to additional genetic defects. Interestingly, the same study also found a correlation between HDL in plasma and amyloid load in brain implying a causative connection between peripheral lipoproteins and Aβ load in CNS.
A plausible hypothesis to explain the above observations could be an effect of ABCA1 on AD pathogenesis (if any), intertwined with its role in cholesterol efflux and generation of HDL in brain and periphery. In CNS the role of ABCA1 is generally attributed to its effect on apoE lipidation and stability that ultimately controls apoE level. Numerous data suggest that apoE affects Aβ aggregation and clearance in isoform-dependent manner, however, the molecular mechanism underlying the effect is still not sufficiently clarified [reviewed in (Kanekiyo et al., 2014; Tai et al., 2014)]. It is a matter of debate if APOE4 isoform is deleterious or less protective with evidence supporting both claims [reviewed in (Kanekiyo et al., 2014; Kim et al., 2009; Mahley and Huang, 2012)].
Clinical data demonstrate that apoE protein level in plasma and CSF is lower in APOE4 than in APOE3/APOE2 carriers. A cross sectional study by Gupta et al. demonstrated that plasma total apoE and apoE4 levels were significantly lower in patients with AD and they further decrease with the increase of Aβ load as assessed by PET (Gupta et al., 2011). A recent large-scale case control study of CSF samples demonstrated that apoE protein levels in CSF positively associated with CSF Aβ42 levels independent of APOE4 genotype (Cruchaga et al., 2012). These data suggest that apoE levels in CSF or plasma may be causatively related to AD. In addition to total apoE level, apoE lipidation could also affect interaction with Aβ and consequently Aβ aggregation [reviewed in Tai et al.(Tai et al., 2014)]. In vitro studies have shown binding of synthetic Aβ to apoE isolated from cell conditioned media (LaDu et al., 1994) CSF (Wisniewski et al., 1993) and plasma (Strittmatter et al., 1993). Using solid-phase binding assays we also found that synthetic Aβ40 binds avidly to apoE2 (KD=15nM) and apoE4 (KD=19nM) (Koldamova et al., 2001). However, a recent study demonstrated that apoE binding to Aβ is not essential for Aβ metabolism (Verghese et al., 2013) clearly advocating for more research on this topic. Published studies are also conflicting in regard to the effect apoE has on Aβ aggregation. In vivo, lack of APOE in APP transgenic mice were shown to dramatically reduce amyloid plaques (Bales et al., 1997), confirmed by more recent studies (Bien-Ly et al., 2012; Kim et al., 2011). In vitro studies are more contradicting with some demonstrating that apoE inhibits Aβ aggregation (Wood et al., 1996a; Wood et al., 1996b) and others that it promotes Aβ aggregation (Castano et al., 1995; Hashimoto et al., 2012).
Our hypothesis is that ABCA1 control over the generation of apoE (or apoA-I)-containing HDL in brain and plasma affects Aβ metabolism on several levels (see Figure 2). In the brain: (a) by decreasing Aβ aggregation and preventing its conversion into toxic oligomers (Lefterov et al., 2009) or plaques (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005a; Wahrle et al., 2005); (b) by maintaining Aβ in soluble state and facilitating Aβ clearance by glia or its degradation by extracellular proteases (Jiang et al., 2008); (c) facilitating Aβ removal from the brain through BBB and drainage into CSF [discussed by Saint-Pol et al. (Gosselet et al., 2013; Saint-Pol et al., 2012)]. A recent randomized clinical trial tested the effect of diet in patients with mild cognitive impairment and demonstrated the significance of apoE and Aβ lipidation to impact cognitive decline (Hanson et al., 2013). However, the role of ABCA1 in maintaining plasma levels of HDL, apoA-I and apoE may be as important for AD as its CNS functions. Since Aβ binds HDL and other plasma lipoproteins (Koudinov et al., 1998; LaDu et al., 2012; LaDu et al., 1995), it is possible that circulating lipoproteins could affect Aβ balance on both sides of BBB and increase its clearance by the mechanism of “peripheral sink” (see (d) on Figure 2). The rarity of TD patients and death from CAD at age earlier than LOAD normally develops, preclude studies examining an association of TD and AD and testing various aspects of the hypothesis as outlined above (Pervaiz et al., 2012; Shahim et al., 2013). Some of the genetics studies as already discussed, however, are in agreement with a concept that higher level of HDL is protective against dementia and AD, or as measured by Global PiB index in a recent study, lower HDL and higher LDL levels are both associated with cerebral amyloidosis (Dias et al., 2014; Reed et al., 2014; Xiao et al., 2012).
Figure 2. A simplified model explaining the role of ABCA1 in Aβ deposition and clearance on both sides of the BBB.

In the brain ABCA1 mediates apoE lipidation that can affect Aβ metabolism on several levels: (a) decreases Aβ aggregation and prevents its conversion into toxic oligomers or fibrils; (b) enables Aβ clearance by glia or degradation by extracellular proteases; (c) facilitates Aβ removal from the brain through BBB or drainage into CSF. In plasma, ABCA1 is essential for HDL level that could affect Aβ clearance by the mechanism of “peripheral sink” (d).
Therapeutic implications
The fact that both ABCA1 and apoE are under the transcriptional control of LXR and RXR transcription factors offers an attractive potential for targeted therapeutic interventions. These approaches are rationalized by the molecular mechanism of Nuclear Receptor activation (see Landreth et al., in this SI: Nuclear Receptors in Neurodegenerative Diseases, Neurobiol. Dis. (2014), http://dx.doi.org/10.1016/j.nbd.2014.04.001), the discovery that LXRα/β have a role in AD pathogenesis and other forms of neurodegeneration (Andersson et al., 2005; Kim et al., 2008; Wang et al., 2002; Zelcer et al., 2007), and positive results of experimental treatment in AD mice. Numerous studies reported data confirming the beneficial effect of LXR agonists on amyloid plaques and cognitive performance in APP mice (Donkin et al., 2010; Fitz et al., 2014; Fitz et al., 2010; Jiang et al., 2008; Katz et al., 2009; Koldamova et al., 2005b; Riddell et al., 2007; Terwel et al., 2011; Vanmierlo et al., 2011). More recently, Cramer et al. reported that FDA approved RXR agonist Bexarotene decreases plaque load, increases Aβ clearance and ameliorates cognitive deficits in APP expressing mice (Cramer et al., 2012). Although the effect on amyloid deposition, as reported, has not been confirmed in follow up studies (Price et al., 2013; Veeraraghavalu et al., 2013), other laboratories reported a significant cognitive improvement even without change in plaque load (Fitz et al., 2013; Tesseur et al., 2013). The effect on memory was challenged in a recent study (LaClair et al., 2013). Yet, recent experimental and clinical data demonstrated that Bexarotene has beneficial effects on other brain disorders such as Parkinson disease (McFarland et al., 2013)) and schizophrenia, and is safe for a chronic use (Lerner et al., 2008; Lerner et al., 2013).
One confounding factor for the use of LXR and RXR agonists in patients is the increase of triglycerides in plasma (Lilley et al., 2013; Tontonoz and Mangelsdorf, 2003). Interestingly, the only published clinical trial with LXR agonist reported adverse effects on CNS and not an increase of serum triglycerides (Katz et al., 2009). Second, because of the uncertainty regarding the molecular mechanisms underlying the association of apoE4 and the increased risk of AD, there is skepticism in the AD scientific community if the effect of LXR and RXR agonists will be equally beneficial for APOE3 and APOE4 carriers. If studies from transgenic mice are any indication, Bexarotene decreases Aβ oligomers (Fitz et al., 2013) and restores cognitive deficits with similar efficiency in both APOE3 and APOE4 expressing mice (Boehm-Cagan, 2014; Fitz et al., 2013). It is obvious that additional research is needed to answer the question if AD therapies based on LXR and RXR activation are worth pursuing.
Conclusion
The significance of ABCA1 for cardiovascular disease is determined by its role as essential regulator of cholesterol efflux and HDL generation. Likewise the probable implication of ABCA1 in AD pathogenesis streams from its main physiological function namely to control the level of lipidated apoE in CNS and plasma HDL level subsequently affecting Aβ metabolism.
Acknowledgments
Supported by NIH: R01AG037481, R01AG037919, R21ES021243, K01AG044490; DOD: W81XWH-13-1-0384.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguilar-Salinas CA, et al. The R230C variant of the ATP binding cassette protein A1 (ABCA1) gene is associated with a decreased response to glyburide therapy in patients with type 2 diabetes mellitus. Metabolism. 2013;62:638–41. doi: 10.1016/j.metabol.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Alaupovic P, et al. Plasma apolipoprotein concentrations in familial apolipoprotein A-I and A-II deficiency (Tangier disease) Metabolism. 1981;30:805–9. doi: 10.1016/0026-0495(81)90027-5. [DOI] [PubMed] [Google Scholar]
- Andersson S, et al. Inactivation of liver X receptor beta leads to adult-onset motor neuron degeneration in male mice. Proc Natl Acad Sci U S A. 2005;102:3857–62. doi: 10.1073/pnas.0500634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17:263–4. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- Bien-Ly N, et al. Reducing human apolipoprotein E levels attenuates age-dependent Abeta accumulation in mutant human amyloid precursor protein transgenic mice. J Neurosci. 2012;32:4803–11. doi: 10.1523/JNEUROSCI.0033-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochem AE, et al. ABCA1 mutation carriers with low high-density lipoprotein cholesterol are characterized by a larger atherosclerotic burden. Eur Heart J. 2013;34:286–91. doi: 10.1093/eurheartj/ehs376. [DOI] [PubMed] [Google Scholar]
- Boehm-Cagan MD. Reversal of apoE4-driven brain pathology and behavioral deficits by Bexarotene. Journal of Neuroscience. 2014 doi: 10.1523/JNEUROSCI.5198-13.2014. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunham LR, et al. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med. 2007;13:340–7. doi: 10.1038/nm1546. [DOI] [PubMed] [Google Scholar]
- Brunham LR, et al. Tissue-specific roles of ABCA1 influence susceptibility to atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:548–54. doi: 10.1161/ATVBAHA.108.182303. [DOI] [PubMed] [Google Scholar]
- Cascorbi I, et al. Association of ATP-binding cassette transporter variants with the risk of Alzheimer’s disease. Pharmacogenomics. 2013;14:485–94. doi: 10.2217/pgs.13.18. [DOI] [PubMed] [Google Scholar]
- Castano EM, et al. Fibrillogenesis in Alzheimer’s disease of amyloid beta peptides and apolipoprotein E. Biochem J. 1995;306(Pt 2):599–604. doi: 10.1042/bj3060599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu LW, et al. A novel intronic polymorphism of ABCA1 gene reveals risk for sporadic Alzheimer’s disease in Chinese. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:1007–13. doi: 10.1002/ajmg.b.30525. [DOI] [PubMed] [Google Scholar]
- Clee SM, et al. Age and residual cholesterol efflux affect HDL cholesterol levels and coronary artery disease in ABCA1 heterozygotes. J Clin Invest. 2000;106:1263–70. doi: 10.1172/JCI10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Cramer PE, et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–6. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, et al. Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer’s disease. Hum Mol Genet. 2012;21:4558–71. doi: 10.1093/hmg/dds296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias IH, et al. Plasma Levels of HDL and Carotenoids are Lower in Dementia Patients with Vascular Comorbidities. J Alzheimers Dis. 2014 doi: 10.3233/JAD-131964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12:105–12. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- Donkin JJ, et al. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2010;285:34144–54. doi: 10.1074/jbc.M110.108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, et al. Improvement of Memory Deficits and Amyloid-beta Clearance in Aged APP23 Mice Treated with a Combination of Anti-Amyloid-beta Antibody and LXR Agonist. J Alzheimers Dis. 2014 doi: 10.3233/JAD-132789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, et al. Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J Neurosci. 2010;30:6862–6872. doi: 10.1523/JNEUROSCI.1051-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924-c. doi: 10.1126/science.1235809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, et al. Abca1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J Neurosci. 2012;32:13125–36. doi: 10.1523/JNEUROSCI.1937-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frikke-Schmidt R. Genetic variation in ABCA1 and risk of cardiovascular disease. Atherosclerosis. 2011;218:281–2. doi: 10.1016/j.atherosclerosis.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Frikke-Schmidt R, et al. Genetic variation in ABC transporter A1 contributes to HDL cholesterol in the general population. J Clin Invest. 2004;114:1343–53. doi: 10.1172/JCI20361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frikke-Schmidt R, et al. Mutation in ABCA1 predicted risk of ischemic heart disease in the Copenhagen City Heart Study Population. J Am Coll Cardiol. 2005;46:1516–20. doi: 10.1016/j.jacc.2005.06.066. [DOI] [PubMed] [Google Scholar]
- Frikke-Schmidt R, et al. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. Jama. 2008;299:2524–32. doi: 10.1001/jama.299.21.2524. [DOI] [PubMed] [Google Scholar]
- Gelissen IC, et al. ABCA1 and ABCG1 synergize to mediate cholesterol export to apoA-I. Arterioscler Thromb Vasc Biol. 2006;26:534–40. doi: 10.1161/01.ATV.0000200082.58536.e1. [DOI] [PubMed] [Google Scholar]
- Gosselet F, et al. Effects of oxysterols on the blood-brain barrier: Implications for Alzheimer’s disease. Biochem Biophys Res Commun. 2013 doi: 10.1016/j.bbrc.2013.11.059. [DOI] [PubMed] [Google Scholar]
- Gupta VB, et al. Plasma apolipoprotein E and Alzheimer disease risk: the AIBL study of aging. Neurology. 2011;76:1091–8. doi: 10.1212/WNL.0b013e318211c352. [DOI] [PubMed] [Google Scholar]
- Hanson AJ, et al. Effect of apolipoprotein E genotype and diet on apolipoprotein E lipidation and amyloid peptides: randomized clinical trial. JAMA Neurol. 2013;70:972–80. doi: 10.1001/jamaneurol.2013.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold D, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid beta peptide. J Neurosci. 2012;32:15181–92. doi: 10.1523/JNEUROSCI.1542-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, et al. The Absence of ABCA1 Decreases Soluble ApoE Levels but Does Not Diminish Amyloid Deposition in Two Murine Models of Alzheimer Disease. Journal of Biological Chemistry. 2005;280:43243–43256. doi: 10.1074/jbc.M508781200. [DOI] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, et al. Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J Biol Chem. 2004;279:41197–207. doi: 10.1074/jbc.M407962200. [DOI] [PubMed] [Google Scholar]
- Ho Hong S, et al. Novel ABCA1 compound variant associated with HDL cholesterol deficiency. Biochim Biophys Acta. 2002;1587:60–4. doi: 10.1016/s0925-4439(02)00066-2. [DOI] [PubMed] [Google Scholar]
- Hobbs HH, Rader DJ. ABC1: connecting yellow tonsils, neuropathy, and very low HDL. J Clin Invest. 1999;104:1015–7. doi: 10.1172/JCI8509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovingh GK, et al. HDL deficiency and atherosclerosis: lessons from Tangier disease. J Intern Med. 2004;255:299–301. doi: 10.1046/j.0954-6820.2003.01256.x. [DOI] [PubMed] [Google Scholar]
- Hughes TM, et al. Markers of cholesterol transport are associated with amyloid deposition in the brain. Neurobiol Aging. 2014;35:802–7. doi: 10.1016/j.neurobiolaging.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, et al. Meta-analysis on association between the ATP-binding cassette transporter A1 gene (ABCA1) and Alzheimer’s disease. Gene. 2012;510:147–53. doi: 10.1016/j.gene.2012.09.009. [DOI] [PubMed] [Google Scholar]
- Jiang Q, et al. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L, et al. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS One. 2010;5:e13950. doi: 10.1371/journal.pone.0013950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, et al. ApoE and Abeta in Alzheimer’s Disease: Accidental Encounters or Partners? Neuron. 2014;81:740–754. doi: 10.1016/j.neuron.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasinska JM, et al. Specific loss of brain ABCA1 increases brain cholesterol uptake and influences neuronal structure and function. J Neurosci. 2009;29:3579–89. doi: 10.1523/JNEUROSCI.4741-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz A, et al. Safety, pharmacokinetics, and pharmacodynamics of single doses of LXR-623, a novel liver X-receptor agonist, in healthy participants. J Clin Pharmacol. 2009;49:643–9. doi: 10.1177/0091270009335768. [DOI] [PubMed] [Google Scholar]
- Katzov H, et al. Genetic variants of ABCA1 modify Alzheimer disease risk and quantitative traits related to beta-amyloid metabolism. Hum Mutat. 2004;23:358–67. doi: 10.1002/humu.20012. [DOI] [PubMed] [Google Scholar]
- Khorram Khorshid HR, et al. The Association between Sporadic Alzheimer’s Disease and the Human ABCA1 and APOE Gene Polymorphisms in Iranian Population. Iran Red Crescent Med J. 2011;13:256–62. [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, et al. Liver X receptor beta (LXRbeta): a link between beta-sitosterol and amyotrophic lateral sclerosis-Parkinson’s dementia. Proc Natl Acad Sci U S A. 2008;105:2094–9. doi: 10.1073/pnas.0711599105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, et al. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, et al. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J Neurosci. 2011;31:18007–12. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WS, et al. Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J Biol Chem. 2007;282:2851–61. doi: 10.1074/jbc.M607831200. [DOI] [PubMed] [Google Scholar]
- Koch S, et al. Characterization of four lipoprotein classes in human cerebrospinal fluid. J Lipid Res. 2001;42:1143–51. [PubMed] [Google Scholar]
- Koldamova R, et al. The role of ATP-Binding cassette transporter A1 in Alzheimer’s disease and neurodegeneration. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbalip.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova R, et al. Lack of ABCA1 Considerably Decreases Brain ApoE Level and Increases Amyloid Deposition in APP23 Mice. Journal of Biological Chemistry. 2005a;280:43224–43235. doi: 10.1074/jbc.M504513200. [DOI] [PubMed] [Google Scholar]
- Koldamova RP, et al. 22R-hydroxycholesterol and 9-cis-retinoic acid induce ATP-binding cassette transporter A1 expression and cholesterol efflux in brain cells and decrease amyloid beta secretion. J Biol Chem. 2003;278:13244–56. doi: 10.1074/jbc.M300044200. [DOI] [PubMed] [Google Scholar]
- Koldamova RP, et al. Apolipoprotein A-I directly interacts with amyloid precursor protein and inhibits A beta aggregation and toxicity. Biochemistry. 2001;40:3553–60. doi: 10.1021/bi002186k. [DOI] [PubMed] [Google Scholar]
- Koldamova RP, et al. The Liver X Receptor Ligand T0901317 Decreases Amyloid {beta} Production in Vitro and in a Mouse Model of Alzheimer’s Disease. Journal of Biological Chemistry. 2005b;280:4079–4088. doi: 10.1074/jbc.M411420200. [DOI] [PubMed] [Google Scholar]
- Koseki M, et al. Impaired insulin secretion in four Tangier disease patients with ABCA1 mutations. J Atheroscler Thromb. 2009;16:292–6. doi: 10.5551/jat.e599. [DOI] [PubMed] [Google Scholar]
- Koudinov AR, et al. Alzheimer’s amyloid beta interaction with normal human plasma high density lipoprotein: association with apolipoprotein and lipids. Clin Chim Acta. 1998;270:75–84. doi: 10.1016/s0009-8981(97)00207-6. [DOI] [PubMed] [Google Scholar]
- LaClair KD, et al. Treatment with bexarotene, a compound that increases apolipoprotein-E, provides no cognitive benefit in mutant APP/PS1 mice. Mol Neurodegener. 2013;8:18. doi: 10.1186/1750-1326-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaDu MJ, et al. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994;269:23403–6. [PubMed] [Google Scholar]
- LaDu MJ, et al. Preferential interactions between ApoE-containing lipoproteins and Abeta revealed by a detection method that combines size exclusion chromatography with non-reducing gel-shift. Biochim Biophys Acta. 2012;1821:295–302. doi: 10.1016/j.bbalip.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaDu MJ, et al. Purification of apolipoprotein E attenuates isoform-specific binding to beta-amyloid. J Biol Chem. 1995;270:9039–42. doi: 10.1074/jbc.270.16.9039. [DOI] [PubMed] [Google Scholar]
- Lambert JC, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Lefterov I, et al. Memory deficits in APP23/Abca1+/− mice correlate with the level of Abeta oligomers. ASN Neuro. 2009:1. doi: 10.1042/AN20090015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner V, et al. Bexarotene as add-on to antipsychotic treatment in schizophrenia patients: a pilot open-label trial. Clin Neuropharmacol. 2008;31:25–33. doi: 10.1097/WNF.0b013e31806450da. [DOI] [PubMed] [Google Scholar]
- Lerner V, et al. The retinoid X receptor agonist bexarotene relieves positive symptoms of schizophrenia: a 6-week, randomized, double-blind, placebo-controlled multicenter trial. J Clin Psychiatry. 2013;74:1224–32. doi: 10.4088/JCP.12m08160. [DOI] [PubMed] [Google Scholar]
- Lilley JS, et al. Oral retinoids and plasma lipids. Dermatol Ther. 2013;26:404–10. doi: 10.1111/dth.12085. [DOI] [PubMed] [Google Scholar]
- Lund-Katz S, Phillips MC. High density lipoprotein structure-function and role in reverse cholesterol transport. Subcell Biochem. 2010;51:183–227. doi: 10.1007/978-90-481-8622-8_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupton MK, et al. The role of ABCA1 gene sequence variants on risk of Alzheimer’s disease. J Alzheimers Dis. 2014;38:897–906. doi: 10.3233/JAD-131121. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Huang Y. Apolipoprotein e sets the stage: response to injury triggers neuropathology. Neuron. 2012;76:871–85. doi: 10.1016/j.neuron.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, et al. Genetic defects in lipoprotein metabolism. Elevation of atherogenic lipoproteins caused by impaired catabolism. Jama. 1991;265:78–83. doi: 10.1001/jama.265.1.78. [DOI] [PubMed] [Google Scholar]
- McFarland K, et al. Low dose bexarotene treatment rescues dopamine neurons and restores behavioral function in models of Parkinson’s disease. ACS Chem Neurosci. 2013;4:1430–8. doi: 10.1021/cn400100f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeish J, et al. High density lipoprotein deficiency and foam cell accumulation in mice with targeted disruption of ATP-binding cassette transporter-1. Proc Natl Acad Sci USA. 2000;97:4245–4250. doi: 10.1073/pnas.97.8.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merched A, et al. Decreased high-density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer’s disease. Neurobiol Aging. 2000;21:27–30. doi: 10.1016/s0197-4580(99)00103-7. [DOI] [PubMed] [Google Scholar]
- Nagata KO, et al. ABCA1 dimer-monomer interconversion during HDL generation revealed by single-molecule imaging. Proc Natl Acad Sci U S A. 2013;110:5034–9. doi: 10.1073/pnas.1220703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oram JF, Vaughan AM. ATP-Binding cassette cholesterol transporters and cardiovascular disease. Circ Res. 2006;99:1031–43. doi: 10.1161/01.RES.0000250171.54048.5c. [DOI] [PubMed] [Google Scholar]
- Panzenboeck U, et al. ABCA1 and scavenger receptor class B, type I, are modulators of reverse sterol transport at an in vitro blood-brain barrier constituted of porcine brain capillary endothelial cells. J Biol Chem. 2002;277:42781–42789. doi: 10.1074/jbc.M207601200. [DOI] [PubMed] [Google Scholar]
- Pervaiz MA, et al. A Non-classical Presentation of Tangier Disease with Three ABCA1 Mutations. JIMD Rep. 2012;4:109–11. doi: 10.1007/8904_2011_81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- Pitas RE, et al. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B, E(LDL) receptors in the brain. J Biol Chem. 1987;262:14352–60. [PubMed] [Google Scholar]
- Poirier J, et al. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993;342:697–9. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- Price AR, et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924-d. doi: 10.1126/science.1234089. [DOI] [PubMed] [Google Scholar]
- Puntoni M, et al. Tangier disease: epidemiology, pathophysiology, and management. Am J Cardiovasc Drugs. 2012;12:303–11. doi: 10.2165/11634140-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Rader DJ, de Goma EM. Approach to the patient with extremely low HDL-cholesterol. J Clin Endocrinol Metab. 2012;97:3399–407. doi: 10.1210/jc.2012-2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed B, et al. Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol. 2014;71:195–200. doi: 10.1001/jamaneurol.2013.5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C. Dyslipidemia and the risk of Alzheimer’s disease. Curr Atheroscler Rep. 2013;15:307. doi: 10.1007/s11883-012-0307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds CA, et al. A survey of ABCA1 sequence variation confirms association with dementia. Hum Mutat. 2009;30:1348–54. doi: 10.1002/humu.21076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell DR, et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol Cell Neurosci. 2007;34:621–8. doi: 10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Ridge PG, et al. Alzheimer’s disease: analyzing the missing heritability. PLoS One. 2013;8:e79771. doi: 10.1371/journal.pone.0079771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Rodriguez E, et al. Association of genetic variants of ABCA1 with Alzheimer’s disease risk. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:964–8. doi: 10.1002/ajmg.b.30552. [DOI] [PubMed] [Google Scholar]
- Saint-Pol J, et al. Brain pericytes ABCA1 expression mediates cholesterol efflux but not cellular amyloid-beta peptide accumulation. J Alzheimers Dis. 2012;30:489–503. doi: 10.3233/JAD-2012-112090. [DOI] [PubMed] [Google Scholar]
- Santamarina-Fojo S, et al. Regulation and intracellular trafficking of the ABCA1 transporter. J Lipid Res. 2001;42:1339–45. [PubMed] [Google Scholar]
- Saunders AM, Roses AD. Apolipoprotein E4 allele frequency, ischemic cerebrovascular disease, and Alzheimer’s disease. Stroke. 1993;24:1416–7. doi: 10.1161/01.str.24.9.1416. [DOI] [PubMed] [Google Scholar]
- Schou J, et al. ABC transporter genes and risk of type 2 diabetes: a study of 40,000 individuals from the general population. Diabetes Care. 2012;35:2600–6. doi: 10.2337/dc12-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Service SK, et al. Re-sequencing expands our understanding of the phenotypic impact of variants at GWAS loci. PLoS Genet. 2014;10:e1004147. doi: 10.1371/journal.pgen.1004147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahim P, et al. Plasma amyloid-beta in patients with Tangier disease. J Alzheimers Dis. 2013;35:307–12. doi: 10.3233/JAD-122425. [DOI] [PubMed] [Google Scholar]
- Shibata N, et al. Association studies of cholesterol metabolism genes (CH25H, ABCA1 and CH24H) in Alzheimer’s disease. Neurosci Lett. 2006;391:142–6. doi: 10.1016/j.neulet.2005.08.048. [DOI] [PubMed] [Google Scholar]
- Simons M, et al. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci U S A. 1998;95:6460–4. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton A, Hardy J. A generalizable hypothesis for the genetic architecture of disease: pleomorphic risk loci. Hum Mol Genet. 2011;20:R158–62. doi: 10.1093/hmg/ddr358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:8098–102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YM, et al. The polymorphism of the ATP-binding cassette transporter 1 gene modulates Alzheimer disease risk in Chinese Han ethnic population. Am J Geriatr Psychiatry. 2012;20:603–11. doi: 10.1097/JGP.0b013e3182423b6a. [DOI] [PubMed] [Google Scholar]
- Sundar PD, et al. Gender-specific association of ATP-binding cassette transporter 1 (ABCA1) polymorphisms with the risk of late-onset Alzheimer’s disease. Neurobiol Aging. 2007;28:856–62. doi: 10.1016/j.neurobiolaging.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Tai LM, et al. Soluble apoE/Abeta complex: mechanism and therapeutic target for APOE4-induced AD risk. Mol Neurodegener. 2014;9:2. doi: 10.1186/1750-1326-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Oram JF. The cell cholesterol exporter ABCA1 as a protector from cardiovascular disease and diabetes. Biochim Biophys Acta. 2009;1791:563–72. doi: 10.1016/j.bbalip.2009.03.011. [DOI] [PubMed] [Google Scholar]
- Terwel D, et al. Critical role of astroglial apolipoprotein E and liver X receptor-alpha expression for microglial Abeta phagocytosis. J Neurosci. 2011;31:7049–59. doi: 10.1523/JNEUROSCI.6546-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslovich TM, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesseur I, et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924-e. doi: 10.1126/science.1233937. [DOI] [PubMed] [Google Scholar]
- Timmins JM, et al. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J Clin Invest. 2005;115:1333–42. doi: 10.1172/JCI23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Mangelsdorf DJ. Liver X receptor signaling pathways in cardiovascular disease. Mol Endocrinol. 2003;17:985–93. doi: 10.1210/me.2003-0061. [DOI] [PubMed] [Google Scholar]
- van Dam MJ, et al. Association between increased arterial-wall thickness and impairment in ABCA1-driven cholesterol efflux: an observational study. Lancet. 2002;359:37–42. doi: 10.1016/S0140-6736(02)07277-X. [DOI] [PubMed] [Google Scholar]
- Vanmierlo T, et al. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol Aging. 2011;32:1262–72. doi: 10.1016/j.neurobiolaging.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Vaughan AM, Oram JF. ABCA1 redistributes membrane cholesterol independent of apolipoprotein interactions. J Lipid Res. 2003;44:1373–1380. doi: 10.1194/jlr.M300078-JLR200. [DOI] [PubMed] [Google Scholar]
- Vedhachalam C, et al. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. Journal of Biological Chemistry. 2007;282:25123–25130. doi: 10.1074/jbc.M704590200. [DOI] [PubMed] [Google Scholar]
- Veeraraghavalu K, et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924-f. doi: 10.1126/science.1235505. [DOI] [PubMed] [Google Scholar]
- Vergeer M, et al. Carriers of loss-of-function mutations in ABCA1 display pancreatic beta-cell dysfunction. Diabetes Care. 2010;33:869–74. doi: 10.2337/dc09-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese PB, et al. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc Natl Acad Sci U S A. 2013;110:E1807–16. doi: 10.1073/pnas.1220484110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal-Molina MT, et al. Association of the ATP-binding cassette transporter A1 R230C variant with early-onset type 2 diabetes in a Mexican population. Diabetes. 2008;57:509–13. doi: 10.2337/db07-0484. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, et al. Deletion of Abca1 Increases A{beta} Deposition in the PDAPP Transgenic Mouse Model of Alzheimer Disease. Journal of Biological Chemistry. 2005;280:43236–43242. doi: 10.1074/jbc.M508780200. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008a;118:671–682. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle SE, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008b;118:671–82. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle SE, et al. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem. 2004;279:40987–93. doi: 10.1074/jbc.M407963200. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, et al. Apolipoprotein E levels in cerebrospinal fluid and the effects of ABCA1 polymorphisms. Mol Neurodegener. 2007;2:7. doi: 10.1186/1750-1326-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter M, et al. The high density lipoprotein- and apolipoprotein A-I-induced mobilization of cellular cholesterol is impaired in fibroblasts from Tangier disease subjects. Biochem Biophys Res Commun. 1994a;205:850–6. doi: 10.1006/bbrc.1994.2742. [DOI] [PubMed] [Google Scholar]
- Walter M, et al. Characterization of atherosclerosis in a patient with familial high-density lipoprotein deficiency. Atherosclerosis. 1994b;110:203–8. doi: 10.1016/0021-9150(94)90205-4. [DOI] [PubMed] [Google Scholar]
- Wang F, Jia J. Polymorphisms of cholesterol metabolism genes CYP46 and ABCA1 and the risk of sporadic Alzheimer’s disease in Chinese. Brain Res. 2007;1147:34–8. doi: 10.1016/j.brainres.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Wang L, et al. Liver X receptors in the central nervous system: From lipid homeostasis to neuronal degeneration. Proc Natl Acad Sci USA. 2002;99:13878–13883. doi: 10.1073/pnas.172510899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, et al. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci U S A. 2004;101:9774–9. doi: 10.1073/pnas.0403506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XF, et al. Quantitative assessment of the effect of ABCA1 gene polymorphism on the risk of Alzheimer’s disease. Mol Biol Rep. 2013;40:779–85. doi: 10.1007/s11033-012-2115-9. [DOI] [PubMed] [Google Scholar]
- Wavrant-De Vrieze F, et al. ABCA1 polymorphisms and Alzheimer’s disease. Neurosci Lett. 2007;416:180–3. doi: 10.1016/j.neulet.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerterp M, et al. ATP-binding cassette transporters, atherosclerosis, and inflammation. Circ Res. 2014;114:157–70. doi: 10.1161/CIRCRESAHA.114.300738. [DOI] [PubMed] [Google Scholar]
- Willer CJ, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–9. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, et al. Apolipoprotein E: binding to soluble Alzheimer’s beta-amyloid. Biochem Biophys Res Commun. 1993;192:359–65. doi: 10.1006/bbrc.1993.1423. [DOI] [PubMed] [Google Scholar]
- Wollmer MA, et al. ABCA1 modulates CSF cholesterol levels and influences the age at onset of Alzheimer’s disease. Neurobiol Aging. 2003;24:421–6. doi: 10.1016/s0197-4580(02)00094-5. [DOI] [PubMed] [Google Scholar]
- Wood SJ, et al. An ApoE-Abeta inhibition complex in Abeta fibril extension. Chem Biol. 1996a;3:949–56. doi: 10.1016/s1074-5521(96)90183-0. [DOI] [PubMed] [Google Scholar]
- Wood SJ, et al. Seeding of A beta fibril formation is inhibited by all three isotypes of apolipoprotein E. Biochemistry. 1996b;35:12623–8. doi: 10.1021/bi961074j. [DOI] [PubMed] [Google Scholar]
- Xiao Z, et al. Association studies of several cholesterol-related genes (ABCA1, CETP and LIPC) with serum lipids and risk of Alzheimer’s disease. Lipids Health Dis. 2012;11:163. doi: 10.1186/1476-511X-11-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka M, et al. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32:17321–31. doi: 10.1523/JNEUROSCI.1569-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, et al. Proposed mechanism for lipoprotein remodelling in the brain. Biochim Biophys Acta. 2010;1801:819–23. doi: 10.1016/j.bbalip.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelcer N, et al. Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc Natl Acad Sci USA. 2007;104:10601–10606. doi: 10.1073/pnas.0701096104. [DOI] [PMC free article] [PubMed] [Google Scholar]