

Abstract
Understanding the biology of cell surface proteins is important particularly when they are utilized as viral receptors for viral entry. By manipulating the expression of cell surface receptors that have been coopted by viruses, the susceptibility of an individual to virus-induced disease or, alternatively, the effectiveness of viral-based gene therapy can be modified. The most commonly studied vector for gene therapy is adenovirus. The majority of adenovirus types utilize the coxsackievirus and adenovirus receptor (CAR) as a primary receptor to enter cells. Species B adenovirus do not interact with CAR, but instead interact with the cell surface proteins desmoglein-2 (DSG-2) and cluster of differentiation 46 (CD46). These cell surface proteins exhibit varying degrees of alternative mRNA splicing, creating an estimated 20 distinct protein isoforms. It is likely that alternative splice forms have allowed these proteins to optimize their effectiveness in a plethora of niches, including roles as cell adhesion proteins and regulators of the innate immune system. Interestingly, there are soluble isoforms of these viral receptors, which lack the transmembrane domain. These soluble isoforms can potentially bind to the surface of a virus in the extracellular compartment, blocking the ability of the virus to bind to the host cell, reducing viral infectivity. Finally, the diversity of viral receptor isoforms appears to facilitate an assortment of interactions between viral receptor proteins and cytosolic proteins, leading to differential sorting in polarized cells. Using adenoviral receptors as a model system, the purpose of this review is to highlight the role that isoform-specific protein localization plays in the entry of pathogenic viruses from the apical surface of polarized epithelial cells.
Introduction
The diversity of viral pathogens that have been identified is tremendous [1, 2]. Viruses have evolved to use many different cell surface proteins as viral receptors or co-receptors. Although the mechanisms behind receptor choice are not well defined, many disparate viral phylogenies have undergone convergent evolution towards the same cell surface receptor. Frequently, the cell-based viral receptor has an essential physiological function within the organism, such as clearance of cytotoxic molecules, or target cell, such as cell-cell adhesion. This limits the ability of the host cell to alter the presence of these essential proteins to circumvent viral entry. However, the aspect potentially under cellular evolutionary control is the protein expression pattern. Through alternative mRNA splicing, viral receptors may possess a multitude of protein isoforms with different physiological implications for both viral tropism and cellular biology. This principle is illustrated below by examining the biology of 3 distinct adenovirus receptors, the Coxsackievirus and adenovirus receptor (CAR), cluster of differentiation 46 (CD46), and desmoglein-2 (DSG-2).
Adenovirus
Adenoviruses (AdV) are common human pathogens that generally cause typical cold symptoms in healthy individuals [3, 4]. However, epidemic AdV outbreaks occur in closed communities and among young military recruits during basic training. Moreover, AdV infections can be lethal in highly susceptible immunosuppressed populations, such as in the transplant setting. Depending on serotype, AdV can also cause gastroenteritis with prolonged fecal shedding or keratoconjunctivitis that can lead to blindness. Human AdV are nonenveloped, icosahedral-shaped viruses that contain an approximately 36-kb double-stranded DNA genome [5, 6]. The outer protein capsid is primarily composed of two distinct regions (Fig. 1). The penton base region is found at the vertices of the icosahedron, while the hexon region connects the penton base regions to each other. Attached to each vertex is an elongated trimeric fiber protein, which attaches to cell surface proteins [6, 7]. The binding of the trimeric fiber knob to a particular cell surface protein subsequently allows the virus to interact with co-receptors and enter the host cell. CAR is the primary attachment receptor for AdV species A and C-G [8–11], while CD46 [12, 13] and DSG-2 [14] are the primary receptors for species B and some species D AdV [15–17]. Virus uptake is mediated by binding of the capsid penton-base with the αvβ3/αvβ5 integrins, which can facilitate cell entry, leading to infection [18–22]. Several other important co-receptors, such as MHC class I [23], sialic acid [24], and coagulation factor X [25], have been described. This review will focus on the protein isoform diversity of the primary AdV receptors.
Figure 1.
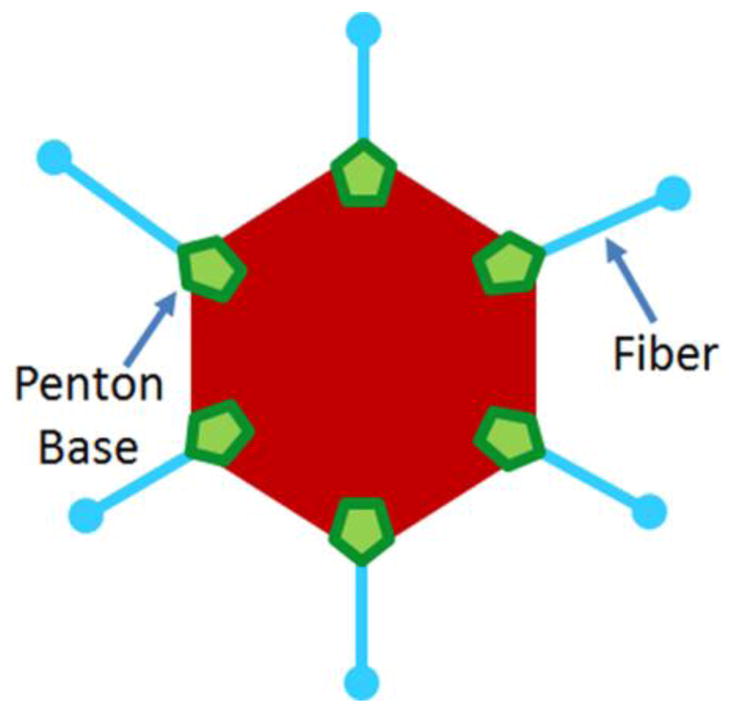
A two-dimensional representation of the adenovirus capsid structure. Although composed of both major and minor capsid proteins, only the major capsid proteins are labeled here. The penton-base region is in green, hexon region in red, and fiber-knobs are blue.
Viral receptors
AdV has been identified as both a pathogen and a potential vector for gene therapy. Thus, the interactions between these viruses and the cell membrane molecules they have adopted as viral receptors have been studied intensely. Through convergent evolution, most adenoviruses and group B coxsackieviruses use CAR as a major receptor [8–10]. In contrast, species B AdV can be divided into three groups based on receptor usage. Group B1 (AdV16, AdV21, AdV35 and AdV50) use CD46 as a primary receptor while group B2 (AdV3, AdV7 and AdV14) primarily utilize DSG-2 and group B3 (AdV11) use both DSG-2 and CD46 as viral receptors [14, 26]. Three AdV-D viruses, AdV-26, 37, and 49 have also been found to use CD46 as a receptor [15–17]. The Edmonston strain of the measles virus and certain serotypes of herpesvirus have also co-evolved to utilize CD46 as a receptor [27, 28]. In addition to being viral receptors, these cell membrane proteins serve other biological functions including roles in cell-cell adhesion and the innate immune system.
Alternative splicing plays a major role behind the incredible diversity in these viral receptors and likely plays an evolutionary role in the selection of these proteins as primary receptors by the viruses. Alternative splicing is a mechanism that allows multiple isoforms of a protein to be synthesized from a single gene by changing which fragments of the messenger RNA (mRNA) are removed as introns and which fragments are sent to the ribosomes for translation into protein as exons. While DSG-2 does not exhibit alternative splicing, its unique interaction with group B2 adenoviruses demonstrates the diversity of viral-cell interactions. The alternative splicing of CAR and CD46 alters their physiological interactions with cytosolic and extracellular proteins, their cellular localization, as well as their functionality as viral receptors. These novel splice forms have major implications for the interactions between these proteins and the viruses that have adopted them as primary receptors.
The Coxsackievirus and Adenovirus Receptor
CAR is a transmembrane cell-adhesion protein that is utilized as a viral receptor by coxsackie B viruses (CVB) and all major genera of adenovirus excluding group B [8–10, 29]. Located on human chromosome 21, genomic CAR, CXADR, consists of 8 separate exons (Fig. 2) [30–33]. Depending on which exons are transcribed, the amino acid sequence of the resultant peptide is between 89 and 365 amino acids (aa) [34, 35]. There are five splice forms of CAR described in humans, but only 3 in mice [34, 36, 37]. The diversity of isoforms has led to great confusion in nomenclature within the literature (Fig. 2B, 3). Three isoforms, CAR β, γ, and δ, lack the transmembrane domain and are soluble proteins secreted into the extracellular space. In addition to the three soluble isoforms, CAR possesses two transmembrane isoforms which we have recently renamed in order to clarify by exon usage [32]. The 7 exon-encoded isoform, CAREx7, is also named hCAR, hCAR1, SIV, or α CAR, and is the protein equivalent of murine CAR2 (mCAR2). The 8 exon-encoded isoform, CAREx8, is also named hCAR5 or TVV, and is the murine CAR1 equivalent (mCAR1). A unified nomenclature for these isoforms would facilitate literacy in this field, particularly in light of the novel “chimeric antigen receptor” (CAR) that is receiving significant clinical interest and the calcium-sensing receptor (CaR) [38, 39].
Figure 2.

Exon map of the 5 protein isoforms of CAR. A) CXADR mRNA exon map relative to protein domains. LS, leader sequence; TM, transmembrane domain. B) The portions of the exons expressed in each isoform are in blue. Note that some isoforms express only part of a particular exon. β CAR and γ CAR express the same portion of exon 7 while δ CAR does not.
Figure 3.
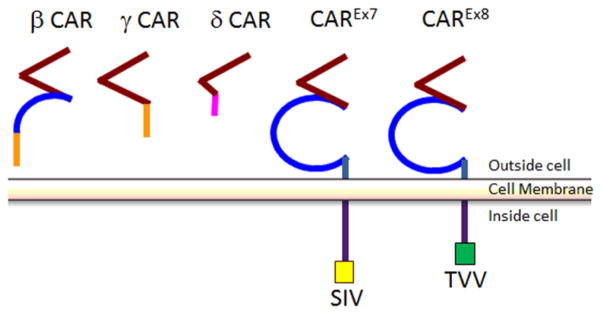
Schematic of morphological differences between the 5 isoforms of human CAR. Red represents the D1 or Ig1-V-like domain, blue represents the D2 or Ig2-C2-like domain, purple represents the cytoplasmic domain, the PDZ-binding domains are highlighted in yellow or green. When the regions of the cytoplasmic tails are homologous, the colors used are the same. Note that δ CAR contains an incomplete copy of the Ig-1 domain whereas β CAR contains an incomplete copy of the Ig2 domain. β CAR and γ CAR have homologous cytoplasmic tails.
Morphology of CAR
CAR is a member of the CTX subfamily of the immunoglobulin superfamily [40]. CAR is composed of 8 separate exons (Fig. 2A). Exon 1 comprises the leader sequence (LS) necessary for translocation to the endoplasmic reticulum (ER) after protein translation is initiated. The next four exons compose the extracellular region, which include two immunoglobulin (Ig)-like domains known as D1 or Ig1 and D2 or Ig2, separated by a J segment [34, 35, 41]. D1 is an immunoglobulin-like variable domain (IgV) that is essential for viral attachment; D2 is structurally similar to an immunoglobulin-like constant domain type 2 (IgC2) [42–44]. D1 is encoded by exons 2 and 3, while D2 is encoded by exons 4 and 5 [41]. These extracellular immunoglobulin-like domains are also essential for CAR’s role as a cell adhesion protein [45, 46]. The first half of exon 6 encodes the transmembrane domain of CAR, bridging the extracellular and cytoplasmic regions of the viral receptor. The remaining exons, the second half of exon 6 and exons 7 and 8, compose the cytoplasmic domain that is not essential for viral infection but is essential for cellular localization and signaling [32, 47–52].
Soluble isoforms of CAR
There are three soluble splice forms of CAR: β, γ, and δ. Soluble CAR originates from the de novo synthesis of alternatively spliced mRNAs from the same segment of genomic DNA as the transmembrane CAR isoforms [34, 35]. All three soluble forms of CAR share the same leader sequence and the complete sequence for exon 1. However, the three splice variants differ significantly in size and morphology. β CAR, the largest of the three, is 252 aa long, while γ CAR is 200 aa, and δ CAR is 89 aa long [35]. The β splice form is also referred to as CAR 4/7 since it is created when small nuclear ribonucleic particles (snRNPs) cleave out a segment from the end of exon 4 to a point in the middle of exon 7. β CAR contains a complete copy of the D1 domain and only part of the D2 domain. As it lacks exons 5 and 6 completely, β CAR does not have a transmembrane region. Due to a splicing event in the middle of exon 7, β CAR experiences a frame shift within its truncated fragment of exon 7, leading to a unique 62 aa C-terminus. γ CAR, also known as CAR 3/7, is the product of a splicing event that cleaves from the end of exon 3 to a site in the middle of exon 7, which it shares with β CAR. γ CAR only contains a complete copy of the D1 domain and the same 62 aa C terminus as β CAR. The smallest of the three splice forms of CAR, δ CAR (CAR 2/7) only contains exons 1 and 2, 21 nucleotides from exon 3, and 19 aa from exon 7. δ CAR has a distinct 19 aa C terminus, resulting from a different splicing site within exon 7 when compared with CAREx7, β CAR, and γ CAR. Although the ability of β CAR to block viral infection is described below [53, 54], the biological importance of these isoforms remains to be fully elucidated.
Soluble isoforms of CAR and adenovirus infection
Alternative splicing results in soluble CAR protein isoforms that are secreted from the cell and can interact with coxsackievirus and AdV. Thus, it is possible that soluble isoforms of CAR could potentially be decoy viral receptors and lead to a decrease in viral infectivity. Possessing a complete D1 domain and a partial D2 domain, β CAR can bind to coxsackievirus B3 (CVB3) and AdV, whereas δ CAR cannot [35]. When β CAR is applied to HeLa cells prior to exposure to CVB3, β CAR binds to the canyon-like receptor binding sites of CVB3, reducing the amount of virus entering the cells [35]. Adding β CAR prior or at the same time as inoculating mice with CVB3 leads to a significant decrease in CVB3 infection in the myocardium and pancreas of mice in vivo, as compared to the control [53–55]. However, using β CAR as a treatment for myocarditis caused by CVB3 in mice leads to increased inflammation of the myocardium and exaggerated tissue damage [53]. The paradoxical increase in tissue damage and severity of the myocarditis in subjects where β CAR was added daily emphasize the importance of the immune response in the severity of myocarditis and limits its potential therapeutic use. In contrast, knock-out of all isoforms of CAR specifically in the adult heart or pancreas ablates CVB3 pathogenesis in the CAR-deficient tissue [56]. To our knowledge, AdV infection has never been evaluated in either model system likely due to the fact that human AdV does not replicate in murine cells and murine AdV does not use CAR as a receptor [57].
Transmembrane isoforms of CAR
CAR has two transmembrane isoforms, which we have named CAREx7 and CAREx8 in order to reflect exon inclusion and simplify nomenclature. CAREx7 is also called α CAR [34] or SIV since it ends with the amino acids SIV [36, 58], and is the human equivalent to murine CAR2 (mCAR2). CAREx8 does not have a Greek letter designation but is also called TVV since it ends in those amino acids, and is the human equivalent to mCAR1. The two mouse isoforms were named based on the order in which they were discovered, whereas the human isoforms have been classified based on the exons expressed in the cytoplasmic tail [32, 36]. Both of the transmembrane isoforms of CAR are identical for the first 339 aa. They share a common extracellular domain. Therefore, both isoforms are predicted to have the same affinities and binding preferences in regards to extracellular molecular interactions, and no differences have been shown to date. These isoforms differ only at the last portion of the cytoplasmic domain. Both exon 7 and 8 have 3′ untranslated regions (UTR) downstream from the stop codon. By virtue of a cryptic splice site within exon 7 that splices to exon 8, CAREx7 contains 26 unique aa that are not found within the 8 exon isoform and exon 8 encodes 13 aa unique to CAREx8 [32]. It is because of their different C termini that CAREx7 and CAREx8 possess differential localizations in mice [58, 59], and radically different physiologies in polarized airway epithelial cells [32]. In polarized epithelial cells, CAREx7 localizes within the basolateral portion of the tight junctions and within the adherens junctions where it behaves as a homophilic or heterophilic adhesion protein between epithelial cells or leukocytes, respectively [46, 60–63]. However, CAREx8 localizes within a subapical compartment and at the apical surface of polarized cells [32, 33]. We have demonstrated that side-stream tobacco smoke, the smoke that emanates from the lit end of a cigarette and comprises the majority of second-hand smoke, specifically upregulates CAREx8, but not CAREx7 [64]. When CAREx8 was upregulated in this study, apical AdV infection of polarized epithelia increased. It is likely that other environmental factors, such as air pollution, also upregulate this apical form of CAR making CAREx8 a major factor that increases the susceptibility of the airway to viral infection upon pollutant exposure.
The differential localization of CAREx7 and CAREx8 in polarized epithelial cells is most likely due to interactions between their last 26 or 13 aa, respectively, and cytosolic proteins. Both CAREx7 and CAREx8 contain type I PDZ-binding domains, a 4 aa protein interaction motif at the extreme end of their respective C-termini (X-(S/T)-X-φ, where X represents any amino acid and φ represents any hydrophobic amino acid; GSIV and ITVV, respectively) [32, 62, 65, 66]. PDZ-binding domains interact with PDZ domains, which are named after the first three proteins discovered to contain these domains, PSD-95, Dlg1, and ZO-1 [67]. PDZ domains are approximately 90 aa in size and occur within of a variety of scaffolding proteins [68, 69]. While both isoforms have the ability to bind to numerous PDZ domain containing proteins, the upstream amino acids, which also differ between the two isoforms, can modify the interactions between the CAR splice forms and a PDZ-domain containing protein [32]. Our data suggest that the differential interaction between CAREx7, CAREx8, and PDZ-domain containing proteins is most likely responsible for the differential localization in polarized epithelial cells [33].
A novel pathway that appears to be responsible for absence of CAREx8 from the basolateral membrane involves the complex relationship between CAREx7, membrane-associated guanylate kinase with inverted domain structure 1 (MAGI-1), and CAREx8 (Fig. 4). CAREx7 is dominant to the PDZ-domain containing cellular-scaffolding protein, MAGI-1, and traffics MAGI-1 to the basolateral cell-cell junctions of polarized epithelial cells (Fig. 4A) [62]. In contrast, MAGI-1 negatively regulates the cellular protein levels of CAREx8 (Fig. 4B) [32, 33]. Whereas upregulation of MAGI-1 decreases CAREx8 levels and apical AdV infection, siRNA-mediated decrease of MAGI-1 allows increased apical CAREx8 and increased AdV infection (Fig. 4C). Our data suggest MAGI-1 renders CAREx8 highly susceptible to endoplasmic reticulum-associated degradation (ERAD) (data not shown). MAGI-1 contains up to 6 PDZ domains (0–5), of which PDZ-1 and PDZ-3 interact with CAREx8. PDZ-3 is able to suppress cell surface CAREx8 protein levels, while paradoxically, PDZ-1 rescues CAREx8 from MAGI-1-mediated degradation [33]. CAREx8 has approximately 17x stronger affinity for PDZ-3 than PDZ1. It is likely this stronger affinity causes CAREx8 to interact more frequently with MAGI-1-PDZ3 and may lead to the ERAD-mediated degradation of CAREx8.
Figure 4.

Schematic representation of MAGI-1-mediated regulation of CAREx8 in a polarized epithelial cell. A) CAREx7 (orange tail) is dominant to MAGI-1 and traffics to the basolateral surface where it behaves as a hemophilic cell adhesion protein. B) MAGI-1 negatively regulates the levels of CAREx8 (green tail). C) A lack of interaction with MAGI-1 allows CAREx8 to traffic to the apical surface and mediate apical AdV infection.
CAR as a component of the tight junction
CAREx7 is a transmembrane protein that is involved in the regulation of tight junctions [60, 61]. Chinese hamster ovary (CHO) cells transfected with CAREx7 results in CAREx7 localization at cell-cell contacts and causes this normally non-polarized cell line to form junctions with other CHO cells [61, 62]. CAREx7 also interacts with numerous tight and adherens junction proteins, such as MAGI-1, PICK1, ZO-1, β-catenin, MUPP-1, and connexin 45 at the intercalated discs within the heart [60–62, 65, 70]. The cytoplasmic tail of CAREx7 is important for the localization of CAREx7 at the tight junctions in polarized cells. When the cytoplasmic tail of CAR is removed, the protein no longer localizes to the tight junction [48, 61]. This abrogation of proper polarization of CAREx7 is likely due to the removal of the YxxΦ tyrosine-based motif, where Y stands for tyrosine, x represents any amino acid, and Φ represents any hydrophobic amino acid [71–73]. In CAREx7, the specific tyrosine based motif, YNQV, interacts with the clathrin adaptor complexes AP-1A and AP-1B, which sorts CAREx7 to the basolateral surface [71–73]. The AP-1B clathrin adaptor complex is unique to epithelial tissue and is involved in the basolateral sorting of other proteins, including the human poliovirus receptor (CD155) [73, 74].
Transmembrane isoforms of CAR as viral receptors
CAR functions as a viral receptor for CVB and for all types of AdV except those in species B. Although there are significant morphological differences between coxsackieviruses and AdV, the two classes of virus interact with the D1 domain of the extracellular domain of CAR [42–44, 75]. In contrast to the protruding trimeric fiber-knobs attached to each of the 12 vertices of AdV, CVB use a depression around the fivefold axes of their capsid, often referred to as a “canyon-like” receptor binding site [7, 29, 75]. However, the site of CVB interaction with CAR is on the opposite side of the D1 domain as compared to the AdV binding site [75]. While the direct interaction between CAR and the two classes of viruses occurs at the D1 domain, neither virus can infect host cells in the absence of D2 domain [45, 75]. This may be due to steric interactions or an effect of CAR glycosylation on binding cooperativity [46, 76].
The two transmembrane isoforms of CAR have identical extracellular domains. Both CAREx7 and CAREx8 bind to AdV fiber knob and the canyon-like receptor binding site of CVB and function similarly as viral receptors [8, 32]. However, the physiological implications of CAREx7 and CAREx8 being at different surfaces in polarized cells have important ramifications for the role of CAR as a viral receptor. Localization of CAREx8 at the apical surface in polarized airway epithelium allows it to be accessible to airborne CVB and AdV, and may be the reason behind tight MAGI-1-mediated regulation. The presence of CAREx8 located on the apical surface of a minority of epithelial cells within a cultured polarized primary human airway epithelium correlates well with infrequent apical AdV transduction (Fig. 5) [32, 64, 77]. In contrast, basolateral localization of CAREx7 presents the challenge of breaking through the tight junctions for viruses entering the lungs, but is able to facilitate basolateral AdV spread and egress from the airway after replication [48]. Experiments performed on polarized human airway epithelial tissue cultures have demonstrated that increasing the expression of CAREx8 results in an increase of apical CAREx8 and the amount of AdV entering the cell [32, 64, 77]. Therefore, by modifying the concentration of CAREx8 at the apical surface, one could modify the susceptibility of the cell to AdV and CVB infection. A mechanism to increase or decrease the levels of apical CAREx8 may increase the effectiveness of AdV-based gene therapy or reduce an individual’s risk of contracting the diseases caused by AdV infection, respectively.
Figure 5.

Polarized primary human airway epithelia immunostained with antibodies specific for CAREx8 (green) or the tight junction protein ZO-1 (red). The X–Y confocal section is taken at the apical tip of the epithelium (60x oil immersion).
CD46: “The Pathogen’s Magnet”
Utilized by a larger and more diverse group of pathogens than CAR, CD46 deserves its epithet as “the pathogen’s magnet”. Able to bind to the Edmonston strain of measles virus, herpesvirus, group B1, B3, and some group D AdV, as well as bacterial pathogens, CD46 is a remarkably versatile gateway for microbial infection [14, 27, 28, 78]. Its nearly ubiquitous nature, appearing in virtually all tissues except for erythrocytes, is likely the impetus that encouraged so many pathogens to evolve mechanisms for utilizing this transmembrane glycoprotein for cell entry [79]. Although it is found in a wide spectrum of human tissues, homologues for CD46 are only found in primate species [80]. In addition to being used for entry into the cytoplasm by a plethora of pathogens, CD46 plays an important role as a negative regulator of the complement system of innate immunity and CD46 dysfunction is associated with several diseases, such as asthma, multiple sclerosis, and rheumatoid arthritis [81, 82].
The 14 isoforms of CD46
CD46, also known as complement regulatory protein and membrane cofactor protein (MCP), is composed of 14 separate exons that are alternatively spliced to create a total of 14 unique isoforms [79, 83] (Fig. 6). Exon 1 encodes the leader peptide sequence (LS) and exons 2–6 are translated into the four extracellular short consensus repeats (SCR), also known as complement control protein repeat domains (CCP). Each SCR contains 3 N-linked glycosylation sites. The SCR region interacts directly with viruses and complement proteins [78, 81, 82, 84–86]. The next 3 exons, exons 7–9, contain 3 extracellular domains rich in serine, threonine, and proline residues, referred to as STP domains [80]. STP domains serve as sites of O-linked glycosylation, which can affect viral binding efficiency. The 3 STP domains are labeled A, B, and C in descending order and are subject to alternative splicing, which can remove any of the three STP domains from the full length sequence [79]. Exon 10 forms a 10 aa bridge between the extracellular domain and the hydrophobic transmembrane region encoded by exons 11 and 12. Finally the last two exons encode for a short cytoplasmic tail (Cyt). If exon 13 is present, a 28 aa cytoplasmic tail (Cyt-1) is present. However, if exon 13 is absent, exon 14 is translated into a 35 aa cytoplasmic tail (Cyt-2) [79, 87].
Figure 6.

Map of the 14 protein isoforms of CD46. A) CD46 mRNA exon map relative to protein domains. LS, leader sequence; SCR, short consensus repeats; STP, serine-threonine-proline repeat domains; TM, transmembrane domain; Cyt, cytoplasmic domain. B) The portions of the exons expressed in each isoform are colored green. Many of the splice forms of CD46 differ only in the last two exons of the cytoplasmic tail region: exons 13 and 14. CD46 isoforms are named for the STP domains, A, B, or C (exons 7, 8, and 9, respectively), and the cytoplasmic tails, Cyt-1 or Cyt-2, that they contain. The isoform BCδCyt-2 contains only part of the transmembrane domain encoded by exon 12. C) Schematic of CD46 showing all domains and the distinct amino acid sequences within the two cytoplasmic domains. Protein isoforms differ in the STP (serine-threonine-proline; A, B, or C) domains and Cyt (cytoplasmic) domains. U, unstructured region with no ascribed function.
The 14 alternatively spliced isoforms of CD46 differ in size, location, and protein expression levels in the human body. There are two major regions of alternative splicing in CD46: the STP region and the cytoplasmic tail [79, 80]. The nomenclature of CD46 splice forms reflects this by first describing which STP regions are present and then which of the two cytoplasmic tails is expressed [83]. While important for intracellular interactions and subcellular localization, the cytoplasmic tail is not a major determinant for the protein size and tissue localization of CD46, in comparison to the STP region [79, 87–89]. The two largest isoforms of CD46 are ABC Cyt-1 and ABC Cyt-2. These two isoforms contain all three STP regions, have a molar mass of 74 kDa, and may be exclusive to EBV-infected B cells and leukemic cells [79]. There are four isoforms found ubiquitously in all CD46 expressing tissues except in sperm: BC Cyt-1/Cyt-2 and C Cyt1/Cyt2. The BC isoforms lack exon 7 and are 66 kDa, while the C isoforms lack both exon 7 and exon 8 and are 56 kDa. Of the remaining CD46 isoforms, AB Cyt-1/Cyt-2 and B Cyt-1/Cyt-2 are found in placenta tissue and are 70 and 63 kDa, respectively. There are two isoforms, øCyt-1 and øCyt-2, which skip the STP region entirely. While they show up in Western blots of placenta tissue, they appear to have low abundance. The two smallest splice forms, BCδCyt-2 and CδCyt-2, are formed from the removal of exon 12 and subsequent shortening of the transmembrane region. BCδCyt-2 contains a small fragment of exon 12 whereas CδCyt-2 lacks exon 12 completely. Both isoforms are found only in spermatozoa, are soluble, and are approximately 35 kDa in length [79].
STP Isoforms
Of the four common isoforms of CD46, two contain both the B and C STP domains and two contain only the C STP domain. The difference in protein sequence inclusion between BC and C STP isoforms affect the number of O-linked glycosylation sites on CD46, which in turn affect its interaction with complement proteins [90]. In contrast, the differences between the BC and C isoforms does not have a significant impact on CD46 interaction with group B AdV [78].
CD46 is a regulator of complement proteins C4b and C3b [78, 81]. The complement system is a part of the innate immune system that supplements the role of macrophages, neutrophils, and natural killer cells. It involves small cytotoxic proteins, including C4b and C3b, which opsonize or tag immune complexes, pathogens, and apoptotic cells for phagocytosis. They also perform “convertase” enzymatic roles when in complex with other complement protein to lyse bacterial cells. These proteins may also bind host cells and must be rapidly neutralized to protect native cells from their cytotoxic and lytic effects. CD46 binds with C4b and C3b and is a cofactor in cleaving these proteins into less harmful substrates [78, 81]. CD46 BC and C isoforms differ in their binding affinities for these two complement proteins. The BC isoform, with its larger O-linked glycosylation STP region, binds C4b more effectively than the C isoform does. The BC isoform also binds with C4b better than it binds with C3b [90]. While the C isoform has a lower affinity for C4b than the BC isoform, the C isoform has equal binding affinity for both C4b and C3b, which is equivalent to the isoform BC binding affinity for C3b, as the size of the O-linked glycosylation region seems to have a minimal effect on C3b binding [90].
Cytoplasmic tail isoforms and polarized cells
The two alternatively spliced cytoplasmic tail regions, Cyt-1 and Cyt-2, affect the interaction between CD46 and intracellular proteins, and CD46 localization in polarized cells [87–89]. The two isoforms show no homology within the cytoplasmic tail region (Fig. 6C). There are conflicting data on CD46 localization in polarized cells, which likely reflect cell-type and cell-line specific sorting mechanisms. Masiner et al. suggest that both Cyt-1 and Cyt-2 cause localization at the basolateral surface of transfected Madin Darby canine kidney (MDCK) epithelial cells [88, 91, 92]. In contrast, Ludford-Menting et al. suggest Cyt-1 can go apical or basolateral depending on the presence of the interacting protein PSD-95/DLG-4 and that Cyt-2 is localized at both the apical and basolateral surfaces [87]. All studies have consistently shown that forms of CD46 either lacking the cytoplasmic tail or with specific residues mutated within the tail appear at both the apical and basolateral surfaces.
The Cyt-1 and Cyt-2 isoforms are not regulated by the same set of cytosolic proteins. Cyt-2 basolateral localization may be regulated by src-kinase Lck (p60c-src)-mediated phosphorylation of tyrosine residues [89]. In contrast, Cyt-1 encodes a PDZ-binding domain (FTSL) that interacts with the third PDZ domain of the PDZ-domain containing scaffold protein PSD-95/DLG4. In transfected MDCK cells, it is proposed that DLG4 regulates the basolateral localization of Cyt-1 and a lack of DLG-4 may allow apical Cyt-1 [87]. Consistent with this idea, endogenous CD46 is observed at the apical surface of the polarized epithelial cells within a normal human kidney [87]. Apical versus basolateral localization is predicted to have significant ramifications for cell and tissue biology, as well as viral infection.
The earliest studies involving measles viral (MV) entry in the polarized human colorectal cancer cell line Caco-2 and the African green monkey kidney cancer cell line Vero C 1008 supported localization of CD46 at the apical surface [93]. However, when those studies were repeated in those same cell lines, Vero C 1008 cells showed that MV entry occurred at both the basolateral or apical membrane with no preference for either surface [94]. Furthermore, although human airway epithelial cells may express CD46 at the apical surface, the basolateral surface is more susceptible to MV entry [94]. The basolateral preference may be due to nectin-4, recently discovered to be a primary receptor for wild-type MV. When human ovarian cancer cells and human breast cancer cell line BT474 were analyzed, CD46 localized to the tight junctions, making it difficult to infect these cells with group B AdV [14, 95]. Taken together, these studies suggest that CD46 localization is tissue and cell-line dependent. The preference for the basolateral surface appears to be regulated by cytosolic proteins interacting with the cytoplasmic tails of CD46. Further studies are required to determine the mechanisms that regulate the localization of CD46 in polarized cells.
The interaction between CD46 and Species B and D Adenoviruses
Species B AdV types 11, 16, 21, 35, and 50 all utilize CD46 as a primary receptor for cell entry [8, 14, 26, 84]. AdV3, 7, and 14 have high affinity for desmoglein-2, but AdV3 and 7 have high avidity for CD46 as a viral receptor [96]. The viral fiber knobs of species B AdV that interact with CD46 directly bind to the second extracellular SCR [84]. Mutation of the 130 to 135 aa segment or the 152 to 156 aa segment ablates AdV35 binding to CD46 and viral infection in transfected CHO cells. Deletion of the first SCR domain also significantly reduces AdV35 binding to CD46 and virtually eliminates AdV35 infection indicating both SCR domains play a crucial role in AdV35 binding and entry. The first SCR domain most likely acts as a scaffold needed to maintain the proper 3-dimensional structure of CD46. AdV35 also does not depend on the N-linked glycosylation sites located in the SCR domains. Adding tunicamycin, which inhibits N-linked glycosylation, does not alter AdV35 infection in transfected CHO cells [84]. Certain species D AdV26, 37 and 49 also utilize CD46 as primary receptor, however, the exact sites of interaction have not yet been mapped [15–17].
Desmoglein-2
Group B2 AdV types 3, 7, and 14 and rely on the desmosomal cadherin known as desmoglein 2 (DSG-2) for viral entry, and AdV11, found in group B3, can interact with both CD46 and DSG-2 [14, 26, 84, 95].
Desmoglein-2: Physiological complexity without splice variants
DSG-2 is a desmosomal glycoprotein composed of 15 exons and essential for the formation of desmosomes (Fig. 7) [30, 97]. Desmosomes are important for cell-cell adhesion in epithelial cells and also serve as attachment points for cytosolic intermediate filaments [98]. DSG-2 is a member of the cadherin family of transmembrane glycoproteins [99]. Cadherins mediate calcium dependent interactions that result in cell-cell adhesion. There are two major subclasses of desmosomal cadherins: desmogleins and desmocollins. Unlike desmocollins, which more closely resemble classical cadherins, desmogleins have an extra C-terminal domain that contains a series of 29±1 aa repeats [100].
Figure 7.
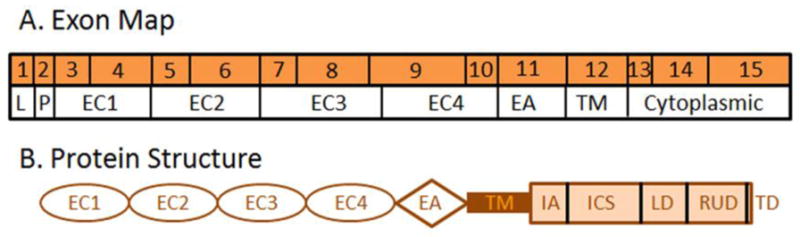
Map of desmoglein 2. A) DSG-2 mRNA exon map of the single DSG-2 protein isoform relative to the protein domains. B) Schematic protein structure of DSG-2. LS, leader sequence; P, preprotein domain; EC1-4, extracellular cadherin domains; EA, extracellular anchor domain; TM, transmembrane domain; IA, intracellular anchor domain; ICS, intracellular cadherin-typical segment domain; LD, linker domain; RUD, repeat unit domain containing 6 repeats; and TD, terminal domain.
The three major desmoglein proteins, DSG-1, DSG-2, and DSG-3, though similar in overarching structure, are encoded by three separate genes. The desmoglein extracellular domain consists of 4 repeated units of roughly 110 aa in length (Fig. 7B). These repeated units, named E1–E4, with E1 being the most distal element, contain Ca2+ binding motifs that facilitate calcium dependent binding between different desmoglein proteins [99, 101, 102]. The extracellular domain of cadherins is fairly well conserved between different cadherin family protein members. For example, there is a 39% aa match between the E2 extracellular regions of DSG-1 and N-cadherin [99]. Desmogleins are also characterized by their extended cytoplasmic tails. The first 59 aa constitute a proline rich region, which is followed by a series of repeats at the C-terminal domain, each containing 29 ± 1 aa residues predicted to form 2 β turns and 2 β strands [99]. After the repeating region, there is a nonpolar glycine rich stretch, terminating in a basic tail at the C-terminus.
While it resembles the two other desmoglein proteins, the amino acid sequence of DSG-2 differs significantly at several key points and defects in DSG-2 are the cause of familial arrhythmogenic right ventricular dysplasia type 10 (ARVD10) [103]. DSG-2 contains 1117 aa; 37% and 40% of those amino acids are identical to DSG-1 and DSG-3 respectively. Interestingly, one of the main areas of dissimilarity is within the extracellular region. At a point in the extracellular domain found to be critical for homologous binding (aa 129–131), DSG-2 contains tyrosine residues [104]. This unique feature more closely resembles desmocollins than the other two desmogleins. The number of C-terminal 29±1 aa repeats varies among desmogleins with DSG-2 containing a total of six tandem repeats [97]. Although the three desmoglein proteins are found in varying concentrations and locations in human tissues, only DSG-2 is present in all desmosome containing tissues [105]. DSG-2 is also present in a variety of non-epithelial cells such as myocardium, Purkinje fiber cells, and in the follicular dendritic reticulum of spleen and lymph nodes [104].
Interactions between desmogleins and desmocollins in the extracellular region as well as their interactions with proteins in the cytosolic desmosomal plaque are essential to the formation of desmosomes. Desmosomal cadherins use desmoplakin and plakoglobulin as direct intermediaries between desmoglein and the intermediate filaments [99]. Interestingly, while desmosomes have stronger binding affinities than adherens junctions, desmoglein homodimers are not as strong as E-cadherin homodimers in adherens junctions [99]. Perhaps desmosomes have a higher binding affinity than adherens junctions because of heterodimers between different desmogleins.
DSG-2 as a viral receptor
In addition to functioning as a cell adhesion protein, DSG-2 is utilized as a viral receptor for group B2 and B3 AdV [14]. DSG-2 is expected to reside on the basolateral surface of polarized epithelial cells. Although the mechanisms that allow airborne group B AdV to access junctional DSG-2 are unknown, it is apparent that viral binding and entry is rapid. When group B2 AdV3 interacts with DSG-2, it weakens cell-cell junctions, allowing further access to proteins associated with intercellular junctions [14]. Interruption of the interactions between these cell-cell adhesion proteins also appear to be important for both viral spread and egress from the epithelium [106, 107].
During viral replication, some species B AdV produce both full size virions as well as smaller virus-like particles that consist solely of the viral penton base and viral fiber knob, referred to as penton dodecahedral particles or PtDds [108]. This PtDd form of AdV blocks AdV infection more efficiently than AdV fiber knob [14]. It is even more effective than a combination of fiber knob and small particles that contain only penton base (BsDds). The dodecahedral shape of the penton base of PtDds provides the correct spatial arrangement to allow the formation of cross-linkages between the viral fiber knobs and shafts that bind DSG-2 [109].
Using PtDds as a non-toxic substitute for group B2 and B3 AdV infection, it is possible to study the effects that AdV-DSG-2 binding have on cellular physiology. When PtDds derived from AdV3 interact with DSG-2 in epithelial cells, they trigger signaling that leads to cell junction changes expected during a epithelial-to-mesenchymal transition (EMT) [14]. EMT is defined by the increased expression of mesenchymal markers, such as vimentin and lipocalin-2, altered intracellular localization of transcription factors and the activation of kinases, such as phosphatidylinositol-3 kinase (PI3K) [110]. PtDds applied to epithelial ovarian cancer cells weakens cell-cell junction integrity and increases access to important proteins trapped within cell-junctions, such as CD46 and the human epidermal growth factor receptor 2 (Her2/Neu) [14]. Her2/Neu is the target protein for the cancer treatment drug Herceptin and increased access to Her2/Neu is predicted to enhance therapeutic efficacy. PtDds applied to the human breast cancer cell line BT474, which polarizes and normally sequesters Her2/Neu within the epithelial junctions, allowed Herceptin to be more effective at killing the cancerous cells [14]. Importantly, pretreating Her2/Neu negative breast cancer cells (MDA-MB-231) with PtDds derived from AdV3 had no obvious cytotoxic effects. Additional studies have investigated a small, recombinant AdV3-derived protein, termed junction opener 1 (JO-1), which binds to the epithelial junction protein desmoglein 2 (DSG-2). The binding of JO-1 with DSG-2 results in cleavage of DSG-2 and activates intracellular signaling pathways which decrease the level E-cadherin at the junctions. Transient epithelial disruption allows chemotherapy drugs access to tissues in vivo [111]. While more research is required, PtDds or JO-1-like peptides derived from group B AdV could become a novel part of drug cocktails for treating certain types of breast and other epithelial cancer.
Conclusion
Alternative splicing creates a plethora of splice forms of CAR and CD46. The diversity of isoforms created by alternative splicing has major implications for how these viral receptors interact with wild-type viruses and their other biological functions. Both proteins contain soluble splice forms. Although β, γ, and δ CAR all possess part of the extracellular domain of CAR, only β CAR efficiently binds to adenovirus fiber knob and can block viral entry. Administration of soluble CAR can reduce the risk of infection from AdV and CVB; however, paradoxically it may increase pathogenesis. Differences in the cytoplasmic tails of splice forms affect the localization and effectiveness of the viral receptors in polarized cells. CAREx7 localizes on the basolateral surface, while CAREx8 localizes to the apical surface of polarized human airway epithelial cells making CAREx8 an ideal target to modify the susceptibility of an epithelium to AdV infection. In CD46 isoforms, Cyt-1 and Cyt-2 may be both apical and basolateral depending on the tissue under examination. This is likely due to the presence or absence of key regulatory proteins. In addition to the mechanisms responsible for differential localization, the implications of alternative localizations of these viral receptors on cell physiology are still unclear. The many different proteins that can be created from the alternative splicing of CAR and CD46 emphasize the diversity of functions these proteins can have in viral infectivity. This diversity also suggests a number of roles for these cell surface proteins beyond being a gateway for viral infection. Additional research is required to elucidate the mechanisms that regulate cellular localization. Knowledge of these mechanisms may lead to novel methods to increase or decrease AdV infection for gene therapy or wild type infection, respectively. They may also lead to novel treatments for cancer and other diseases.
Acknowledgments
This work was supported by the National Institute of Allergy and Infectious Diseases of the NIH Award R15AI090625-01, a Wright State University Research Initiation Award and a Biology Award for Research Excellence (JRB).
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or WSU.
References
- 1.Oude Munnink BB, et al. PLoS One. 2013;8:e78454. doi: 10.1371/journal.pone.0078454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Minot S, Bryson A, Chehoud C, Wu GD, Lewis JD, Bushman FD. Proc Natl Acad Sci U S A. 2013;110:12450–12455. doi: 10.1073/pnas.1300833110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.William SM, Wold MSH. Adenoviruses. In: David M, Knipe PMH, editors. Fields Virology. Philadelphia: Lippincott Williams and Wilkins; 2007. pp. 2395–2436. [Google Scholar]
- 4.Lynch JP, 3rd, Fishbein M, Echavarria M. Semin Respir Crit Care Med. 2011;32:494–511. doi: 10.1055/s-0031-1283287. [DOI] [PubMed] [Google Scholar]
- 5.Davison AJ, Benko M, Harrach B. J Gen Virol. 2003;84:2895–2908. doi: 10.1099/vir.0.19497-0. [DOI] [PubMed] [Google Scholar]
- 6.Nemerow GR, Stewart PL, Reddy VS. Current opinion in virology. 2012;2:115–121. doi: 10.1016/j.coviro.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu H, Wu L, Zhou ZH. Journal of molecular biology. 2011;406:764–774. doi: 10.1016/j.jmb.2010.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergelson JM, et al. Science. 1997;275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 9.Carson SD, Chapman NN, Tracy SM. Biochem Biophys Res Commun. 1997;233:325–328. doi: 10.1006/bbrc.1997.6449. [DOI] [PubMed] [Google Scholar]
- 10.Tomko RP, Xu R, Philipson L. Proc Natl Acad Sci U S A. 1997;94:3352–3356. doi: 10.1073/pnas.94.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roelvink PW, et al. J Virol. 1998;72:7909–7915. doi: 10.1128/jvi.72.10.7909-7915.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Segerman A, Atkinson JP, Marttila M, Dennerquist V, Wadell G, Arnberg N. J Virol. 2003;77:9183–9191. doi: 10.1128/JVI.77.17.9183-9191.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaggar A, Shayakhmetov DM, Lieber A. Nature medicine. 2003;9:1408–1412. doi: 10.1038/nm952. [DOI] [PubMed] [Google Scholar]
- 14.Wang H, et al. Nature medicine. 2011;17:96–104. doi: 10.1038/nm.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu E, et al. J Virol. 2004;78:3897–3905. doi: 10.1128/JVI.78.8.3897-3905.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemckert AA, et al. J Gen Virol. 2006;87:2891–2899. doi: 10.1099/vir.0.82079-0. [DOI] [PubMed] [Google Scholar]
- 17.Li H, Rhee EG, Masek-Hammerman K, Teigler JE, Abbink P, Barouch DH. J Virol. 2012;86:10862–10865. doi: 10.1128/JVI.00928-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wickham TJ, Mathias P, Cheresh DA, Nemerow GR. Cell. 1993;73:309–319. doi: 10.1016/0092-8674(93)90231-e. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Bergelson JM. J Virol. 2005;79:12125–12131. doi: 10.1128/JVI.79.19.12125-12131.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nemerow GR. Mol Ther. 2009;17:1490–1491. doi: 10.1038/mt.2009.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzalez-Mariscal L, Garay E, Lechuga S. Front Biosci. 2009;14:731–768. doi: 10.2741/3276. [DOI] [PubMed] [Google Scholar]
- 22.Wolfrum N, Greber UF. Cellular microbiology. 2013;15:53–62. doi: 10.1111/cmi.12053. [DOI] [PubMed] [Google Scholar]
- 23.Hong SS, Karayan L, Tournier J, Curiel DT, Boulanger PA. The EMBO journal. 1997;16:2294–2306. doi: 10.1093/emboj/16.9.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arnberg N, Edlund K, Kidd AH, Wadell G. J Virol. 2000;74:42–48. [PMC free article] [PubMed] [Google Scholar]
- 25.Doronin K, et al. Science. 2012;338:795–798. doi: 10.1126/science.1226625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marttila M, et al. J Virol. 2005;79:14429–14436. doi: 10.1128/JVI.79.22.14429-14436.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenstone HL, Santoro F, Lusso P, Berger EA. J Biol Chem. 2002;277:39112–39118. doi: 10.1074/jbc.M206488200. [DOI] [PubMed] [Google Scholar]
- 28.Naniche D, et al. J Virol. 1993;67:6025–6032. doi: 10.1128/jvi.67.10.6025-6032.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freimuth P, Philipson L, Carson SD. Curr Top Microbiol Immunol. 2008;323:67–87. doi: 10.1007/978-3-540-75546-3_4. [DOI] [PubMed] [Google Scholar]
- 30.Kent WJ, et al. Genome research. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Excoffon KJ, et al. Hear Res. 2006;215:1–9. doi: 10.1016/j.heares.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 32.Excoffon KJ, Gansemer ND, Mobily ME, Karp PH, Parekh KR, Zabner J. PLoS One. 2010;5:e9909. doi: 10.1371/journal.pone.0009909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolawole AO, et al. J Virol. 2012;86:9244–9254. doi: 10.1128/JVI.01138-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thoelen I, Magnusson C, Tagerud S, Polacek C, Lindberg M, Van Ranst M. Biochem Biophys Res Commun. 2001;287:216–222. doi: 10.1006/bbrc.2001.5535. [DOI] [PubMed] [Google Scholar]
- 35.Dorner A, Xiong D, Couch K, Yajima T, Knowlton KU. J Biol Chem. 2004;279:18497–18503. doi: 10.1074/jbc.M311754200. [DOI] [PubMed] [Google Scholar]
- 36.Bergelson JM, et al. J Virol. 1998;72:415–419. doi: 10.1128/jvi.72.1.415-419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen JW, Ghosh R, Finberg RW, Bergelson JM. DNA Cell Biol. 2003;22:253–259. doi: 10.1089/104454903321908647. [DOI] [PubMed] [Google Scholar]
- 38.Sadelain M, Brentjens R, Riviere I. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brennan SC, et al. Bba-Mol Cell Res. 2013;1833:1732–1744. [Google Scholar]
- 40.Chretien I, et al. European journal of immunology. 1998;28:4094–4104. doi: 10.1002/(SICI)1521-4141(199812)28:12<4094::AID-IMMU4094>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 41.Raschperger E, Engstrom U, Pettersson RF, Fuxe J. J Biol Chem. 2004;279:796–804. doi: 10.1074/jbc.M308249200. [DOI] [PubMed] [Google Scholar]
- 42.Kirby I, et al. J Virol. 2000;74:2804–2813. doi: 10.1128/jvi.74.6.2804-2813.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomko RP, Johansson CB, Totrov M, Abagyan R, Frisen J, Philipson L. Experimental cell research. 2000;255:47–55. doi: 10.1006/excr.1999.4761. [DOI] [PubMed] [Google Scholar]
- 44.Freimuth P, Springer K, Berard C, Hainfeld J, Bewley M, Flanagan J. J Virol. 1999;73:1392–1398. doi: 10.1128/jvi.73.2.1392-1398.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Excoffon KJ, Traver GL, Zabner J. Am J Respir Cell Mol Biol. 2005;32:498–503. doi: 10.1165/rcmb.2005-0031OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Excoffon KJ, Gansemer N, Traver G, Zabner J. J Virol. 2007;81:5573–5578. doi: 10.1128/JVI.02562-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Bergelson JM. J Virol. 1999;73:2559–2562. doi: 10.1128/jvi.73.3.2559-2562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walters RW, Grunst T, Bergelson JM, Finberg RW, Welsh MJ, Zabner J. J Biol Chem. 1999;274:10219–10226. doi: 10.1074/jbc.274.15.10219. [DOI] [PubMed] [Google Scholar]
- 49.Cohen CJ, Gaetz J, Ohman T, Bergelson JM. J Biol Chem. 2001;276:25392–25398. doi: 10.1074/jbc.M009531200. [DOI] [PubMed] [Google Scholar]
- 50.Ashbourne Excoffon KJ, Moninger T, Zabner J. J Virol. 2003;77:2559–2567. doi: 10.1128/JVI.77.4.2559-2567.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Farmer C, Morton PE, Snippe M, Santis G, Parsons M. Experimental cell research. 2009;315:2637–2647. doi: 10.1016/j.yexcr.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 52.Yuen S, Smith J, Caruso L, Balan M, Opavsky MA. J Mol Cell Cardiol. 2011;50:826–840. doi: 10.1016/j.yjmcc.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 53.Dorner A, et al. J Mol Med. 2006;84:842–851. doi: 10.1007/s00109-006-0076-y. [DOI] [PubMed] [Google Scholar]
- 54.Goodfellow IG, et al. J Virol. 2005;79:12016–12024. doi: 10.1128/JVI.79.18.12016-12024.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yanagawa B, et al. J Infect Dis. 2004;189:1431–1439. doi: 10.1086/382598. [DOI] [PubMed] [Google Scholar]
- 56.Shi Y, et al. J Am Coll Cardiol. 2009;53:1219–1226. doi: 10.1016/j.jacc.2008.10.064. [DOI] [PubMed] [Google Scholar]
- 57.Lenaerts L, Daelemans D, Geukens N, De Clercq E, Naesens L. FEBS Lett. 2006;580:3937–3942. doi: 10.1016/j.febslet.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 58.Raschperger E, Thyberg J, Pettersson S, Philipson L, Fuxe J, Pettersson RF. Experimental cell research. 2006;312:1566–1580. doi: 10.1016/j.yexcr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 59.Shaw CA, et al. BMC Cell Biol. 2004;5:42. doi: 10.1186/1471-2121-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walters RW, Freimuth P, Moninger TO, Ganske I, Zabner J, Welsh MJ. Cell. 2002;110:789–799. doi: 10.1016/s0092-8674(02)00912-1. [DOI] [PubMed] [Google Scholar]
- 61.Cohen CJ, Shieh JT, Pickles RJ, Okegawa T, Hsieh JT, Bergelson JM. Proc Natl Acad Sci U S A. 2001;98:15191–15196. doi: 10.1073/pnas.261452898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Excoffon KJ, Hruska-Hageman A, Klotz M, Traver GL, Zabner J. J Cell Sci. 2004;117:4401–4409. doi: 10.1242/jcs.01300. [DOI] [PubMed] [Google Scholar]
- 63.Zen K, et al. Mol Biol Cell. 2005;16:2694–2703. doi: 10.1091/mbc.E05-01-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharma P, Kolawole AO, Core SB, Kajon AE, Excoffon KJ. PLoS One. 2012;7:e49930. doi: 10.1371/journal.pone.0049930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coyne CB, Voelker T, Pichla SL, Bergelson JM. J Biol Chem. 2004;279:48079–48084. doi: 10.1074/jbc.M409061200. [DOI] [PubMed] [Google Scholar]
- 66.Sollerbrant K, et al. J Biol Chem. 2003;278:7439–7444. doi: 10.1074/jbc.M205927200. [DOI] [PubMed] [Google Scholar]
- 67.Kennedy MB. Trends Biochem Sci. 1995;20:350. doi: 10.1016/s0968-0004(00)89074-x. [DOI] [PubMed] [Google Scholar]
- 68.Javier RT, Rice AP. J Virol. 2011;85:11544–11556. doi: 10.1128/JVI.05410-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee HJ, Zheng JJ. Cell Commun Signal. 2010;8:8. doi: 10.1186/1478-811X-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lim BK, et al. J Clin Invest. 2008;118:2758–2770. doi: 10.1172/JCI34777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Diaz F, et al. Proc Natl Acad Sci U S A. 2009;106:11143–11148. doi: 10.1073/pnas.0811227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carvajal-Gonzalez JM, et al. Proc Natl Acad Sci U S A. 2012;109:3820–3825. doi: 10.1073/pnas.1117949109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gravotta D, et al. Dev Cell. 2012;22:811–823. doi: 10.1016/j.devcel.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ohka S, Ohno H, Tohyama K, Nomoto A. Biochem Biophys Res Commun. 2001;287:941–948. doi: 10.1006/bbrc.2001.5660. [DOI] [PubMed] [Google Scholar]
- 75.He Y, et al. Nature structural biology. 2001;8:874–878. doi: 10.1038/nsb1001-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim JW, Glasgow JN, Nakayama M, Ak F, Ugai H, Curiel DT. PLoS One. 2013;8:e55533. doi: 10.1371/journal.pone.0055533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sharma P, Kolawole AO, Wiltshire SM, Frondorf K, Excoffon KJ. J Gen Virol. 2012;93:155–158. doi: 10.1099/vir.0.036269-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Persson BD, et al. PLoS Pathog. 2010;6:e1001122. doi: 10.1371/journal.ppat.1001122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Russell SM, Sparrow RL, McKenzie IF, Purcell DF. European journal of immunology. 1992;22:1513–1518. doi: 10.1002/eji.1830220625. [DOI] [PubMed] [Google Scholar]
- 80.Manchester M, Liszewski MK, Atkinson JP, Oldstone MB. Proc Natl Acad Sci U S A. 1994;91:2161–2165. doi: 10.1073/pnas.91.6.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barilla-LaBarca ML, Liszewski MK, Lambris JD, Hourcade D, Atkinson JP. J Immunol. 2002;168:6298–6304. doi: 10.4049/jimmunol.168.12.6298. [DOI] [PubMed] [Google Scholar]
- 82.Ni Choileain S, Astier AL. Immunobiology. 2012;217:169–175. doi: 10.1016/j.imbio.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Purcell DF, Russell SM, Deacon NJ, Brown MA, Hooker DJ, McKenzie IF. Immunogenetics. 1991;33:335–344. doi: 10.1007/BF00216692. [DOI] [PubMed] [Google Scholar]
- 84.Gaggar A, Shayakhmetov DM, Liszewski MK, Atkinson JP, Lieber A. J Virol. 2005;79:7503–7513. doi: 10.1128/JVI.79.12.7503-7513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hsu EC, Dorig RE, Sarangi F, Marcil A, Iorio C, Richardson CD. J Virol. 1997;71:6144–6154. doi: 10.1128/jvi.71.8.6144-6154.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sakurai F, et al. J Control Release. 2006;113:271–278. doi: 10.1016/j.jconrel.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 87.Ludford-Menting MJ, et al. J Biol Chem. 2002;277:4477–4484. doi: 10.1074/jbc.M108479200. [DOI] [PubMed] [Google Scholar]
- 88.Maisner A, Liszewski MK, Atkinson JP, Schwartz-Albiez R, Herrler G. J Biol Chem. 1996;271:18853–18858. doi: 10.1074/jbc.271.31.18853. [DOI] [PubMed] [Google Scholar]
- 89.Wang G, Liszewski MK, Chan AC, Atkinson JP. J Immunol. 2000;164:1839–1846. doi: 10.4049/jimmunol.164.4.1839. [DOI] [PubMed] [Google Scholar]
- 90.Liszewski MK, Atkinson JP. J Immunol. 1996;156:4415–4421. [PubMed] [Google Scholar]
- 91.Maisner A, Zimmer G, Liszewski MK, Lublin DM, Atkinson JP, Herrler G. J Biol Chem. 1997;272:20793–20799. doi: 10.1074/jbc.272.33.20793. [DOI] [PubMed] [Google Scholar]
- 92.Teuchert M, Maisner A, Herrler G. J Biol Chem. 1999;274:19979–19984. doi: 10.1074/jbc.274.28.19979. [DOI] [PubMed] [Google Scholar]
- 93.Blau DM, Compans RW. Virology. 1995;210:91–99. doi: 10.1006/viro.1995.1320. [DOI] [PubMed] [Google Scholar]
- 94.Sinn PL, Williams G, Vongpunsawad S, Cattaneo R, McCray PB., Jr J Virol. 2002;76:2403–2409. doi: 10.1128/jvi.76.5.2403-2409.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Strauss R, et al. Cancer Res. 2009;69:5115–5125. doi: 10.1158/0008-5472.CAN-09-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Trinh HV, et al. J Virol. 2012;86:1623–1637. doi: 10.1128/JVI.06181-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nava P, et al. Mol Biol Cell. 2007;18:4565–4578. doi: 10.1091/mbc.E07-05-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Coulombe PA. Nature structural biology. 2002;9:560–562. doi: 10.1038/nsb0802-560. [DOI] [PubMed] [Google Scholar]
- 99.Kowalczyk AP, et al. Biophysical chemistry. 1994;50:97–112. doi: 10.1016/0301-4622(94)85023-2. [DOI] [PubMed] [Google Scholar]
- 100.Buxton RS, et al. J Cell Biol. 1993;121:481–483. doi: 10.1083/jcb.121.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kemler R, Ozawa M, Ringwald M. Curr Opin Cell Biol. 1989;1:892–897. doi: 10.1016/0955-0674(89)90055-0. [DOI] [PubMed] [Google Scholar]
- 102.Takeichi M. Annual review of biochemistry. 1990;59:237–252. doi: 10.1146/annurev.bi.59.070190.001321. [DOI] [PubMed] [Google Scholar]
- 103.Awad MM, et al. Am J Hum Genet. 2006;79:136–142. doi: 10.1086/504393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schafer S, Koch PJ, Franke WW. Experimental cell research. 1994;211:391–399. doi: 10.1006/excr.1994.1103. [DOI] [PubMed] [Google Scholar]
- 105.Schafer S, Stumpp S, Franke WW. Differentiation; research in biological diversity. 1996;60:99–108. doi: 10.1046/j.1432-0436.1996.6020099.x. [DOI] [PubMed] [Google Scholar]
- 106.Fender P, Hall K, Schoehn G, Blair GE. J Virol. 2012;86:5380–5385. doi: 10.1128/JVI.07127-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lu ZZ, et al. PLoS Pathog. 2013;9:e1003718. doi: 10.1371/journal.ppat.1003718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Norrby E, Nyberg B, Skaaret P, Lengyel A. J Virol. 1967;1:1101–1108. doi: 10.1128/jvi.1.6.1101-1108.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang H, et al. J Virol. 2011;85:6390–6402. doi: 10.1128/JVI.00514-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Turley EA, Veiseh M, Radisky DC, Bissell MJ. Nature clinical practice Oncology. 2008;5:280–290. doi: 10.1038/ncponc1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Beyer I, et al. Cancer Res. 2011;71:7080–7090. doi: 10.1158/0008-5472.CAN-11-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]