

Abstract
Purpose
The urokinase receptor (uPAR) plays a critical role in breast cancer (BC) progression and metastases, and is a validated target for novel therapies. The current study investigates the effects of MV-uPA, an oncolytic measles virus fully retargeted against uPAR in syngeneic and xenograft BC metastases models.
Methods
In vitro replication and cytotoxicity of MVs retargeted against human (MV-h-uPA) or mouse (MV-m-uPA) uPAR were assessed in human and murine cancer and non-cancer mammary epithelial cells. The in vivo effects of species-specific uPAR retargeted MVs were assessed in syngeneic and xenograft models of experimental metastases, established by intravenous administration of luciferase expressing 4T1 or MDA-MD-231 cells. Metastases progression was assessed by in vivo bioluminescence imaging. Tumor targeting was evaluated by qRT-PCR of MV-N, rescue of viable viral particles and immunostaining of MV particles in lungs from tumor bearing mice.
Results
In vitro, MV-h-uPA and MV-m-uPA selectively infected, replicated and induced cytotoxicity in cancer compared to non-cancer cells in a species-specific manner. In vivo, MV-m-uPA delayed 4T1 lung metastases progression and prolonged survival. These effects were associated with identification of viable viral particles, viral RNA and detection of MV-N by immunostaining from lung tissues in treated mice. In the human MDA-MB-231 metastases model, intravenous administration of MV-h-uPA markedly inhibited metastases progression and significantly improved survival, compared to controls. No significant treatment related toxicity was observed in treated mice.
Conclusions
The above preclinical findings strongly suggest that uPAR retargeted measles virotherapy is a novel and feasible systemic therapy strategy against metastatic breast cancer.
Keywords: Urokinase receptor, measles virus, tumor targeting, metastasis
INTRODUCTION
According to the American Cancer Society, approximately 232,670 new cases of invasive breast cancer and 40,000 breast cancer deaths are expected to occur among US women in 2014 [1]. Even though significant progress has been made in the management of early and locally advanced breast cancer, metastatic breast cancer is an incurable disease, associated with a poor prognosis [2].
The plasminogen activator system plays a critical role in breast cancer progression and metastases [3]. Both urokinase and its inhibitor plasminogen activator inhibitor-1 (PAI-1) are clinically validated prognostic and predictive biomarkers for disease free survival and overall survival in breast cancer patients [4–7]. The urokinase receptor (uPAR), a glycosylphosphoinositol (GPI) anchored cell surface receptor, binds uPA with high affinity and is critical for protease mediated cancer cell invasion, as well as protease independent signaling pathways involved with the metastatic process, such as proliferation, migration and epithelial to mesenchymal transition (EMT) [8–10]. It is overexpressed in a variety of human and murine cancers, compared to non-cancer tissues, and its presence has been associated with high metastatic potential and poor prognosis [11–17].
The oncolytic virotherapy field has significantly expanded in the last decade, and recently, several novel viral vectors have reached late phase clinical evaluation [18]. Among the novel oncolytic viruses under development, the Edmonston vaccine strain of measles virus (MV-Edm) is a promising one, whose in vitro and in vivo safety and efficacy are well established [19, 20]. Recombinant, non-targeted oncolytic MVs are currently being evaluated in ovarian cancer, brain tumors, multiple myeloma and mesothelioma [18, 21, 22], with promising preliminary reports of clinical antitumor efficacy [23]. Redirecting viral tropism to tumor specific targets is an active area of research in the field of oncolytic viruses, which has the potential to improve safety and delivery of viral vectors to sites of distant metastases.
Based on the above, our group has successfully engineered and rescued an oncolytic measles virus fully retargeted against the urokinase receptor (MV-uPA) [24]. Species specific MV-uPA vectors were engineered by displaying the aminoterminal fragment (ATF) of either human (MV-h-uPA) or mouse (MV-m-uPA) urokinase in the C-terminus of a CD46 and SLAM “blind” MV-H glycoprotein (HAALS) [24]. Recombinant oncolytic MVs retargeted against that human or mouse uPAR induce antitumor effects in primary (mammary fat pad) breast cancer models in vivo [24]. In this report, the in vivo effects of uPAR retargeted oncolytic measles viruses in human and murine experimental breast cancer metastases models were investigated.
RESULTS
In vitro tumor selectivity and species specificity of uPAR dependent MV-h-uPA and MV-m-uPA
The engineering and rescue of fully retargeted oncolytic measles viruses against human (MV-h-uPA) or murine (MV-m-uPA) uPAR was reported by our group [24]. To determine the in vitro tumor selectivity and species specificity of the above viruses in breast cancer, human and murine mammary cancer (known to express uPAR [24–26], as well as normal (human and murine) mammary epithelial cells were infected with MV-GFP (non-targeted measles virus), MV-h-uPA and MV-m-uPA. The human MDA-MB-231, MCF-7 and MDA-MB-436 breast cancer cell lines were sensitive to both MV-GFP and MV-h-uPA infection, as demonstrated by strong virally induced GFP expression and syncytia formation, but not to the murine uPAR retargeted virus (Fig. 1. A, B, C). Infection of non-cancerous human mammary epithelial cells (HMEC) was less prominent than cancer cells. However the permissivity of HMEC was markedly less for MV-h-uPA compared to MV-GFP (Fig. 1. D). Murine 4T1 mammary cancer cells were only permissive to the species specific MV-m-uPA and not to the human retargeted viruses (Fig. 1. E). Neither virus was able to infect non-cancer murine mammary epithelial cells (NMuMG, Fig. 1. F). The above findings clearly confirm species and tumor cell specificity of the human and murine uPAR retargeted MVs against breast cancer in vitro.
Figure 1. In vitro viral infection by MV.

Human breast cancer cells (A. MDA-MB-231, B. MCF-7, C. MDA-MB-436), human mammary epithelial cells (D. HMEC), murine mammary cancer cells (E. 4T1) and murine mammary epithelial cells (F. NMuMG) were infected with MV-GFP, MV-h-uPA, or MV-m-uPA as indicated at an MOI= 1 and photographed 48 h after infection. Scale bar = 500 μm.
Next, to determine whether MV-uPA preferentially replicates in breast cancer cell lines compared to normal breast epithelial cells, MDA-MB-231, MCF-7, MDA-MB-436 and 4T1 (cancer cells), as well as HMEC and NMuMG (normal mammary epithelial cells) were infected with the human or murine uPAR retargeted virus. Titers of virus were determined at 24, 48 and 72h by the one-step growth curve. As shown in Figure 2 A-C, MV-h-uPA successfully replicated in human breast cancer cells (viral titers -TCID50- at 72 hours: MDA-MB-231= 1.5 × 106; MCF-7= 4 × 106; MDA-MB-436: 6.9 × 105), but significantly less in HMEC (1.2 × 103 and 2.4 × 101 TCID50 at 48 and 72 hours, respectively). MV-m-uPA successfully replicated in murine cancer cells 4T1 (1.1 × 104 TCID50 at 72 hours) but not in mouse mammary epithelial cells (NMuMG) (Fig 2. E, F).
Figure 2. In vitro replication.

Human breast cancer cells MDA-MB-231 (A), MCF-7 (B), MDA-MB-436 (C) and human mammary epithelial cells HMEC (D) were infected with MV-h-uPA (MOI = 3). Murine mammary epithelial cells NMuMG (E) and mammary cancer cells 4T1 (F) were infected with MV-m-uPA (MOI = 3). Titers of virus were determined at different time points by the one-step growth curve.
In vitro cytotoxicity
The cytopathic effects of MV-h-uPA and MV-m-uPA were determined at different time points after infection, by trypan blue exclusion. MV-h-uPA induced significant (p 0.0001) cytotoxicity at 48 and 72 hours after infection in all of human breast cancer cells (Fig 3. A, B, C). MV-m-uPA, on the other hand, induced significant cytotoxicity in the murine tumorigenic cell lines 4T1 at 72 hours (p = 0.0292; 3. F). MV-h-uPA and MV-m-uPA caused minimal cytopathic effects against normal human (HMEC; Fig. 3, D) or murine (NMuMG) mammary epithelial cells (Fig. 3, E).
Figure 3. In vitro cytopathic effects of MV.
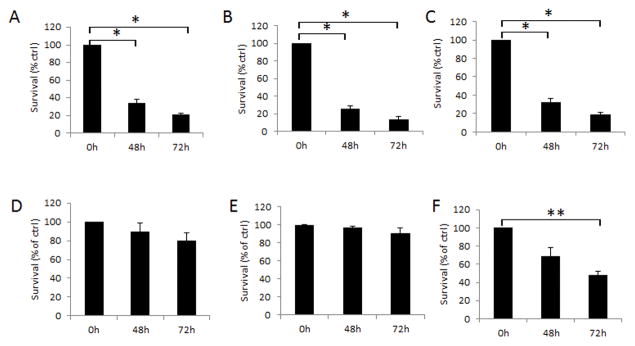
Human breast cancer cells MDA-MB-231 (A), MCF-7 (B), MDA-MB-436 (C) and human mammary epithelial cells HMEC (D) were infected with MV-h-uPA (MOI = 1). Murine mammary epithelial cells MMuMG (E) and mammary cancer cells 4T1 (F) were infected with MV-m-uPA (MOI = 1). Viability was determined at different time points (48h, 72h) by trypan blue exclusion and presented as percentage of controls. Bars represent averages +/− SD of triplicate experiments. * p < 0.0001; ** p = 0.0292.
In vivo effects of MV-m-uPA on syngeneic mammary cancer metastases
The highly aggressive 4T1 experimental lung metastases model was established as described in methods. At day 7 after tumor cell injection (when significant lung bioluminescence was detected), mice were treated with three IV injections (every other day) of vehicle (PBS), or MV-m-uPA. Metastases progression was measured by in vivo luciferase imaging. As shown in figure 4. A, C. Treatment with MV-m-uPA was associated with significant delay in tumor metastases (p= 0.0303) as well as a modest, but statistically significant prolongation of survival (p =0.0149) compared to control mice (Fig. 4. B).
Figure 4. In vivo anti-metastasic effects and tumor targeting in mouse cancer models.

(A) The lung metastases model was established as described in methods. At day 7 after tumor cell injection, mice (n=10/group) were treated with three IV injections (every other day) of vehicle (PBS), or MV-m-uPA. Lung metastases progression was assessed by in vivo bioluminescence imaging at days (−3), 1, and 8 after MV-m-uPA treatment. * p=0.0303. (B). Kaplan-Mier analysis of survival of tumor bearing mice treated with vehicle control or MV-m-uPA. ** p=0.0149. (C) Representative pictures of differences in lung metastases progression by in vivo Bioluminescence imaging. I: Mock. II: MV-m-uPA. (D, E, F) Imunocompetent (Balb/c) mice (n = 3 per group) bearing 4T1 tumors received two intravenous injections of either PBS or MV-m-uPA (1.5×106 TCID50). Lung tissues were harvested 3 days later and frozen sections were used for detection of measles virus. (D) Total RNA was extracted from lung tissues for qRT-PCR analysis of MV-N mRNA. Results were expressed as copies of MV-RNA/μg of total RNA in each organ/tissue. (E) Infectious virus recovery from lung tissues after MV-m-uPA administration. Viral titers are displayed as TCID50/ gram of tissue. (F) Representative pictures of syncytia formation induced by lung tissue lysates of MV-m-uPA treated mice (II) vs. Controls (I). Scale bar = 500 μm. (G) Immunofluorescence (I, II) and immunohistochemical (III, IV) staining of MV-N protein in MV-m-uPA treated vs. control metastases bearing mice. Upper panel: control, lower panel: MV-m-uPA. Scale bar = 50 μm.
Virus rescue and tumor targeting
To determine successful tumor targeting of the recombinant viruses after systemic administration, additional 4T1 tumor bearing mice were treated with MV-m-uPA or vehicle at days 10 and 17 after tumor cell innoculation, and lungs were resected 72 hours after virus treatment. Total RNA was extracted from lung specimens and qRT-PCR for MV-N mRNA was performed. Tumor viral RNA was detected in 3 of 3 mice from days 10 and day 17 after 4T1 cell injections (Fig. 4. D). Next, virus recovery assays were performed from lungs of treated and control mice, as described in methods. Viable MV-m-uPA virions were rescued from 2 of 3 tumor samples from each time point (Fig. 4. E, F II), while no virus was rescued from the control group (not shown). Finally, MV-N viral protein was determined from lung tissues by immunofluorescence and immunohistochemistry. MV-N was detected in the lungs of treated animals (Fig 4. G II, IV), by both methods, but not in the controls (Fig. 4. I, III). IHC studies showed localization of MV-N staining in areas of metastatic nodules (rich in tumor cells, Fig. 4 G. IV).
In vivo anti-metastasic effects in a xenograft model of experimental metastases
To further extend the above findings in a human model of breast cancer metastasis, MDA-MB 231-luc2 cells were intravenously administered to female NOD/SCID mice. Three weeks after tumor cell administration, mice were treated with three IV injections of vehicle or the human uPAR retargeted MV (MV-h-uPA). Metastases progression was measured weekly in vivo luciferase imaging of lung metastases for 6 weeks after treatment. As shown in figure 5. A-C, treatment with MV-h-uPA was associated with significant delay in tumor metastases (p= 0.0093) compared to control mice. This was translated into significant prolongation of survival in mice treated with MV-h-uPA (p =0.0057), compared to controls (Fig. 5. D).
Figure 5. In vivo anti-metastasic effects in Xenograft metastases models.

(A). The MDA-MB-231 experimental lung metastases model was established as described in methods. Mice were (n= 8/group) treated with three IV injections (every other day) of vehicle or MV-h-uPA. Lung metastases progression was assessed by in vivo bioluminescence imagingat weeks 1, 2, 3, 4, 5 and 6 after treatment. *p=0.0093 (week 6). (B, C) Representative pictures of differences in lung metastases progression at week 1 (B) and 6 (C) by in vivo Bioluminescence imaging. I: Control. II: MV-h-uPA. (D). Kaplan-Mier analysis of survival of tumor bearing mice treated with vehicle control or MV-h-uPA. MV-h-uPA vs. control ** p=0.0057. (E) Total RNA was extracted from lung tissues for qRT-PCR analysis of MV-N mRNA. Results were expressed as copies of MV-RNA/μg of total RNA in each organ/tissue. (F) Infectious virus recovery from lung tissues after MV-m-uPA administration. Viral titers are displayed as TCID50/ gram of tissue.
Determination of viral RNA and viable virus from lungs of treated mice
Additional tumor bearing mice were treated with MV-h-uPA intravenously at days 30 and 60 after tumor cell inoculation and lungs were harvested at day 3 after treatment. Viral RNA was detected in all treated mice at both time points (Fig. 6. E), similar to our findings in the syngeneic model. Virus recovery assays were performed from lungs at day 3 after MV-h-uPA treatment. Infectious viral particles were rescued from 1 of 3 lungs at each time point (Fig. 6. F), while no virus was rescued from the control group (not shown).
DISCUSSION
The urokinase receptor plays a critical role in tumor progression and metastases in breast and other cancers [10, 13, 27–31], and represents an ideal target for novel biotherapies. As this receptor is predominantly expressed in cancer compared to non-cancer tissues, uPAR directed agents, like anti-uPAR antibodies or uPAR targeted nanoparticles have the potential to successfully target primary tumors and metastases in vivo, without affecting non-tumor bearing organs [27, 32]. Other uPAR directed strategies that have demonstrated in vivo antitumor efficacy include uPAR antisense oligonucleotides [33] and radiolabeled anti-uPAR antibodies [34].
The Edmonston strain of measles virus is a promising oncolytic platform with an established track record of safety and preclinical antitumor activity. The clinical antitumor potential of this viral vector was recently shown in studies by Russel et al [23], and Galanis et al [22], who reported antitumor effects in patients with multiple myeloma and ovarian cancer, after intravenous and intraperitoneal administration of MV-NIS, an oncolytic MV expressing the sodium iodide symporter, respectively.
Redirecting the virus tropism has the potential to improve tumor selectivity, especially to sites of distant metastases. We have previously demonstrated that MV-uPA, a fully retargeted oncolytic measles against the urokinase receptor is safe and induces antitumor effects against primary (mammary fat pad) human breast cancer [24]. Intravenous administration of MV-m-uPA, retargeted to murine uPAR, was associated with tumor selective replication and lack of organ toxicity in a syngeneic, immunocompetent model of primary mammary cancer [35].
The current report demonstrates for the first time the promising antimetastatic activity of MV-m-uPA and MV-h-uPA. In vitro, our studies clearly showed that uPAR mediated targeting preferentially affects breast/mammary cancer and not normal mammary epithelial cells in a species specific manner. We observed that MV-h-uPA replication in human breast cancer cell lines was more efficient than that in murine 4T1 cells, as reflected by higher viral titers at 48 and 72 hours, and more potent cytotoxic effects in human, compared to murine breast cancer than in murine 4T1 cells.
The 4T1 experimental metastases model is highly aggressive, where mice develop lung metastases rapidly (within 7 days), and the majority of mice die within the first 3 weeks. Intravenous administration of MV-m-uPA significantly delayed the rapid progression of lung metastases (Fig. 4. A), and even with only 3 treatments, given in the first week of therapy, survival was improved. We clearly showed evidence of viral targeting of metastases, as well as presence of both viable, replicating viral vectors, and viral RNA in lungs from treated, tumor bearing mice (Figs. 4. D, E, F, G). These results indicate successful targeting as well as the biological and antitumor effects of this agent in this highly aggressive model. Importantly, these findings were validated in the experimental human MDA-MB-231 metastases model. The metastases delaying effects were markedly reduced in the treated animals, and survival was significantly prolonged. Similar to the syngeneic model, viable virus and viral RNA was found in the lungs of treated animals. As MV-h-uPA only targets human (and not murine) uPAR, the presence of viral particles and RNA 3 days after virus administration demonstrates effective in vivo viral targeting of human tumor cells.
While marked antimetastatic effects and survival prolongation were observed in the xenograft model, no long term survival was observed in the 4T1 metastases model. There are several reasons that may explain these findings, including the highly aggressive nature of the 4T1 model (development of large nodules early after tumor cell inoculation), limited number of treatments, and the less permissive nature of 4T1 cells found in vitro, compared to human breast cancer cell lines. In addition, the development of antiviral immunity in mice with an intact immune system likely plays an important role in reducing viral propagation over time. The results obtained from our syngeneic model prove the concept that uPAR targeting of metastatic lesions in immunocompetent models of cancer metastases is safe and feasible, and will lead to future studies aimed at further characterizing the best schedule and timing of treatment with MV-m-uPA. As the majority of previously reported targeted oncolytic MVs are directed against human, and not murine targets [36–40], MV-m-uPA offers the advantage of testing safety and antitumor effects in in syngeneic, immunocompetent animals, without the need to use transgenic mice or modified mouse cell line models. An important, but unexplored area of research in oncolytic virotherapy, which can be tested with our agents in immunocompetent and immunodeficient models, is the evaluation of the role of this fully retargeted viral agent in the prevention of micrometastases, either in experimental or spontaneous metastases. This could open a novel strategy for future studies of retargeted oncolytic viruses in general, and uPAR targeted viral agents in particular, in the postoperative setting in high risk patients with breast and other malignancies.
In summary, our studies demonstrate successful uPAR mediated targeting and delay in metastases progression in vivo, in both immunocompetent and immunodeficient mammary cancer metastasis models. Our observations validate at a preclinical level the feasibility and activity of MV-uPA in the metastatic setting after systemic administration. Further characterization of this strategy in the prevention of metastases after surgery and combination therapies with agents to improve the efficacy and replication of the viral vector in vivo are underway.
MATERIALS AND METHODS
Virus preparation and cell culture
Construction of MV-m-uPA and MV-h-uPA, virus rescue, propagation and titration were performed as previously reported [24, 36]. 4T1 cells (murine mammary carcinoma), MDA-MB-231 cells (human breast cancer), MCF-7 cells (human breast cancer), MDA-MB-436 cells (human breast cancer) were purchased from the American Type Culture Collection (ATCC, Manassas, VA). 4T1-luc2 cells stably expressing highly efficient luciferase were from PerkinElmer (Santa Clara CA). Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), penicillin and streptomycin at 37 °C and 5% CO2. HMEC cells (human mammary epithelial cells) were purchased from Lonza (Walkersville, MD) and maintained in MEGM according to manufacture’s instruction at 37 °C and 5% CO2. NMuMG (murine mammary epithelial cells) were purchased from the ATCC and maintained in DMEM containing 10% fetal bovine serum (FBS), penicillin and streptomycin at 37 °C and 5% CO2. Vero-αHis cells [36] were grown in DMEM containing 10% FBS at 37 °C and 5% CO2.
In vitro infection
104 of each cell line in a 24-well plate were incubated with each MV at an MOI of 1 in Opti-MEM for 2 h at 37 °C. At the end of the incubation period, free viruses were removed and cells were maintained in the appropriate medium. At 48 h after infection, cells were photographed under fluorescence microscopy (Nikon, Melville, NY).
Assessment of in vitro cytopathic effects
Cells were plated in six-well plates at a density of 105 per well. Twenty-four hours after seeding, the cells were infected at MOI = 1 in 1 mL of Opti-MEM for 2 hours at 37°C. At the end of the incubation period, the virus was removed, and cells were maintained in their standard medium. At 48h, 72h after infection, the number of viable cells (determined by tryptan blue exclusion) in each well was counted using Vi-Cell cell viability analyzer (Beckman Coulter, Fullerton, CA).
In vitro replication
Each cell line (in duplicate) was infected with measles virus at an MOI of 3 and incubating at 37°C, 5% CO2 for 2 h, after which the virus was removed, and cells were maintained in 5% FBS of DMEM at 37°C. Virus titers were obtained by titration on Vero-αHis cells and expressed as 50% tissue culture infectious dose (TCID50)/ml at the specified timepoints.
Animal studies
Animal studies were approved by Institutional Animal Care and Use Committee of University of Miami.
Characterization of MV-m-uPA’s in vivo oncolytic effects in immunocompetent cancer murine models
The lung metastases model was established by tail vein injection of 2×105 4T1-Luc2 cells (Caliper) in 100 μl of PBS in female BALB/C mice. At day 7 after tumor cell injection, mice were (n=10/group) treated with three IV injections (every other day) of vehicle (PBS), or MV-m-uPA (1×106 TCID50 in 100 μl PBS). Endpoints of the study were tumor progression and survival. Metastases progression will be measured by in vivo luciferase imaging.
Characterization of MV-h-uPA’s in vivo oncolytic effects in Xenograft metastases model
Female NOD/SCID mice (7–8 weeks) were received 2×105 MDA-MB231-luc2 cells (Caliper) in 100 μl of PBS via tail vein. 3 weeks after tumor cell injection, mice were (n=8/group) treated with three IV injections (every other day) of either vehicle (PBS), or MV-h-uPA (1×106 TCID50 in 100 μl PBS). Endpoints of the study were tumor progression and survival. Metastases progression will be measured by in vivo luciferase imaging of lung metastases at days 0, 7, 14, 21, 28 after treatment. In vivo luciferase imaging was evaluated as follows: mice were intraperitoneally injected with D-luciferin (Caliper) at a dose of 150 mg/kg per mouse and anesthetized. Bioluminescence images were then acquired using Xenogen IVIS imaging system. Bioluminescence signal was quantified as photon flux (photons/s/cm2) in defined regions of interest using Living Image software (Xenogen).
Immunohistochemistry and Immunofluorescence studies
Tumor bearing mice (n = 3) were treated with two intravenous injections of 1.5×106 TCID50 of MV-m-uPA at days 10 and 17 after tumor cell inoculation. Mice were sacrificed and tumors resected after 72 hours of the last injection (in each time point), for immunohistochemistry and immunofluorescence studies for MV-N protein. Tissue samples were collected and frozen, and cryostat sections were fixed in cold acetone for 10 min and endogenous peroxidase activity were quenched with 0.3% H2O2 for 10 min. The slides were washed in PBS and incubated with biotinylated mouse anti-MV-nucleoprotein antibody (Chemicon International, Temecula, CA) for 30 min at 37°C. After washing in PBS, the slides were developed with VECTASTAIN ABC horseradish peroxidase (HRP) kit (Vector Laboratories) and 3, 3, 9-diaminobenzidine (DAB) HRP substrate (Vector Laboratories) according to the manufacturer’s instructions. For anti-MV immunofluorescence staining, cryostat sections were fixed in cold acetone for 10 min. The slides were washed in PBS and incubated with anti-MV-nucleoprotein-FITC antibody (Chemicon International) for 30 min at 37°C.
Virus Recovery
Tissues were weighed and homogenized in three volumes (w/v) of Opti-MEM utilizing mechanical crushing and a single freeze thaw cycle. The supernatant was clarified by centrifugation and ten-fold serial dilutions of samples were prepared in Opti-MEM. Aliquots (50 μL) of each dilution were placed in 96 well plates containing Vero-his cells and TCID50 titrations were performed. TCID50 calculations were normalized per gram of tissue.
Viral RNA quantification
Total RNA was extracted from frozen specimens using the RNeasy tissue mini kit (Qiagen, Valencia, CA) following the manufacturer’s recommendations. qRT-PCR for MV-N mRNA was performed as previously reported [41].
Statistical analysis
In vitro data are presented as means +/− standard deviations. Results from in vivo studies are shown as means +/− standard error of the mean. All in vitro experiments were performed in triplicate unless otherwise specified. Statistical analysis among groups was performed by analysis of variance. Sub-group comparisons were made after the overall analyses using the Student t-test, Tukey-Kramer, Fisher’s or Wilcoxon rank sum test, as appropriate. Overall survival was analyzed by the Kaplan Meier method and differences were analyzed by the log-rank test. Statistical significance was set at p = 0.05, with adjustements for multiple comparisons as appropriate. All statistical tests were two-sided.
Acknowledgments
This work was supported by a research grant from the National Cancer Institute (1R01CA149659-01 to JRM, JZ), and by the Sylvester Comprehensive Cancer Center (JRM).
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: a cancer journal for clinicians. 2014;64(1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Chia SK, Speers CH, D’Yachkova Y, Kang A, Malfair-Taylor S, Barnett J, Coldman A, Gelmon KA, O’Reilly SE, Olivotto IA. The impact of new chemotherapeutic and hormone agents on survival in a population-based cohort of women with metastatic breast cancer. Cancer. 2007;110(5):973–979. doi: 10.1002/cncr.22867. [DOI] [PubMed] [Google Scholar]
- 3.Tang L, Han X. The urokinase plasminogen activator system in breast cancer invasion and metastasis. Biomed Pharmacother. 2013;67(2):179–182. doi: 10.1016/j.biopha.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Foekens JA, Peters HA, Look MP, Portengen H, Schmitt M, Kramer MD, Brunner N, Janicke F, Meijer-van Gelder ME, Henzen-Logmans SC, et al. The urokinase system of plasminogen activation and prognosis in 2780 breast cancer patients. Cancer research. 2000;60(3):636–643. [PubMed] [Google Scholar]
- 5.Harbeck N, Kates RE, Schmitt M. Clinical relevance of invasion factors urokinase-type plasminogen activator and plasminogen activator inhibitor type 1 for individualized therapy decisions in primary breast cancer is greatest when used in combination. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2002;20(4):1000–1007. doi: 10.1200/JCO.2002.20.4.1000. [DOI] [PubMed] [Google Scholar]
- 6.Janicke F, Prechtl A, Thomssen C, Harbeck N, Meisner C, Untch M, Sweep CG, Selbmann HK, Graeff H, Schmitt M. Randomized adjuvant chemotherapy trial in high-risk, lymph node-negative breast cancer patients identified by urokinase-type plasminogen activator and plasminogen activator inhibitor type 1. Journal of the National Cancer Institute. 2001;93(12):913–920. doi: 10.1093/jnci/93.12.913. [DOI] [PubMed] [Google Scholar]
- 7.Look MP, van Putten WL, Duffy MJ, Harbeck N, Christensen IJ, Thomssen C, Kates R, Spyratos F, Ferno M, Eppenberger-Castori S, et al. Pooled analysis of prognostic impact of urokinase-type plasminogen activator and its inhibitor PAI-1 in 8377 breast cancer patients. J Natl Cancer Inst. 2002;94(2):116–128. doi: 10.1093/jnci/94.2.116. [DOI] [PubMed] [Google Scholar]
- 8.Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002;3(12):932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 9.Jo M, Lester RD, Montel V, Eastman B, Takimoto S, Gonias SL. Reversibility of epithelial-mesenchymal transition (EMT) induced in breast cancer cells by activation of urokinase receptor-dependent cell signaling. The Journal of biological chemistry. 2009;284(34):22825–22833. doi: 10.1074/jbc.M109.023960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romer J, Nielsen BS, Ploug M. The urokinase receptor as a potential target in cancer therapy. Curr Pharm Des. 2004;10(19):2359–2376. doi: 10.2174/1381612043383962. [DOI] [PubMed] [Google Scholar]
- 11.Bianchi E, Cohen RL, Thor AT, Todd RF, 3rd, Mizukami IF, Lawrence DA, Ljung BM, Shuman MA, Smith HS. The urokinase receptor is expressed in invasive breast cancer but not in normal breast tissue. Cancer Res. 1994;54(4):861–866. [PubMed] [Google Scholar]
- 12.Boyd D, Florent G, Kim P, Brattain M. Determination of the levels of urokinase and its receptor in human colon carcinoma cell lines. Cancer Res. 1988;48(11):3112–3116. [PubMed] [Google Scholar]
- 13.Dass K, Ahmad A, Azmi AS, Sarkar SH, Sarkar FH. Evolving role of uPA/uPAR system in human cancers. Cancer Treat Rev. 2008;34(2):122–136. doi: 10.1016/j.ctrv.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 14.Lester RD, Jo M, Montel V, Takimoto S, Gonias SL. uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J Cell Biol. 2007;178(3):425–436. doi: 10.1083/jcb.200701092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohba K, Miyata Y, Kanda S, Koga S, Hayashi T, Kanetake H. Expression of urokinase-type plasminogen activator, urokinase-type plasminogen activator receptor and plasminogen activator inhibitors in patients with renal cell carcinoma: correlation with tumor associated macrophage and prognosis. J Urol. 2005;174(2):461–465. doi: 10.1097/01.ju.0000165150.46006.92. [DOI] [PubMed] [Google Scholar]
- 16.Rabbani SA, Xing RH. Role of urokinase (uPA) and its receptor (uPAR) in invasion and metastasis of hormone-dependent malignancies. Int J Oncol. 1998;12(4):911–920. doi: 10.3892/ijo.12.4.911. [DOI] [PubMed] [Google Scholar]
- 17.Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11(1):23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- 18.Patel MR, Kratzke RA. Oncolytic virus therapy for cancer: the first wave of translational clinical trials. Translational research : the journal of laboratory and clinical medicine. 2013;161:355–364. doi: 10.1016/j.trsl.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura T, Russell SJ. Oncolytic measles viruses for cancer therapy. Expert Opin Biol Ther. 2004;4(10):1685–1692. doi: 10.1517/14712598.4.10.1685. [DOI] [PubMed] [Google Scholar]
- 20.Russell SJ. RNA viruses as virotherapy agents. Cancer Gene Ther. 2002;9(12):961–966. doi: 10.1038/sj.cgt.7700535. [DOI] [PubMed] [Google Scholar]
- 21.Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA, Kaur JS, Haluska PJ, Jr, Aderca I, Zollman PJ, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70(3):875–882. doi: 10.1158/0008-5472.CAN-09-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galanis E, Atherton PJ, Maurer MJ, Knutson KL, Dowdy SC, Cliby WA, Halulska P, Jr, Long HJ, Oberg A, Aderca I, et al. Oncolytic measles virus expressing the sodium iodide symporter to treat drug-resistant ovarian cancer. Cancer research. 2014 doi: 10.1158/0008-5472.CAN-14-2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Russell SJ, Federspiel MJ, Peng KW, Tong C, Dingli D, Morice WG, Lowe V, O’Connor MK, Kyle RA, Leung N, et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc. 2014;89(7):926–933. doi: 10.1016/j.mayocp.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jing Y, Tong C, Zhang J, Nakamura T, Iankov I, Russell SJ, Merchan JR. Tumor and vascular targeting of a novel oncolytic measles virus retargeted against the urokinase receptor. Cancer Res. 2009;69(4):1459–1468. doi: 10.1158/0008-5472.CAN-08-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sliutz G, Eder H, Koelbl H, Tempfer C, Auerbach L, Schneeberger C, Kainz C, Zeillinger R. Quantification of uPA receptor expression in human breast cancer cell lines by cRT-PCR. Breast cancer research and treatment. 1996;40(3):257–263. doi: 10.1007/BF01806814. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen DH, Hussaini IM, Gonias SL. Binding of urokinase-type plasminogen activator to its receptor in MCF-7 cells activates extracellular signal-regulated kinase 1 and 2 which is required for increased cellular motility. The Journal of biological chemistry. 1998;273(14):8502–8507. doi: 10.1074/jbc.273.14.8502. [DOI] [PubMed] [Google Scholar]
- 27.Rabbani SA, Gladu J. Urokinase receptor antibody can reduce tumor volume and detect the presence of occult tumor metastases in vivo. Cancer Res. 2002;62(8):2390–2397. [PubMed] [Google Scholar]
- 28.Mazar AP, Henkin J, Goldfarb RH. The urokinase plasminogen activator system in cancer: implications for tumor angiogenesis and metastasis. Angiogenesis. 1999;3(1):15–32. doi: 10.1023/a:1009095825561. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Cozzi PJ. Targeting uPA/uPAR in prostate cancer. Cancer Treat Rev. 2007;33(6):521–527. doi: 10.1016/j.ctrv.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 30.Jo M, Takimoto S, Montel V, Gonias SL. The urokinase receptor promotes cancer metastasis independently of urokinase-type plasminogen activator in mice. The American journal of pathology. 2009;175(1):190–200. doi: 10.2353/ajpath.2009.081053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grondahl-Hansen J, Peters HA, van Putten WL, Look MP, Pappot H, Ronne E, Dano K, Klijn JG, Brunner N, Foekens JA. Prognostic significance of the receptor for urokinase plasminogen activator in breast cancer. Clin Cancer Res. 1995;1(10):1079–1087. [PubMed] [Google Scholar]
- 32.Yang L, Peng XH, Wang YA, Wang X, Cao Z, Ni C, Karna P, Zhang X, Wood WC, Gao X, et al. Receptor-targeted nanoparticles for in vivo imaging of breast cancer. Clin Cancer Res. 2009;15(14):4722–4732. doi: 10.1158/1078-0432.CCR-08-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Alessio S, Margheri F, Pucci M, Del Rosso A, Monia BP, Bologna M, Leonetti C, Scarsella M, Zupi G, Fibbi G, et al. Antisense oligodeoxynucleotides for urokinase-plasminogen activator receptor have anti-invasive and anti-proliferative effects in vitro and inhibit spontaneous metastases of human melanoma in mice. Int J Cancer. 2004;110(1):125–133. doi: 10.1002/ijc.20077. [DOI] [PubMed] [Google Scholar]
- 34.LeBeau AM, Duriseti S, Murphy ST, Pepin F, Hann B, Gray JW, VanBrocklin HF, Craik CS. Targeting uPAR with antagonistic recombinant human antibodies in aggressive breast cancer. Cancer research. 2013;73(7):2070–2081. doi: 10.1158/0008-5472.CAN-12-3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jing Y, Zaias J, Duncan R, Russell SJ, Merchan JR. In vivo safety, biodistribution and antitumor effects of uPAR retargeted oncolytic measles virus in syngeneic cancer models. Gene therapy. 2014;21(3):289–297. doi: 10.1038/gt.2013.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamura T, Peng KW, Harvey M, Greiner S, Lorimer IA, James CD, Russell SJ. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat Biotechnol. 2005;23(2):209–214. doi: 10.1038/nbt1060. [DOI] [PubMed] [Google Scholar]
- 37.Liu C, Hasegawa K, Russell SJ, Sadelain M, Peng KW. Prostate-specific membrane antigen retargeted measles virotherapy for the treatment of prostate cancer. Prostate. 2009;69(10):1128–1141. doi: 10.1002/pros.20962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hasegawa K, Nakamura T, Harvey M, Ikeda Y, Oberg A, Figini M, Canevari S, Hartmann LC, Peng KW. The use of a tropism-modified measles virus in folate receptor-targeted virotherapy of ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(20 Pt 1):6170–6178. doi: 10.1158/1078-0432.CCR-06-0992. [DOI] [PubMed] [Google Scholar]
- 39.Allen C, Vongpunsawad S, Nakamura T, James CD, Schroeder M, Cattaneo R, Giannini C, Krempski J, Peng KW, Goble JM, et al. Retargeted oncolytic measles strains entering via the EGFRvIII receptor maintain significant antitumor activity against gliomas with increased tumor specificity. Cancer Res. 2006;66(24):11840–11850. doi: 10.1158/0008-5472.CAN-06-1200. [DOI] [PubMed] [Google Scholar]
- 40.Allen C, Paraskevakou G, Iankov I, Giannini C, Schroeder M, Sarkaria J, Puri RK, Russell SJ, Galanis E. Interleukin-13 displaying retargeted oncolytic measles virus strains have significant activity against gliomas with improved specificity. Molecular therapy : the journal of the American Society of Gene Therapy. 2008;16(9):1556–1564. doi: 10.1038/mt.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peng KW, Frenzke M, Myers R, Soeffker D, Harvey M, Greiner S, Galanis E, Cattaneo R, Federspiel MJ, Russell SJ. Biodistribution of oncolytic measles virus after intraperitoneal administration into Ifnar-CD46Ge transgenic mice. Hum Gene Ther. 2003;14(16):1565–1577. doi: 10.1089/104303403322495070. [DOI] [PubMed] [Google Scholar]