

Abstract
Purpose
Quantitative mass spectrometry assays for immunoglobulins (Igs) are compared with existing clinical methods in samples from patients with plasma cell dyscrasias, e.g. multiple myeloma.
Experimental design
Using LC-MS/MS data, Ig constant region peptides and transitions were selected for liquid chromatography-multiple reaction monitoring mass spectrometry (LC-MRM). Quantitative assays were used to assess Igs in serum from 83 patients.
Results
LC-MRM assays quantify serum levels of Igs and their isoforms (IgG1–4, IgA1–2, IgM, IgD, and IgE, as well as kappa(κ) and lambda(λ) light chains). LC-MRM quantification has been applied to single samples from a patient cohort and a longitudinal study of an IgE patient undergoing treatment, to enable comparison with existing clinical methods. Proof-of-concept data for defining and monitoring variable region peptides are provided using the H929 multiple myeloma cell line and two MM patients.
Conclusions and Clinical Relevance
LC-MRM assays targeting constant region peptides determine the type and isoform of the involved immunoglobulin and quantify its expression; the LC-MRM approach has improved sensitivity compared with the current clinical method, but slightly higher interassay variability. Detection of variable region peptides is a promising way to improve Ig quantification, which could produce a dramatic increase in sensitivity over existing methods, and could further complement current clinical techniques.
Keywords: Immunoglobulin quantification, Liquid Chromatography-Multiple Reaction Monitoring Mass Spectrometry, Multiple Myeloma
Introduction
Multiple myeloma (MM) is a plasma cell cancer characterized by bone marrow clonal plasmacytosis, monoclonal immunoglobulin expression in the serum and/or urine, lytic bone lesions, hypercalcemia, anemia, and renal failure. MM patients initially respond to therapy, but relapse with drug-resistant disease. Therefore, early detection and effective monitoring are critical for management of MM patients [1]. Current clinical assays focus on detection and quantification of the monoclonal immunoglobulin (or M-protein) secreted by the tumor cells, which is essential for diagnosis and monitoring of patients with MM and other plasma cell dyscrasias (PCD) [2]. The Durie and Salmon staging system and the International Staging System (ISS) are based on the correlation between the expression of the monoclonal immunoglobulin and the disease burden [1,3]. The International Myeloma Working Group guidelines also describe the assessment of treatment outcomes based on the changes in expression of the M-protein [4]. Patient monitoring strategies present significant challenges, particularly in the diagnosis of premalignant monoclonal gammopathy of undetermined significance (MGUS), prediction of progression from MGUS to MM, assessment of response to therapy, and detection of relapse [5].
Evaluation of disease is accomplished by serial measurements of the M-protein in serum and urine using a variety of techniques [6,7,8,9,10,11]. Typically, initial measurements are made using serum protein electrophoresis (SPEP), which is limited in sensitivity to approximately 0.1 gram per deciliter (g/dl) [12]. The monoclonal immunoglobulin produced in high concentration by MM cells can be visualized as a narrow, discrete, dark band usually in the γ region of the gel or electropherogram. SPEP densitometry and total serum protein concentration are used to estimate the amount of the immunoglobulin secreted by the tumor. Patients can be further characterized using immunofixation electrophoresis (IFE). IFE screens test for immunoglobulin G, A, and M heavy chains, as well as kappa (κ) and lambda (λ) light chains. Immunoglobulin D and E myelomas are rare; when suspected, IFE is repeated to detect IgD or IgE. The combination of SPEP and IFE establishes an estimated level in the serum and type of the immunoglobulin that is secreted by the tumor. These traditionally gel-based techniques have recently been replaced by capillary array instruments [13]. For immunoglobulin heavy chains with high expression, SPEP is the current clinical standard for detecting tumor burden, because the disease-specific immunoglobulin is directly monitored.
However, several factors limit SPEP in monitoring tumor burden in patients [14,15,16] Therefore, quantification of the involved immunoglobulin by nephelometry is also used to monitor tumor burden [17], and it has particular value for immunoglobulins with lower abundances in serum (e.g. IgD and IgE), particularly because the background expression of these immunoglobulins is low. Serum free light chain assays (SFLC) are also implemented using nephelometry to provide an expression ratio between the light chains, which supplements other techniques for the detection of light chain only disease [18,19,20]. Antibody-based methods for protein quantification are also influenced by the complexity of the immunoglobulin system of the biologically derived antiserum and variation in its reactivity, as well as changes in the levels of proteins in the standard reagents (typically pooled serum) [21] The presence of immunological subclasses (i.e. IgG1–4 and IgA1–2) also adds to the complexity of the analysis [21]. Therefore, quantitative mass spectrometry methods could complement existing techniques by producing measurements of the total expression of each immunoglobulin and their isoforms in a single analysis. In addition, development of quantitative proteomic assays for each individual patient will enable direct measurement of the disease-specific immunoglobulin, with the potential to significantly increase the sensitivity of detection over SPEP.
Liquid chromatography-multiple reaction monitoring mass spectrometry (LC-MRM) using stable isotope dilution has enabled the assessment of protein biomarkers [22,23,24,25,26,27,28,29,30,31]. In addition, collaborative groups have standardized LC-MRM assays at multiple sites [32,33]. Based on these advances, this technology holds great promise for patient assessment, and LC-MRM is being used in translational research programs [5]. This technology has also been used to measure clinically-relevant protein biomarkers, including troponin I and interleukin-33 [34], apolipoproteins [22,35], and thyroglobulin [36].
Quantification of immunoglobulins can be achieved at two levels. Peptides from the constant regions can be quantified to evaluate levels of total immunoglobulin expression. The comparison of LC-MRM of constant region peptides to immunoglobulin quantification with current clinical techniques provides information about the utility, advantages, and disadvantages of the technique. In addition, development of assays for peptides from the variable region enables a measurement of the disease-specific immunoglobulin, similar in specificity to SPEP detection. Using the previously published strategy of informing proteomics with RNA-sequencing [37], proof-of-concept data are provided for personalized detection of myeloma tumor burden using RNA-sequencing and LC-MRM variable region peptide detection for both an in vitro system (i.e. H929 cells) and patients.
Materials and Methods
Chemicals and reagents were acquired from Sigma-Aldrich (Milwaukee, WI); HPLC solvents were purchased from Burdick and Jackson (Honeywell, Muskegon, MI). Standard peptides were synthesized, HPLC-purified, characterized with MALDI MS, QqQ MS, and amino acid analysis, as previously described [38].
Sample Collection and Summary of Patient Data
De-identified serum was collected from patients in accordance with protocols approved by the University of South Florida Institutional Review Board. Blood was collected in serum separator tubes (BD, Franklin Lakes, NJ), clotted for 30 minutes, spun down at 3,600 rpm for 10 minutes (5702, Eppendorf), and refrigerated until analysis (t < 3 weeks). Samples (n = 83) were collected between 07/05/2009 and 05/31/2010. The study population contained 46 males and 37 females between ages 34 and 87 (median age 63) with diagnoses including MM (45), smoldering MM (3), light chain only MM (1), non-secretory MM (1) MGUS (6), plasmacytoma (5), plasma cell leukemia (2), Waldenstrom’s macroglobinemia (8), Non-Hodgkin’s Lymphoma (6), other leukemias or lymphomas (4), amyloidosis (2), and prostate cancer (1). Samples were selected to represent all types of PCDs with varying levels of immunoglobulin expression and age-matched patients with other diseases. Of these patients, 71 had elevated levels of immunoglobulin expression detected by SPEP compared to reference values for healthy controls. Twelve patients had immunoglobulin expression levels comparable to healthy controls and were not detectable by SPEP (SPEP-). Clinical details are provided in Supplementary Table 1.
Bone marrow aspirates were collected from 2 patients (not included in the cohort used for the constant region analysis). MM tumor cells were obtained by Ficoll and CD138+ plasma cell selection (Miltenyi, Auburn, CA). An aliquot of 0.5 million cells was used for RNA-sequencing.
Clinical Measurements
SPEP was performed using capillary zone electrophoresis (Capillarys2, Sebia). Immunoglobulin concentrations were calculated using measurements of total serum protein (Fusion 5.1FS Chemistry Analyzer, Ortho Clinical Diagnostics, NJ). Immunotyping was performed using monospecific antisera for IgG, IgA, and IgM heavy chains as well as κ and λ light chains (Capillarys2, Sebia). Immunofixation electrophoresis (SPIFE 3000, Helena Laboratories) was used to confirm serum immunotyping results or to test for IgD and IgE. Nephelometry of the immunoglobulins was performed (Vitros 5.1 FS Chemistry System, Ortho-Clinical Diagnostics) to determine the concentration of IgA, IgG, and IgM using goat antisera as the primary active reagents.
LC-MRM Quantification of Proteins in Serum
Peptide targets were selected from LC-MS/MS data (see Figure S1). Serum proteins were denatured with 8M urea, reduced with tris(2-carboxyethyl) phosphine, and alkylated with iodoacetamide prior to a ten-fold dilution in aqueous 30 mM ammonium bicarbonate and in-solution tryptic digestion (Promega, Madison, WI). Internal standards were spiked into each sample (Table 1) [6,7,8,9,10,11]. The equivalent of 0.5 nanoliters of tryptically digested serum was injected for each LC-MRM analysis.
Table 1. Endogenous Peptides and Labeled Standards used for LC-MRM Quantification of Immunoglobulins.
Peptides representative for individual proteins and groups of isoforms are quantified.
| Protein | Average Expression & Reference Range (mg/ml) |
Peptide | IS | Transitions | Median Intra- Assay CV (%) |
Median Inter- Assay CV (%) |
Outliers |
|---|---|---|---|---|---|---|---|
| IgG1 | 5.917 3.19–10.2 |
GPSVFPLAPSSK | 10.2 | y8–y10 | 7.5 | 7.2 | 3 |
| IgG2 | 3.047 1.23–6.63 |
GLPAPIEK | 6.63 | y4–y6 | 6.0 | 5.9 | 2 |
| IgG3 | 0.617 0.16–1.94 |
WYVDGVEVHNAK | 1.94 | y6,y8,y9 | 16 | 18.3 | 12 |
| IgG4 | 0.247 0.03–1.33 |
TTPPVLDSDGSFFLYSR | NS | y8,y10,y12 | -- | -- | -- |
| IgA1 | 1.886 1.36–2.5 |
TPLTATLSK | NS | y5–y7 | -- | -- | -- |
| IgA2 | 0.546 0.28–0.61 |
DASGATFTWTPSSGK* | 0.6 | y7–y10 | 21 | 16 | 5 |
| IgA1-2 | 2.42 1.64–3.11 |
WLQGSQELPR | 3.1 | y6–y8 | 6.8 | 9.6 | 2 |
| IgM | 0.708 0.4–2.3 |
DGFFGNPR | 2.3 | y4–y6 | 11 | 7.6 | 1 |
| κ LC | 2.319 1.55–3.08 |
TVAAPSVFIFPPSDEQLK* | 3.0 | y8,y9,y11 | 7.3 | 6.6 | 6 |
| λ LC | 1.549 0.83–2.24 |
AGVETTTPSK | 2.24 | y5–y7 | 19 | 9.0 | 16 |
| IgD | 0.013910 0.001–0.024 |
EPAAQAPVK | 0.50 | y5–y7 | 0.52 | -- | 0 |
| IgE | 0.000111 0–0.002 |
GSGFFVFSR* | 0.50 | y5–y7 | 6.7 | -- | 0 |
| Albumin | 3511 30–40 |
LVNEVTEFAK* | 3.5 | y5,y7,y8 | 5.0 | 6.3 | 0 |
Standard peptides were synthesized with either isotope-labeled amino acids or with a structural analog created by single conservative amino acid replacement (as noted by *) that differs only by a methylene group in the side chain of the underlined residue. The limit of detection based on internal standards is also shown along with other monitored peptides that do not have quantitative assays developed. Eleven additional peptides are monitored to improve confidence in quantification by evaluation of consistency, but SIS peptides have not been synthesized. Reference ranges provided by ARUP Laboratories.
LC-MRM was performed using a nanoLC interfaced with a triple quadrupole mass spectrometer (EasyNanoLC and TSQ Quantum Ultra or Vantage, Thermo, San Jose, CA), as previously described (see supplement for additional details) [38]. Briefly, peptides were desalted on a reversed phase pre-column prior to reversed phase chromatography using10 minute gradients (C18 Pepmap100, Thermo, San Jose, CA). Peptide precursors were selected with 0.4 Q1resolution; fragment ions were then selected with 0.7 Q3 resolution. Scan width was 0.002, and transitions were acquired for 5–20 milliseconds. If the peptides were not optimized by manual infusion, collision energy values were calculated using Equation 1 based on the mass-to-charge ratio (m/z) of the doubly protonated peptide precursor [39,40].
| Equation 1 |
Batches of ten samples were analyzed on the instrument with a reverse calibration curve of the following three standards: a common serum standard was spiked with stable isotope-labeled standard peptides at the average normal abundance, maximum normal abundance, and maximum normal abundance plus 1 mg/ml. Samples were analyzed one through ten, followed by standard samples, and then this batch was repeated for triplicate measurements (see Figure S2A for example data from the IgA peptide, WLQGSQELPR).
Data Analysis
Peak areas (PA) were calculated using MRMer [41] implemented in GenePattern [42]. Raw data files were converted into mzxml; LC-MRM peaks were extracted and visualized for transition evaluation. Resulting data were assessed for quality control and compared between patient groups using Post-MRMer (http://proteome.moffitt.org/). After data review, protein concentrations (in mg/ml) were calculated using the PA ratio of the endogenous peptide to its corresponding standard. The resulting data were evaluated using existing reference ranges for protein-based measurements and compared with nephelometry measurements. Intra-assay CV values were determined using triplicate LC-MRM analysis; inter-assay CV values were calculated from ten LC-MRM analyses of different preparations of the same serum samples (n = 3). Median values are reported. Batch-to-batch variation was also examined (see supplement for description and Figure S2 for an example).
Statistical Analyses
To evaluate assay robustness, the data set was filtered for outliers, when CV > 0.5 and at least one value calculated to be above the previously defined range of healthy controls. Samples below the defined normal maximum value were not examined for outliers because the low protein expression level often contributed to increased CV values. In the cases with high CV values, an individual measurement was discarded when its distance from the median was twice (or greater) the distance of the other data point from the median. Out of 249 data points for each peptide, zero to fifteen outliers were removed (see Table 1). Batch effects were also evaluated using the reverse calibration curves and visual inspection of the entire dataset for all 83 patient specimens (see Supplemental Figure S2B). From the remaining sample points, the mean, median, and standard deviation were calculated and a two-sided Wilcoxon rank-sum test was used to determine if a statistical difference existed between the expression of each protein in patients diagnosed with each specific type of PCD (as determined by clinical diagnoses for the different immunoglobulins or LC-MRM analysis for specific isoforms of IgA and IgG) and all other patient samples. The Holm-Bonferroni method [43] was used to adjust for multiple hypotheses testing with type I error (α), which was set to 0.05.
RNA Sequencing and LC-MRM Detection of Variable Region Peptides
H929 multiple myeloma cells and two patient specimens (all n = 0.5 × 106 cells) were selected for a proof-of-concept experiment in immunoglobulin variable region sequencing and detection of variable region peptides. mRNA sequencing (RNA-seq) was performed from 100 ng of total RNA using the Encore Complete Library System (NuGEN, San Carlos, CA). Strand-specific cDNA generated from this kit was used to prepare a barcoded library appropriate for multiplexed massively parallel sequencing. Paired-end 100 base reads (n ~2 × 107) were generated using the HiScan SQ sequencer (Illumina, San Diego, CA). Demultiplexing and data quality evaluation were performed using CASAVA 1.8.2 (Illumina). RNA-seq reads were aligned to the human hs37d5 reference genome and the Gencode v14 gene model using Tophat2 [44,45]. The expressed transcripts were assembled and evaluated using the de novo assembly software, Trinity [46]. Contiguous sequences are aligned back to the human genome with BLAST, and best hits are manually examined. NCBI ORF Finder [47] identifies potential protein constructs. After generating the protein sequence for the immunoglobulin secreted by the tumor cells, we screened for the detection of tryptic peptides from the Igκ constant and variable regions in H929 conditioned culture media with LC-MRM, as described above. For patient specimens, both samples were analyzed for LC-MRM detection of variable region peptides from Patient 1 to examine their detection in low levels of disease and against a control background.
Results and Discussion
LC-MRM Quantification of Heavy Chains using Constant Region Peptides
Triplicate LC-MRM analysis was performed on 83 patient samples. For most standards, spike-in concentrations equivalent to the maximum value for expression in healthy controls to enabled rapid patient evaluation (higher endogenous peptide signal indicates disease burden). Spiked amounts of the internal standards for albumin and those common to multiple IgG isoforms were decreased due to their high abundance in serum. Due to the low abundance of IgD and IgE in healthy controls, their internal standards were spiked at 0.5 mg/ml (or 0.05 g/dl), which is well below the typical limit for starting treatment after disease relapse and half the limit of detection for SPEP. With the exception of peptides monitoring IgA isoforms, median CV values were below 20%.
Comparison of SPEP/IFE results to LC-MRM is useful for verification that the new technique is able to accurately discern involved immunoglobulins for PCD patients. As expected, LC-MRM detection of increased expression of immunoglobulins matched the clinical diagnoses made by serial SPEP/IFE measurements in all cases when the involved immunoglobulin abundance exceeded the reference range for healthy controls. Samples were taken from 19 IgG patients (average SPEP 14.9 mg/ml), 20 IgA patients (average SPEP 14.0 mg/ml), and 17 patients with elevated IgM (average SPEP 10.8 mg/ml). LC-MRM was able to identify increased expression in 18/19 patients with IgG disease. For SPEP+ patients with IgA disease, 18/20 could be detected by LC-MRM of the peptide, WLQGSQELPR. The relative quantification of the second peptide from IgA1detected elevated expression in one of these patients, but the total IgA expression was in the normal range for the other patient. When SPEP detects low levels the monoclonal immunoglobulin but the total immunoglobulin expression level was still in the reference range for healthy controls, both LC-MRM and nephelometry were unable to detect the disease. Longitudinal monitoring of these IgG and IgA patients may still indicate the presence of disease and increases in tumor burden. All 17SPEP+ IgM cases could be detected. All IgD (n=5) and IgE (n=1) patients were also correctly identified. Seven patients diagnosed with free light chain disease expressed no elevated concentrations of any heavy chain immunoglobulin. Additional details for the SPEP+ patients measured in the normal range by LC-MRM are included in Supplementary Table 1. LC-MRM measurements of immunoglobulins were in the normal range for all SPEP-samples. Samples from two patients with biclonal MM diagnoses made by SPEP/IFE were analyzed with LC-MRM (one IgG/IgM and one IgA/IgG). Both immunoglobulin chains were detected above the normal range for the IgA/IgG biclonal patient, but only the elevated IgM expression was detected in the IgM/IgG biclonal patient.
The LC-MRM heavy chain measurements correlated well to the values determined by nephelometry, the current clinical standard for quantification. Comparison of IgG measurements using the two methods is shown in Figure 1A. LC-MRM IgG data was compiled by summing the IgG1, IgG2, and IgG3 values. Because the IgG4 could not be quantified absolutely (due to poor synthesis of the SIS peptide), that value is excluded, and LC-MRM measurements should be slightly less than those from nephelometry (i.e. slope < 1). This calculation may also limit the correlation (R2 = 0.82). Correlation was poorer in patients with high IgG expression levels, perhaps due to LC-MRM saturation from nanoLC column loading limits. Removal of the samples with saturated IgG detection increased the correlation between LC-MRM and nephelometry (R2 = 0.98). IgA and IgM values were also highly correlated (Figure 1B and Figure 1C). In addition, both LC-MRM and nephelometry measurements were compared to SPEP data (Supplementary Figure S3). As expected, LC-MRM and nephelometry correlate better with each other than with SPEP, due to the fact that they both measure the total immunoglobulin rather than the disease-specific M-protein.
Figure 1. Correlation of Immunoglobulin Measurements by Nephelometry and LC-MRM Quantification of Peptides Selected from the Constant Region of Each Immunoglobulin.

Data from patients whose MM secrete each immunoglobulin (black diamonds) and from other types of patients (open circles) are plotted for IgG (A), IgA (B), and IgM (C) measurements. LC-MRM could also quantify immunoglobulin expression levels that were not detectable by nephelometry; data are presented for IgG (D), IgA (E), and IgM (F).
LC-MRM has better sensitivity than nephelometry, but has slightly poorer precision. LC-MRM was able to quantify immunoglobulin concentrations in patients when nephelometry reported that the values were below the stated limits of quantification: 2.71 mg/ml IgG, 0.41 mg/ml IgA, and 0.26 mg/ml IgM. Data are shown for IgG (n = 11) in Figure 1D, IgA (n = 29) in Figure 1E, and IgM (n = 34) in Figure 1F. This improvement in sensitivity may enable better evaluation of immune paresis, reduction of the population of other plasma cells due to the clonal expansion of the tumor cells, which could have a bearing on patient prognosis. Intra-assay and inter-assay CV values are approximately 5% and 10% for nephelometry [48,49]; LC-MRM intra-assay CV values are competitive, but inter-assay CV values are ~2-fold higher due to the additional processing steps (Table 1). Batch-to-batch variability in LC-MRM measurements of heavy chain expression is shown in Supplementary Figure S4.
LC-MRM of constant region peptides also enables the quantification of individual immunoglobulin heavy chain isoforms, which builds on existing clinical methods. This additional information could enable more sensitive detection of disease for patients with MM tumors that secrete lower abundance isoforms (e.g. IgG4). In order to assess the value of this additional level of detail provided by LC-MRM, the expression levels of each immunoglobulin were compared between the patients expressing each monoclonal immunoglobulin and those with other types of PCDs. Box plots are used for visualization (Figure 2), and statistical results are included in Table 2. In Figure 2A, the IgG patients are compared to other samples (as described above). IgA patients were differentiated using a peptide representing total IgA, WLQGSQELPR (Figure 2B). IgM levels were detected using the peptide, DGFFGNPR (Figure 2C). Box plots are not shown for IgD or IgE, because of the low sample size of those patients and limited detection of those endogenous peptides. As expected, statistical significance improves as the normal range of protein expression decreases; in other words, quantification of total immunoglobulin levels is more effective at detecting disease when the background levels of that immunoglobulin are lower. Therefore, LC-MRM detection of isoforms can further separate IgG patients (defined by serial SPEP/IFE measurements) into IgG1 (n = 11), IgG2 (n = 2), IgG3 (n = 4), and IgG4 (n = 1) based on the elevation of isoform-specific peptides. Two examples are shown in box plots in Figure 2; statistical results are included in Table 2 to indicate the increased separation of the groups and subsequently better detection of elevated immunoglobulin levels for IgG isoforms with lower total expression. Patients identified by LC-MRM with IgG1 disease (n = 11) had significantly higher IgG1 expression levels than other patients (Figure 2D). Patients with IgG3 disease (n = 4) had significantly higher levels of that isoform than other patients (Figure 2E). Due to insufficient sample size, data are not plotted for IgG2 and IgG4. IgA patients could be separated by isoforms into IgA1 and IgA2, but no IgA2 patients were detected in this cohort.
Figure 2. Box plots Comparing Monoclonal Immunoglobulin Expression in Serum for MM Patients with IgG, IgA, and IgM Diagnoses to Other Patients.

Patients with positive diagnosis for specific immunoglobulin types, IgG (A), IgA (B), and IgM (C), are compared with other patients, including both patients not diagnosed with MM and patients diagnosed with MM that express other types of monoclonal immunoglobulins. Data are not included for IgD (n = 5) and IgE (n = 1), because the separation is clear between those patients and the rest of the population. Current clinical measurements are not able to determine the expression of IgG (or IgA) isoforms, but specific peptides from each sequence can be monitored by LC-MRM to differentiate the isoforms of these heavy chains. Patients with each IgG subtype, IgG1 (D) and IgG3 (E) are compared with all other patients in the study. Data are not included for IgA2 (n = 0), IgG2 (n = 2), or IgG4 (n = 1), because the separation is clear between the patients expressing those isoforms and the rest of the population. In each plot, dashed lines indicate the maximum normal immunoglobulin level.
Table 2. Statistical Evaluation of LC-MRM Performance in Separating Patient Groups.
Each immunoglobulin measurement is compared between the population of SPEP positive patients and patients with other diagnoses. For isoform-specific measurements, the diagnosis of the involved immunoglobulin is achieved solely by LC-MRM. In each case, the number of patients as well as the mean, standard deviation, and the median of the protein expression are listed.
| Monitored Protein(s) |
Corresponding Ig Diagnosis |
Other Diagnoses | Statistical Results |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | Mean | SD | Median | n | Mean | SD | Median | z- value |
p- value |
|
| IgA1,2 | 21 | 14.83 | 16.53 | 6.20 | 62 | 0.66 | 0.61 | 0.46 | 5.10 | 3.5E−7 |
| IgA2 | 21 | 0.03 | 0.04 | 0.01 | 62 | 0.08 | 0.18 | 0.03 | −1.36 | 0.17 |
| IgG1,2,3 | 21 | 16.59 | 18.57 | 12.40 | 62 | 4.26 | 3.06 | 3.78 | 4.92 | 8.8E−7 |
| IgG1 | 10 | 15.19 | 5.38 | 13.74 | 73 | 2.40 | 1.91 | 1.81 | 5.09 | 3.7E−7 |
| IgG3 | 4 | 30.95 | 41.11 | 17.62 | 79 | 0.66 | 0.60 | 0.53 | 2.24 | 0.02 |
| IgM | 18 | 7.26 | 9.05 | 3.69 | 65 | 0.10 | 0.10 | 0.06 | 6.45 | 1.4E−10 |
| κ LC | 36 | 11.99 | 11.09 | 8.83 | 47 | 2.37 | 1.72 | 2.04 | 6.16 | 7.2E−10 |
| λ LC | 36 | 12.00 | 30.00 | 1.75 | 47 | 0.44 | 0.63 | 0.22 | 5.66 | 1.6E−8 |
| IgD | 5 | 6.57 | 8.37 | 2.30 | 78 | 0.00 | 0.03 | 0.00 | 7.23 | 4.8E−13 |
Statistical significance was assessed by the Holm-Bonferroni method; z-values and p-values are listed.
This additional information has the potential to improve patient monitoring, especially in cases of IgG MM where some isoforms are naturally high in abundance (such as IgG1 and IgG2) and others are significantly lower (IgG3 and IgG4). The patient’s total IgG concentration may fall within the reference range for healthy controls (4.5 – 20 mg/ml), but a single isoform can be detected by LC-MRM and shown to be significantly overexpressed. For example, the total IgG was measured to be within normal limits for three patients, but elevated levels of IgG3 were detected with LC-MRM. This additional capability enables more sensitive detection of disease, and it also may implicate involvement of multiple immunoglobulins, which could indicate another clonal tumor population. As an example, 3 patients were found by LC-MRM quantification to have elevated IgG3 levels that were previously undetected (two in IgM patients and one in a patient not presenting with MM).
LC-MRM Quantification of Light Chains using Constant Region Peptides
In addition to monitoring heavy chains and their isoforms, LC-MRM was used to measure constant regions peptides as surrogates for the expression of the light chains, κ and λ, and to calculate a κ:λ ratio. Assays were developed for TVAAPSVFIFPPSDEQLK (κ) and AGVETTTPSK (λ). These measurements could be used to detect dysregulation of light chain expression not only as overexpression but also by the κ:λ ratio. LC-MRM data was not compared to the results of SFLC assays, because the values are expected to differ (due to the comparison of total light chain by LC-MRM to free light chain by SFLC). However, LC-MRM was effective in detecting light chain-only disease in all cases (4/4). Patients with light chain-only disease that could not be monitored by SFLC were not included in this study, so it is unclear if LC-MRM would have utility there.
To evaluate the separation of patients with κ and λ diagnoses by LC-MRM, box plots are shown in Figure 3A and Figure 3B, respectively. Statistical results are listed in Table 2. Protein expression levels in patients diagnosed with elevated κ were significantly higher than in other patients; λ levels were also significantly different in λ patients when compared to other patients. The κ:λ ratio values have been plotted in log-scale (Figure 3C). Data from non-MM patients and those expressing biclonal light chains (i.e. both κ and λ) were not included, nor were samples (n = 5) where only one light chain could be quantified. Out of the 30 κ samples, only one patient was found to have a lower than expected κ:λ ratio (the reference range for total κ:λ in healthy controls is 1.3–2.5, which is significantly different from the reference values for the free light chain ratio, 0.26–1.65)[50], and one patient had a ratio within the normal range. Out of the 30 λ patients, two samples were found to have κ:λ ratios above the normal range, and two samples fell within the normal limits. Additional discussion on the characteristics of these patients is included in the supplement.
Figure 3. LC-MRM Analysis of Total Light Chain Expression in Serum.

Box plots are shown to compare patients expressing kappa light chain in their monoclonal to other patients (A) and those expressing lambda light chain to other patients (B). Average κ:λ ratios determined from triplicate LC-MRM measurements are also shown for each patient (C), with the exception of 5 patients with κ monoclonals that produced detectable, but not quantifiable, amounts of λ light chain. The normal ratio for total expression of κ:λ light chains in healthy adults (1.3–2.5) [50] is outlined by dotted lines in the inset (C); three patients with low monoclonal immunoglobulin expression had κ:λ ratios on the opposite side of the normal range from the light chain indicated in their diagnosis. In the inset, the numbers are included for reference to the supplemental patient information.
Longitudinal Patient Monitoring
An IgE MM patient was monitored using LC-MRM for comparison with existing protein-based methods (Figure 4); total IgE quantification is more sensitive for disease detection than SPEP due to the low levels of background IgE expression (at µg/ml levels). Serum samples were acquired at diagnosis and each time the patient received treatment. Initially, LC-MRM and SPEP show a distinct M-protein correlating to IgE myeloma, assigned by IFE to confirm the SPEP diagnosis and by endogenous IgE peptide intensity in LC-MRM (Figure 4A). However, analyses during the course of treatment illustrate the similarity of nephelometry and LC-MRM for monitoring IgE. SPEP results show no elevated IgE protein in the serum after the fourth and fifth cycles of treatment at 15and19 weeks after diagnosis (Figure 4B and 4C); nephelometry and LC-MRM were still able to detect elevated IgE levels at those time points (Figure 4D). Compared with SPEP, nephelometry and LC-MRM can better characterize the disease in this IgE patient, demonstrating another decrease in tumor burden after the fifth cycle of treatment. After the sixth cycle of treatment and during the subsequent follow–up, neither method could detect IgE. Based on the clinical evaluation (and not LC-MRM data), treatment was discontinued after six cycles, and the patient has been in remission.
Figure 4. LC-MRM and Nephelometry Have Similar Sensitivity for Detection of Treatment Response in an IgE MM Patient.
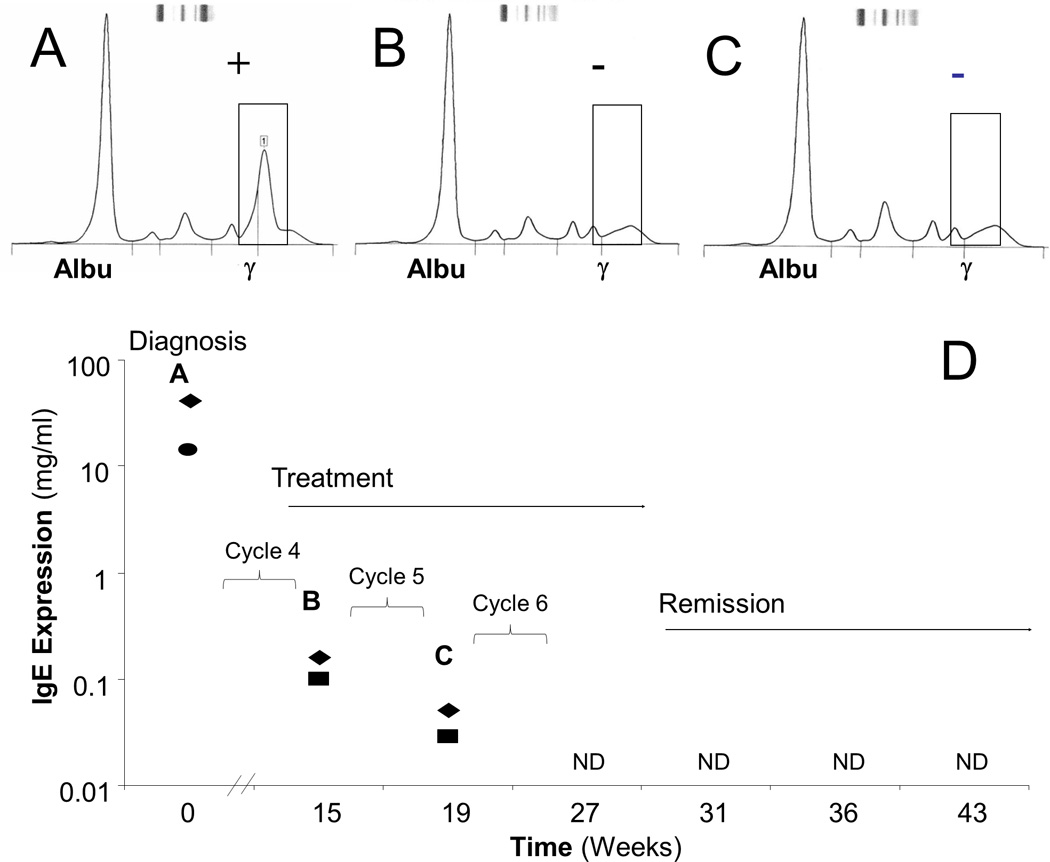
The patient was monitored over time with SPEP (A–C) as well as nephelometry and LC-MRM (D) starting at the time of diagnosis and continuing through the treatment regimen of a clinical trial combining a proteasome inhibitor and an immunomodulator. Both SPEP (circle) and LC-MRM (diamonds) results show IgE elevation at diagnosis (A). During treatment (B and C), the SPEP is negative, but elevated IgE is still detected by nephelometry (squares) and LC-MRM (D, letters A–C indicate quantification from the same serum samples as the SPEP data panels above) are still able to detect elevated IgE. Nephelometry and LC-MRM can observe the decrease in tumor burden after the 5th cycle of treatment, and IgE is not detected (ND) in the patient serum by either method after the 6th cycle of therapy. Tumor burden is reduced by three orders of magnitude, before the immunoglobulin is no longer detected.
Detection of Variable Region Peptides for M-Protein Quantification
LC-MRM of constant region peptides has similar performance for detection of disease with similar intra-assay variability but higher inter-assay variability as compared with current immunoglobulin quantification techniques. One way to improve the LC-MRM method is to define the sequence of the variable region, which could lead to detection of the specific monoclonal immunoglobulin, similar to SPEP. In a proof-of-concept experiment, de novo assembly was performed from RNA-seq data to determine the Ig sequence secreted by the H929 cell line (Figure S5). Although NCBI ORF Finder [47] identified several potential constructs, a single protein sequence had the expected size and the expected constant region primary amino acid sequence (Figure 5A). This amino acid sequence matched that expected from a translation of the IGKC, IGKJ1, and IGKV3–15 regions. Differences at the junctions were detected as can be expected during Ig recombination: proline was inserted between IGKV3–15 and IGKJ1, and arginine was inserted between IGKJ1 and IGKC. For the Igκ secreted by H929 cells, three constant region peptides (C) and 6 variable region peptides (V) could be detected by LC-MRM (Figure 5C). The peptides are: C1: VDNALQSGNSQESVTEQDSK, C2: DSTYSLSSTLTLSK, C3: TVAAPSVFIFPPSDEQLK, V1: ATGIPAR, V2: VTLSCR, V3: LSVSPGER, V4: LL(ox)MYDASTR, V5: ASQSVSSHLVWYQQKPGQAPR, and V6: LLMYDASTR; the identity of each peptide was verified by comparison of RT and fragmentation with LC-MS/MS and standards. Because the specific amino acid sequences of the variable region peptides from monoclonal immunoglobulin may be contained in other antibodies secreted by different cells, the quantification of the disease-specific VRPs will be limited by the amount of the peptide produced from the background of immunoglobulins secreted by normal plasma cells. Detection of the Igκ immunoglobulin from H929 cells by LC-MRM of the LSVSPGER sequence in a background of serum from a healthy control could be accomplished over more than two orders of magnitude and down to a spiked level of 0.6 fmol/µl of serum, which corresponds to detection of 0.0015 g/dl spiked H929 kappa immunoglobulin above the background for that peptide in the control serum (Figure 5D). This value represents ~67-fold improvement over the SPEP detection limit at 0.1 g/dl.
Figure 5. RNA-Seq, Protein Sequence, and LC-MRM Verification of Constant and Variable Region Peptides for H929 MM Cells.
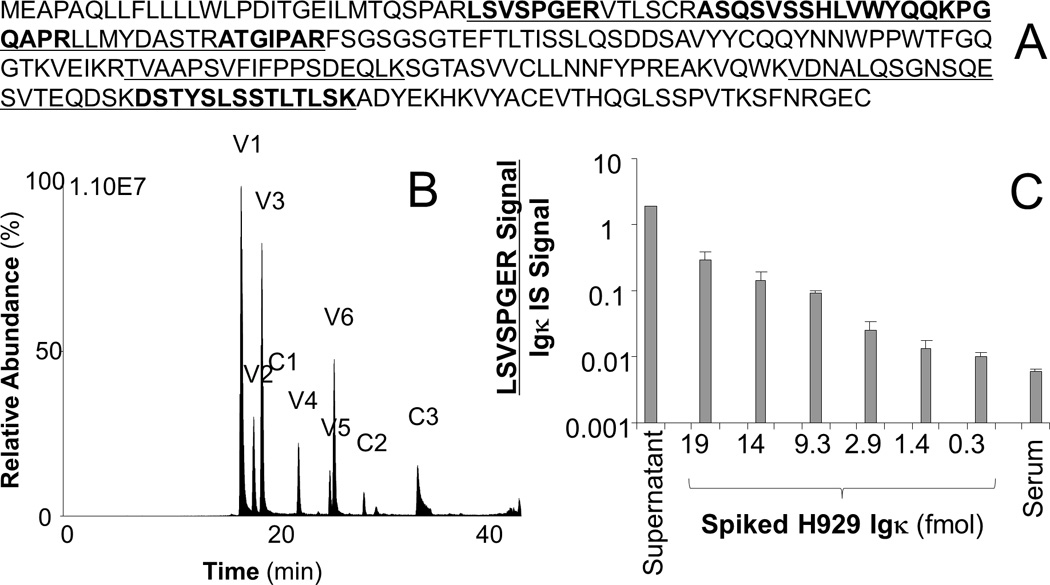
RNA-seq and in silico translation produced a protein sequence (A), which was used to generate tryptic peptides for monitoring (underlined sequences). The peptides from the variable and constant regions are alternately placed in bold font to show their sequences. LC-MRM could detect three constant (C1–C3) and six variable region (V1–V6) peptides from digests of conditioned media (B). Peptide sequences are provided again in the text. Spiked H929 Igκ could also be detected in control serum (C).
In addition, RNA-sequencing and in silico translation also provided the protein sequences of two κ immunoglobulins from patients. Both patients are currently in remission, but serum samples were collected and analyzed with LC-MRM for variable region peptides. The first patient was IgA/κ with a current M-protein measurement by SPEP of 0.2 g/dl, which is below the 0.5–1 g/dl level used to initiate treatment for relapse; the second patient has IgG/κ disease with no detectable M-protein at the time the sample was taken. Therefore, three peptides from the κ light chain of Patient 1 were tested in both patients to see the specificity of assays for the variable region peptides (Figure 6). In all cases, the level of each variable region peptide was at least 1000-fold higher in Patient 1 than in Patient 2. While these data are promising, the value of each individual peptide would have to be evaluated in time course measurements in the individual patient. Based on these preliminary data, this approach could generate personalized assays for detection of tumor burden, though selection of unique peptides with suitable characteristics for LC-MRM quantification may prove challenging (see sequence alignments in FiguresS5 and S6 and further discussion in supplement).
Figure 6. Detection of Peptides from the Disease-Specific Igκ Light Chain.
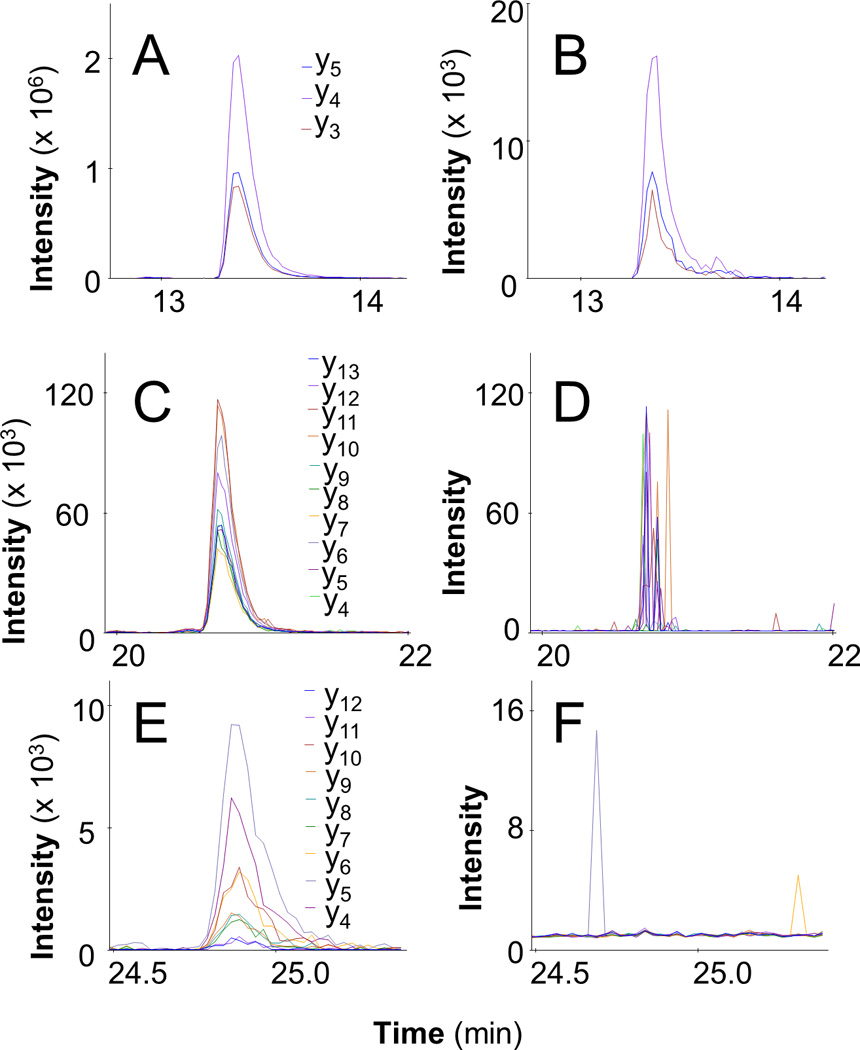
Three peptides derived from RNA-seq and κ light chain assembly using tumor cells from Patient 1 were observed with LC-MRM in a serum sample with 0.2g/dl M-protein: VTITCR (A), SLIYAASSLQSGVPSK (B), and ANQDITNSLVWFQQK (C). Data are provided from Patient 2 to illustrate the uniqueness of the three peptides (B, D, and F, respectively).
Conclusions
Using reference ranges for current protein-based assays, LC-MRM quantification of immunoglobulins was compared against current clinical methods using a sample size of 83 patients. While the LC-MRM assay is slower to run (overnight versus a few minutes), the therapeutic window for these patients is long, so treatment decisions do not have to be made immediately and the additional time required for this assay is not detrimental to the patients [5]. The ability of LC-MRM to monitor all immunoglobulins (and albumin as well as other proteins) allows for an increase in efficiency, with MM immunoglobulin and isoform quantification accomplished with one experiment (and one instrument). The LC-MRM method may improve analysis of the rarer MM types (IgD, IgE, and light chain only disease) that currently require multiple tests for monitoring. Peptide-based LC-MRM quantification of total immunoglobulin expression levels offers slight improvements in sensitivity over nephelometry, as well as the ability to quantify isoforms, with a trade-off in precision (~2-fold higher CV values). For higher abundance immunoglobins (e.g. IgG1), the precision is the critical variable for detection of disease because of the background level of protein expression, which is a disadvantage for LC-MRM. For lower abundance isoforms and other immunoglobulins (e.g. IgD or IgE), an increase in sensitivity would be critical to detect low levels of tumor burden during treatment and relapse. The increase in sensitivity can also be used to monitor the amount of immune paresis (reduction in other immunoglobulin levels due to reduction of normal plasma cells from the bone marrow by the growth of the tumor), but this parameter is not currently factored into patient treatment decisions.
To examine the robustness of these assays, triplicate measurements were analyzed for outliers, which were observed at levels of 6.4% or less; some peptides did not have any outliers detected. Reduction of CV values and elimination of outliers will be critical for clinical implementation without triplicate analysis. Automation of sample preparation may provide improvements in precision, and injection of larger sample amounts for analytical scale chromatography would be expected to improve on the nanoLC analysis here by eliminating saturation effects, improving precision, and eliminating outliers. If technology shifts to mass spectrometry-based methods, this set of assays could be clinically utilized. In order to pursue its implementation, a large cohort of healthy controls would need to be analyzed to define the appropriate reference ranges for peptide-based LC-MRM assays. Furthermore, additional data should be collected for longitudinal patient monitoring to define the levels of natural variation in immunoglobulin abundance in serum and to determine the threshold for increases in immunoglobulin level that should trigger additional treatment.
Finally, the determination of variable region sequences and the resulting tryptic peptides is a promising method for improving patient monitoring with quantitative mass spectrometry. Although this requires an initial RNA-seq experiment to identify the highly expressed rearrangements, these transcripts can be identified with as few as 20 million RNA-seq reads (see supplement) due to the high expression of Ig in multiple myeloma tumor cells. This method allows for a highly specific tracking of the disease-specific monoclonal protein. This personalized approach could offer a better understanding of the unique aspects of an individual patient’s disease, but the effectiveness of the assay would vary between patients because of the sequence-specific performance of the variable region peptides and the patient-specific background of other immunoglobulins with the same sequence.
We envision an assay platform that measures both constant region peptides for all immunoglobulins and variable region peptides for the patient’s disease-specific immunoglobulin, which could significantly improve on sensitivity. The detection of the disease-specific biomarker is then paired with the ability to detect changes in the other immunoglobulins, which could indicate immune paresis (describing disease severity) or expansion of another MM clone that secretes a different immunoglobulin (i.e. relapse with a different MM tumor). This platform would be of use in monitoring depth of response to therapy, which has prognostic value, and determining disease progression either from premalignant conditions (e.g. MGUS) or in relapse after treatment. Improvements in the ability to quantify the tumor burden are likely to shape novel clinical approaches to MM treatment.
Supplementary Material
Statement of Clinical Relevance.
Because plasma cells secrete immunoglobulins, uncontrolled replication of clonal population of plasma cells creates an elevated concentration of a monoclonal immunoglobulin, which is used as a biomarker of disease burden for a patient with a plasma cell dyscrasia, including multiple myeloma (MM). Detection of increasingly elevated immunoglobulin levels in serum and/or urine triggers additional patient assessment to establish a MM diagnosis. For ongoing monitoring, serum and urine immunoglobulin measurements are used at intervals of 2 to 12 weeks to assess the level of disease to measure therapeutic response and detect disease recurrence. Currently, serum protein electrophoresis, immunofixation, nephelometry, and the serum free light chain assay are used to detect, type, and quantify the monoclonal immunoglobulin involved with each patient’s disease. Liquid chromatography-multiple reaction monitoring mass spectrometry assays for immunoglobulin quantification using peptides from the constant regions have been applied to patient samples to enable comparison with current clinical techniques. Finally, proof-of-principle experiments are shown for personalized detection of tumor burden using RNA sequencing of the monoclonal immunoglobulin and LC-MRM detection of peptides from the variable region.
Statement of Novelty.
Liquid chromatography-multiple reaction monitoring mass spectrometry has been used to quantify immunoglobulin expression in the serum of controls and patients with plasma cell dyscrasias (e.g. MM) in order to compare peptide-based quantification against protein-based measurements currently employed in the clinic. In addition, RNA-sequencing has been used to enable development of disease-specific assays that have the potential to significantly improve on current clinical methods.
Acknowledgments
The authors would like to thank Larry Dangott and Ginny Johnson at the Texas A&M University Protein Chemistry Laboratory for amino acid analysis, enabling absolute quantification of the peptide standards. Patients’ samples and medical data were collected and processed using protocols approved by the Institutional Review Board at the University of South Florida.
Grant Support
This research is supported by the National Cancer Institute (R21-CA141285). The Moffitt Proteomics Core Facility is supported by the US Army Medical Research and Materiel Command under Award No. DAMD17-02-2-0051, continuing as W81XWH-08-2-0101, for a National Functional Genomics Center, the National Cancer Institute under Award No. P30-CA076292 as a Cancer Center Support Grant, and the Moffitt Foundation. The triple quadrupole mass spectrometers were purchased with shared instrument grants from the Bankhead-Coley Cancer Research program of the Florida Department of Health (06BS-02-9614 and 09BE-04).
Abbreviations
- IFE
immunofixation electrophoresis
- Ig
immunoglobulin
- MGUS
monoclonal gammopathy of undetermined significance
- MM
multiple myeloma
- PCD
plasma cell dyscrasia
- SFLC
serum free light chain assay
- SPEP
serum protein electrophoresis
Footnotes
Conflict of Interest: A provisional patent for the methods for measuring constant region peptides to quantify immunoglobulin expression with reaction monitoring mass spectrometry has been issued (to EW, MH, KB, and JK).
References
- 1.Greipp PR, San Miguel J, Durie BG, Crowley JJ, et al. International staging system for multiple myeloma. J Clin Oncol. 2005;23:3412–3420. doi: 10.1200/JCO.2005.04.242. [DOI] [PubMed] [Google Scholar]
- 2.International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121:749–757. [PubMed] [Google Scholar]
- 3.Durie BG, Salmon SE. A clinical staging system for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer. 1975;36:842–854. doi: 10.1002/1097-0142(197509)36:3<842::aid-cncr2820360303>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 4.Durie BG, Harousseau JL, Miguel JS, Bladé J, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20:1467–1473. doi: 10.1038/sj.leu.2404284. [DOI] [PubMed] [Google Scholar]
- 5.Koomen JM, Haura EB, Bepler G, Sutphen R, et al. Proteomic contributions to personalized cancer care. Mol Cell Proteomics. 2008;7:1780–1794. doi: 10.1074/mcp.R800002-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berth M, Delanghem J, Langlois M, Buyzere M. Reference values of serum IgA subclasses in caucasian adults by immuno nephelometry. Clin Chem. 1999;45:309–310. [PubMed] [Google Scholar]
- 7.French MAH, Harrison G. Serum IgG subclass concentrations in healthy adults: a study using monoclonal antisera. Clin ExpImmunol. 1984;56:473–475. [PMC free article] [PubMed] [Google Scholar]
- 8.Haraldsson A, Kock-Jansen M, Jaminon M, van Eck-Arts PB, et al. Determination of kappa and lambda light chains in serum immunoglobulins G, A and M. Ann Clin Biochem. 1991;28:461–466. doi: 10.1177/000456329102800507. [DOI] [PubMed] [Google Scholar]
- 9.Chen K, Jan Y, Chen C, Cheng C, et al. Multiple myeloma-associated cast nephropathy with crystal structure: Case report and review of the literature. Nephrology. 2005;10:594–596. doi: 10.1111/j.1440-1797.2005.00499.x. [DOI] [PubMed] [Google Scholar]
- 10.Buckley R, Fiscus S. Serum IgD and IgE concentrations in immunodeficiency diseases. J Clin Invest. 1975;55:157–165. doi: 10.1172/JCI107906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunder G, Gleich G. Immunoglobulin E (IgE) Levels in serum and synovial fluid in rheumatiod arthritis. Arthritis Rheum. 1974;17:955–963. doi: 10.1002/art.1780170606. [DOI] [PubMed] [Google Scholar]
- 12.O’Connell TX, Horita TJ, Kasravi B. Understanding and interpreting serum protein electrophoresis. Am Fam Phys. 2005;71:105–112. [PubMed] [Google Scholar]
- 13.Jolliff CR, Blessum CR. Comparison of serum protein electrophoresis by agarose gel and capillary zone electrophoresis in a clinical setting. Electrophoresis. 1997;18:1781–1784. doi: 10.1002/elps.1150181012. [DOI] [PubMed] [Google Scholar]
- 14.Morita K, Okamoto Y. The discrepancy between electrophoretic and nephelometric determinations of serum gamma globulin level. Clin Lab. 2004;50:415–418. [PubMed] [Google Scholar]
- 15.Mussap M, Pietrogrande F, Ponchia S, Stefani PM, et al. Measurement of serum monoclonal components: comparison between densitometry and capillary zone electrophoresis. Clin Chem Lab Med. 2006;44:609–611. doi: 10.1515/CCLM.2006.112. [DOI] [PubMed] [Google Scholar]
- 16.Schreiber WE, Chiang E, Tse SS. Electrophoresis underestimates the concentration of polyclonal immunoglobulins in serum. Am J Clin Pathol. 1992;97:610–613. doi: 10.1093/ajcp/97.5.610. [DOI] [PubMed] [Google Scholar]
- 17.Clark R, Katzmann JA, Kyle RA, Fleisher M, Landers JP. Differential diagnosis of gammopathies by capillary electrophoresis and immunosubtraction: analysis of serum samples problematic by agarose gel electrophoresis. Electrophoresis. 1998;19:2479–2484. doi: 10.1002/elps.1150191421. [DOI] [PubMed] [Google Scholar]
- 18.Jagannath S. Value of serum free light chain testing for the diagnosis and monitoring of monoclonal gammopathies in hematology. Clin Lymphoma Myeloma. 2007;7:518–523. doi: 10.3816/clm.2007.n.036. [DOI] [PubMed] [Google Scholar]
- 19.Tate JR, Mollee P, Dimeski G, Carter AC, Gill D. Analytical performance of serum free light-chain assay during monitoring of patients with monoclonal light-chain diseases. Clin Chim Acta. 2007;376:30–36. doi: 10.1016/j.cca.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 20.Pika T, Minarik J, Schneiderka P, Budikova M, et al. The correlation of serum immunoglobulin free light chain levels and selected biological markers in multiple myeloma. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2008;152:61–64. doi: 10.5507/bp.2008.009. [DOI] [PubMed] [Google Scholar]
- 21.Reimer CB, Maddison SE. Standardization of human imunoglobulin quantitation: A review of current status and problems. Clin Chem. 1976;22:577–582. [PubMed] [Google Scholar]
- 22.Barr JR, Maggio VL, Patterson DG, Jr, Cooper GR. Isotope dilution--mass spectrometric quantification of specific proteins: model application with apolipoprotein A-I. Clin Chem. 1996;42:1676–1682. [PubMed] [Google Scholar]
- 23.Barnidge DR, Dratz EA, Martin T, Bonilla LE, et al. Absolute quantification of the G protein-coupled receptor rhodopsin by LC/MS/MS using proteolysis product peptides and synthetic peptide standards. Anal Chem. 2003;75:445–451. doi: 10.1021/ac026154+. [DOI] [PubMed] [Google Scholar]
- 24.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci USA. 2003;100:6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5:573–588. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Kuzyk MA, Smith D, Yang J, Cross TJ, et al. Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol Cell Proteomics. 2009;8:1860–1877. doi: 10.1074/mcp.M800540-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keshishian H, Addona T, Burgess M, Kuhn E, Carr SA. Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol Cell Proteomics. 2007;6:2212–2229. doi: 10.1074/mcp.M700354-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirsch S, Widart J, Louette J, Focant JF, De Pauw E. Development of an absolute quantification method targeting growth hormone biomarkers using liquid chromatography coupled to isotope dilution mass spectrometry. J Chromatogr A. 2007;1153:300–306. doi: 10.1016/j.chroma.2007.03.058. [DOI] [PubMed] [Google Scholar]
- 29.Kuhn E, Wu J, Karl J, Liao H, et al. Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics. 2004;4:1175–1186. doi: 10.1002/pmic.200300670. [DOI] [PubMed] [Google Scholar]
- 30.Barnidge DR, Goodmanson MK, Klee GG, Muddiman DC. Absolute quantification of the model biomarker prostate-specific antigen in serum by LC-MS/MS using protein cleavage and isotope dilution mass spectrometry. J Proteome Res. 2004;3:644–652. doi: 10.1021/pr049963d. [DOI] [PubMed] [Google Scholar]
- 31.Yocum AK, Khan AP, Zhao R, Chinnaiyan AM. Development of selected reaction monitoring-MS methodology to measure peptide biomarkers in prostate cancer. Proteomics. 2010;10:3506–3514. doi: 10.1002/pmic.201000023. [DOI] [PubMed] [Google Scholar]
- 32.Addona TA, Abbatiello SE, Schilling B, Skates SJ, et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol. 2009;27:633–641. doi: 10.1038/nbt.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prakash A, Rezai T, Krastins B, Sarracino D, et al. Platform for establishing interlaboratory reproducibility of selected reaction monitoring-based mass spectrometry peptide assays. J Proteome Res. 2010;9:6678–6688. doi: 10.1021/pr100821m. [DOI] [PubMed] [Google Scholar]
- 34.Kuhn E, Addona T, Keshishian H, Burgess M, et al. Developing multiplexed assays for troponin I and interleukin-33 in plasma by peptide immunoaffinity enrichment and targeted mass spectrometry. Clin Chem. 2009;55:1108–1117. doi: 10.1373/clinchem.2009.123935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agger SA, Marney LC, Hoofnagle AN. Simultaneous quantification of apolipoprotein A-I and apolipoprotein B by liquid-chromatography-multiple- reaction-monitoring mass spectrometry. Clin Chem. 2010;56:1804–1813. doi: 10.1373/clinchem.2010.152264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoofnagle AN, Becker JO, Wener MH, Heinecke JW. Quantification of thyroglobulin, a low-abundance serum protein, by immunoaffinity peptide enrichment and tandem mass spectrometry. Clin Chem. 2008;54:1796–1804. doi: 10.1373/clinchem.2008.109652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans VC, Barker G, Heesom KJ, Fan J, et al. De novo derivation of proteomes from transcriptomes for transcript and protein identification. Nat Methods. 2012;9:1207–1211. doi: 10.1038/nmeth.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Remily-Wood ER, Liu RZ, Xiang Y, Chen Y, et al. A database of reaction monitoring mass spectrometry assays for elucidating therapeutic response in cancer. Proteomics Clin Appl. 2011;5:383–396. doi: 10.1002/prca.201000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacLean B, Tomazela DM, Shulman N, Chambers M, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang G, Fang B, Liu RZ, Lin H, et al. Mass spectrometry mapping of epidermal growth factor receptor phosphorylation related to oncogenic mutations and tyrosine kinase inhibitor sensitivity. J Proteome Res. 2011;10:305–319. doi: 10.1021/pr1006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin DB, Holzman T, May D, Peterson A, et al. MRMer, an interactive open source and cross-platform system for data extraction and visualization of multiple reaction monitoring experiments. Mol Cell Proteomics. 2008;7:2270–2278. doi: 10.1074/mcp.M700504-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. http://genecruiserbroadinstituteorg/cancer/software/genepattern/ [Google Scholar]
- 43.Holm S. A simple sequentially rejective multiple test procedure. Scandinavian Journal of Statistics. 1979;6:65–70. [Google Scholar]
- 44.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grabherr MG, Haas BJ, Yassour M, Levin JZ, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi.
- 48.Alexander RL., Jr Comparison of radial immunodiffusion and laser Nephelometry for quantitating some serum proteins. Clin. Chem. 1980;26:314–317. [PubMed] [Google Scholar]
- 49.Guiguet M, Padieu P, Mack G. Laser nephelometric measurement of seven serum proteins compared with radial immunodiffusion. J. Clin. Chem. Biochem. 1983;21:217–221. doi: 10.1515/cclm.1983.21.4.217. [DOI] [PubMed] [Google Scholar]
- 50.Katzmann JA, Clark RJ, Abraham RS, Bryant S, et al. Serum reference intervals and diagnostic ranges for free κ and free λ immunoglobulin light chains: relative sensitivity for detection on monoclonal light chains. Clin Chem. 2002;48:1437–1444. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.