

Abstract
Background
Mutations affecting the Na+/ K+ATPase (a.k.a. the sodium-potassium pump) genes cause conditional locomotor phenotypes in flies and three distinct complex neurological diseases in humans. More than 50 mutations have been identified affecting the human ATP1A2 and ATP1A3 genes that are known to cause rapid-onset Dystonia Parkinsonism, familial hemiplegic migraine, alternating hemiplegia of childhood, and variants of familial hemiplegic migraine with neurological complications including seizures and various mood disorders. In flies, mutations affecting the ATPalpha gene have dramatic phenotypes including altered longevity, neural dysfunction, neurodegeneration, myodegeneration, and striking locomotor impairment. Locomotor defects can manifest as conditional bang-sensitive (BS) or temperature-sensitive (TS) paralysis: phenotypes well-suited for genetic screening.
Results
We performed a genome-wide deficiency screen using three distinct missense alleles of ATPalpha and conditional locomotor function assays to identify novel modifier loci. A secondary screen confirmed allele-specificity of the interactions and many of the interactions were mapped to single genes and subsequently validated. We successfully identified 64 modifier loci and used classical mutations and RNAi to confirm 50 single gene interactions. The genes identified include those with known function, several with unknown function or that were otherwise uncharacterized, and many loci with no described association with locomotor or Na+/K+ ATPase function.
Conclusions
We used an unbiased genome-wide screen to find regions of the genome containing elements important for genetic modulation of ATPalpha dysfunction. We have identified many critical regions and narrowed several of these to single genes. These data demonstrate there are many loci capable of modifying ATPalpha dysfunction, which may provide the basis for modifying migraine, locomotor and seizure dysfunction in animals.
Electronic supplementary material
The online version of this article (doi:10.1186/s13041-014-0089-3) contains supplementary material, which is available to authorized users.
Keywords: Drosophila melanogaster, ATPalpha, Sodium pump, Temperature-sensitive paralysis, Conditional paralysis, Seizure, Migrane, Screen, Genome-wide, Seizure suppressor
Background
In many organisms, highly conserved Na+/K+ ATPases are responsible for maintaining ion gradients across the plasma membrane through ATP-dependent asymmetric translocation of Na+ and K+ ions. These ion gradients maintain the resting potential of cells, which facilitates neural signaling and many essential secondary processes. Mature Na+/K+ ATPase complexes are heteromultimers of alpha, beta, and gamma subunits in mammals. Flies express only the alpha and beta subunits, the former of which is known as ATPalpha. Like its mammalian homologue, ATPalpha contains ten transmembrane domains and has the ATP-dependent catalytic activity essential for pump function [1-3].
Mutations affecting the alpha subunit of the Na+/K+ ATPase in humans are associated with at least three human diseases: Rapid-onset Dystonia Parkinsonism (RDP), Familial Hemiplegic Migraine (FHM), and Alternating Hemiplegia of Childhood (AHC; [4]). RDP is a severe DOPA non-responsive form of dystonia the etiology of which is poorly understood [5]. FHM, possibly the most severe form of migraine, is associated with a debilitating partial paralysis, and currently is largely untreatable [6]. AHC is a severe childhood locomotor disease associated with recurring acute bouts of paralysis and muscle weakness, and general developmental delays (reviewed by [7]). Recently Sasaki and colleagues have described several children who seem to have a disease intermediate to AHC and RDP [8]. All of these diseases are complex neuromuscular conditions associated with marked locomotor dysfunction and for which the underlying pathogenesis is poorly understood.
Drosophila conditional mutants have been isolated based upon temperature-sensitive (TS) or bang-sensitive (BS) paralysis phenotypes over the past many decades. TS mutants generally become paralyzed in less than five minutes at 38°C and BS mutants paralyze in response to 20 seconds of mechanical stress. These classes of mutants have proven informative and have defined many essential components of neural signaling [9-15]. Conditional TS mutations typically affect critical neural proteins and include well-studied genes such as para (voltage-dependent NaCH), NapTS (RNA helicase affecting para transcripts), cacophony (a voltage-gated calcium channel), ATPalpha (Na+/K+ ATPase), comatose (dNSF1), shibire (Dynamin), syntaxin, synaptobrevin, and dao (regulator of Erg-type K-channels), to name a few [15-24]. Conditional BS mutations can also affect important neural signaling and ion homeostasis proteins, such as para and ATPalpha [23,25]. They also affect many proteins with integral roles in bioenergetics and mitochondrial function, such as sesB, ATP6, kdn, eas, and SOD2 [26-30]. Interestingly, numerous BS mutants have been shown to exhibit seizures and model epilepsies (e.g. paraBSS1, ATP61, and Kazachoc; [25,31,32]). BS and TS conditional mutants have proven incredibly important to our understanding of neurobiology and previous studies have successfully used them to identify genes that modify these behaviors (e.g. [33-35]). However, there are no reports of genome-wide screens for modifier loci using these behavioral phenotypes in Drosophila or studying ATPalpha in any model system. This suggests that such an approach could yield novel loci involved in regulating ion homeostasis or neural excitability.
It has previously been shown that mutations in ATPalpha result in profound neural and locomotor dysfunction in Drosophila [23,36-40]. Hypomorphic ATPalpha alleles, such as ATPalpha2206, display BS paralysis and phenocopy injection of the selective Na+/K+ ATPase inhibitor, ouabain [39]. The ATPalphaDTS1 mutation is a dominant, conditional, gain-of-function, missense mutation [23]. The mutation results in an E982K substitution near the protein’s C-terminus (short isoform numbering). ATPalphaDTS1 heterozygotes exhibit rapid paralysis at 38°C with complete penetrance. This is thought to be a result of conditional neuronal hyperexcitability caused by the mutation [23]. ATPalphaCJ5 and ATPalphaCJ10 are also dominant missense mutations affecting evolutionarily conserved amino acids [36]. However, they each exhibit unique locomotor phenotypes. ATPalphaCJ5 behaves like a loss-of-function allele of ATPalpha, exhibiting haploinsufficiency and BS paralysis [36]. ATPalphaCJ10 exhibits BS and progressive TS phenotypes, suggesting this is a loss-of-function allele that exhibits weak gain-of-function features, which are uncovered with age [36]. Thus, ATPalphaDTS1, ATPalphaCJ5, and ATPalphaCJ10 are all dominant, phenotypically well-characterized, and possibly functionally distinct, conditional locomotor mutants. Such alleles are ideally suited for a modifier screen. Using multiple alleles of ATPalpha increases the power of the screen and affords the likelihood of identifying allele-specific modifiers. Furthermore, to our knowledge, this is the first report of a genome wide genetic screen in any animal system using three distinct alleles of the same gene in parallel to identify allele-specific interactions.
Deficiency screens have been effectively used for elucidating novel gene interactions in Drosophila using various phenotypes [41-43]. Deficiency (Df) strains each have a unique deletion of a segment of the genome. Phenotypically screening for genetic interactions between defined point mutations and an individual defined deficiency is an efficient way to identify modifier loci. Using a collection of Dfs covering a high percentage of the genome (95-98%), one can identify critical modifier loci anywhere in the genome. This provides an efficient yet powerful and unbiased forward genetic approach. Critical loci can often be narrowed to single genes using smaller deficiencies and single gene disruptions. We have performed such a screen using ATPalphaDTS1, ATPalphaCJ5 and ATPalphaCJ10, identified 64 critical modifier intervals, and successfully confirmed 50 single-gene modifiers, including numerous novel loci of interest. These data suggest the existence of many susceptibility loci capable of modifying migraine, locomotor and seizure dysfunction in animals and provide a rich data set from which new targets for anti-migraine or anti-epileptic drugs could be drawn.
Results
Primary genetic modifier screen
To identify new genes that interact with ATPalpha we performed a deficiency screen using three characterized alleles: ATPalphaDTS1, ATPalphaCJ5 and ATPalphaCJ10. We used the Bloomington Stock Center deficiency (Df) kit that covers approximately 98% of the Drosophila genome. All of the 467 Df strains we received were tested with at least one ATPalpha mutant allele and the vast majority of strains were tested with multiple alleles (see Table 1). Each of the three ATPalpha mutants was mated to each Df line. F1 progeny bearing ATPalphaDTS1 and each deficiency were subjected to TS assays while progeny bearing ATPalphaCJ5 or ATPalphaCJ10 and each Df were assayed for BS. The average response for ATPalphaDTS1, ATPalphaCJ5 and ATPalphaCJ10Df double mutants was 34.8+/−25.3, 89.9+/−53.6 and 41.5+/−34.8 seconds, respectively (Additional file 1). We used these values to identify putative genetic interactions. Df(3R)BSC819 contains a deletion of the ATPalpha locus and failed to complement each mutant allele, as expected.
Table 1.
Primary screen summary
| DTS1 | CJ5 | CJ10 | |
|---|---|---|---|
| Number tested in primary screen | 386 | 393 | 358 |
| % of Kit tested | 83% | 84% | 77% |
| Avg. Response (Sec.) | 34 | 88 | 42 |
| St. Dev. (Sec.) | 26.4 | 74.6 | 46.1 |
| Normal Range (Sec.) | 20-60 | 20-190 | 10-150 |
| Number selected for verification | 89 | 69 | 78 |
| Screened phenotype | TS | BS | BS |
The data from the primary screen were organized graphically by average time to recovery or paralysis for each double mutant (Figure 1). In each case, the resulting data formed a largely normal distribution. Double mutants that deviated significantly from the mean were termed putative enhancers or suppressors and were tested again in a verification screen. The workflow for the genetic screen is depicted in Figure 2. In the primary screen, 1137 interactions were examined for the three conditional locomotor mutants identifying 117 putative enhancer, suppressor, or synthetic lethal regions. These interactions were examined further in the verification screen.
Figure 1.
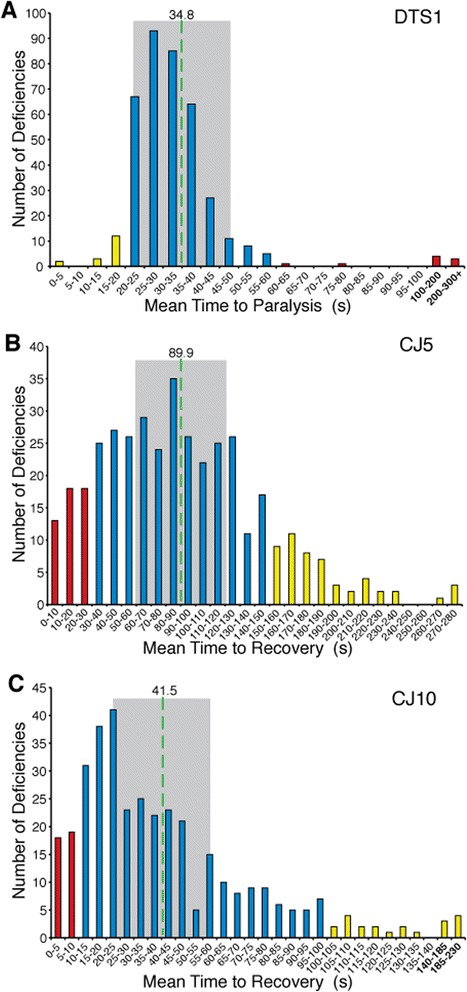
Distribution of phenotypic modifiers identified through a deficiency screen. A-C) ATPalpha mutant animals also bearing individual unique chromosomal deficiencies (Df) were assayed for conditional locomotor function to identify modifiers. The data reveal a largely normal distribution centered around a typical response (blue) for each mutant. Those deviating from the typical response were termed putative enhancers (yellow) or suppressors (red). A) ATPalpha CJ5 , Df double mutants and B) ATPalpha CJ10 , Df double mutants were assayed for recovery from mechanical stress at adult day 15. C) ATPalpha DTS1 , Df double mutants were assayed for time to TS paralysis on adult day 1. A-C) The mean response is shown as a dashed green line. +/− 0.5 Std. Dev. are indicated by gray shading.
Figure 2.
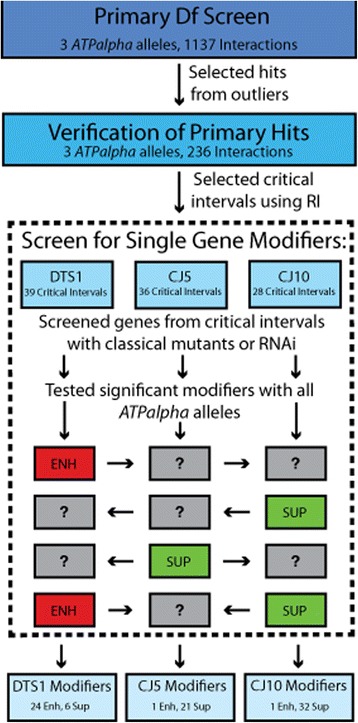
Schematic of the deficiency screen workflow. Using the Bloomington deficiency kit, 1137 initial interactions were screened using ATPalpha CJ5, ATPalpha CJ10, or ATPalpha DTS1. Putative enhancers and suppressors were selected for verification with a larger sample size. Any verified interacting deficiencies were deemed critical intervals. Once critical intervals were selected a screen for single gene modifiers from within the intervals was performed using available classical mutants and transgenic RNAi strains. If a modifier was found it was retested with other ATPalpha alleles to determine whether the interaction was allele-specific.
Verification screen
To mitigate the effect of false positives and confirm that interactions were reproducible before pursuing them further, we performed a verification screen (an independent experiment) with the putative modifiers. We began the verification with 89 ATPalphaDTS1 modifiers (Figure 1A), 69 ATPalphaCJ5 modifiers (Figure 1B), and 78 ATPalphaCJ10 modifiers (Figure 1C). After verification, we took advantage of having two data sets (primary and verification screen) and created a formula to determine the reproducibility of each putative genetic interaction (see Materials and Methods). We calculated a reproducibility index (RI) and used it to help us identify the most promising critical intervals. Dfs with the highest RIs were prioritized for mapping and secondary screening. This approach yielded 7 putative ATPalphaDTS1 enhancers, 12 suppressors, and five synthetic lethal (enhanced to lethality) combinations (Table 2). The ATPalphaCJ5 screen yielded 13 enhancers, 10 suppressors, and four synthetic lethal combinations (Table 3). The ATPalphaCJ10 primary screen yielded 17 enhancers, 11 suppressors, and one synthetic lethal (Table 4).
Table 2.
Confirmed ATPalpha DTS1 interacting deficiencies
| Df Name | Enh/Sup | Mean +/− SEM | Total N | RI | Hits in region | Coincidence |
|---|---|---|---|---|---|---|
| DTS1 | Control | 37.8 +/− 2.6 | 23 | - | - | - |
| Df(3 L)Exel6092 | Sup | 76.4 +/− 33.4 | 11 | 6.27 | spz5, FMRFaR, scramb2, aly | CJ10 |
| Df(2R)ED1725 | Sup | 209.8 +/− 57.0 | 6 | 4.87 | ||
| Df(2R)BSC361 | Sup | 87.8 +/− 10.6 | 16 | 4.68 | Stj | CJ10 |
| Df(3 L)BSC33 | Sup | 103.9 +/− 34.4 | 14 | 4.64 | ||
| Df(3 L)Exel8104 | Enh | 27.3 +/− 27.3 | 11 | 3.18 | ||
| Df(3R)BSC486 | Enh | 17.2 +/− 1.7 | 19 | 2.65 | CJ10 | |
| Df(2 L)BSC180 | Sup | 85.3 +/− 16.5 | 25 | 2.13 | Rbp9 | CJ10 |
| Df(3R)Exel6210 | Sup | 151.3 +/− 42.9 | 11 | 2.02 | ||
| Df(2R)BSC383 | Sup | 129.1 +/− 40.9 | 11 | 1.80 | ||
| Df(2 L)BSC278 | Sup | 52.3 +/− 14.9 | 25 | 1.54 | ||
| Df(3 L)BSC23 | Enh | 11.2 +/− 1.1 | 14 | 1.53 | spz5, scramb2, rasp, aly | CJ5, CJ10 |
| Df(2 L)Exel6005 | Sup | 73.9 +/− 23.1 | 19 | 1.51 | ||
| Df(3R)BSC650 | Enh | 22.1 +/− 2.3 | 13 | 1.20 | ||
| Df(2 L)ED1203 | Enh | 21.2 +/− 2.1 | 13 | 0.92 | Ham | CJ5 |
| Df(3R)ED2 | Enh | 20.0 +/− 2.6 | 25 | 0.88 | ||
| Df(2R)ED3728 | Sup | 46.3 +/− 8.2 | 15 | 0.86 | ||
| Df(1)BSC767 | Sup | 138.2 +/− 26.7 | 13 | 0.82 | ||
| Df(2R)M60E | Sup | 48.1 +/− 5.9 | 25 | 0.74 | Rpl19, pain | |
| Df(2 L)ED629 | Enh | 27.1 +/− 2.9 | 13 | 0.49 | Glutactin, sema-1a | |
| Df(3R)ED7665 | Enh/Leth | - | - | CJ10 | ||
| Df(3R)ED6361 | Enh/Leth | - | - | |||
| Df(3 L)BSC375 | Enh/Leth | - | - | |||
| Df(3R)BSC467 | Enh/Leth | - | - | CJ10 | ||
| Df(1)BSC708 | Enh/Leth | - | - | |||
| Df(3R)BSC819 | Enh/Leth | - | - | ATPalpha | All Enh/Leth |
Table 3.
Confirmed ATPalpha CJ5 interacting deficiencies
| Df Name | Enh/Sup | Mean ± SEM | Total N | RI | Hits in region | Coincidence |
|---|---|---|---|---|---|---|
| CJ5 | Control | 100.0 +/− 11.4 | 28 | - | - | - |
| Df(3 L)BSC797 | Enh | 240.6 +/− 17.3 | 14 | 4.03 | ||
| Df(2 L)BSC214 | Enh | 178.7 +/− 22.6 | 15 | 2.38 | ||
| Df(3 L)ED4475 | Enh | 172.4 +/− 21.4 | 16 | 1.97 | CJ10 | |
| Df(2 L)BSC781 | Sup | 16.2 +/− 4.2 | 25 | 1.93 | Cact, CG5888 | |
| Df(3R)BSC547 | Enh | 165.0 +/− 24.0 | 17 | 1.81 | Sro, Dop1r2, ppk21 | |
| Df(3 L)M21 | Enh | 182.5 +/− 26.2 | 13 | 1.80 | ||
| Df(2R)BSC199 | Enh | 168.8 +/− 24.2 | 14 | 1.63 | ||
| Df(3R)ED5495 | Enh | 182.0 +/− 24.0 | 16 | 1.62 | ||
| Df(2R)PK1 | Sup | 26.9 +/− 8.7 | 20 | 1.57 | Pu | |
| Df(2 L)Exel6005 | Enh | 235.9 +/− 22.6 | 13 | 1.55 | ||
| Df(2 L)H20 | Sup | 29.2 +/− 6.4 | 25 | 1.52 | ||
| Df(2 L)ED1203 | Sup | 31.0 +/− 4.8 | 23 | 1.52 | ham, ddc | DTS1 |
| Df(3 L)BSC23 | Sup | 31.6 +/− 8.0 | 17 | 1.50 | rasp, spz5, scramb2, aly | DTS1, CJ10 |
| Df(2 L)J39 | Sup | 23.0 +/− 4.7 | 21 | 1.43 | FKBP59 | CJ10 |
| Df(2R)BSC267 | Enh | 144.7 +/− 24.7 | 3 | 1.38 | ||
| Df(1)BSC825 | Sup | 36.7 +/− 7.2 | 9 | 1.37 | ||
| Df(2 L)BSC213 | Enh | 146.3 +/− 46.2 | 8 | 1.35 | ||
| Df(3 L)Exel6112 | Enh | 143.1 +/− 27.7 | 14 | 1.35 | CJ10 | |
| Df(2 L)ED489 | Sup | 41.4 +/− 16.2 | 13 | 1.28 | Ndae1 | CJ10 |
| Df(2 L)ED8142 | Sup | 38.9 +/− 7.9 | 24 | 1.20 | ||
| Df(2R)BSC429 | Sup | 40.1 +/− 16.4 | 16 | 1.15 | ||
| Df(2 L)BSC295 | Enh | 181.7 +/− 20.2 | 15 | 1.09 | ||
| Df(2 L)BSC149 | Enh | 107.1 +/− 41.2 | 12 | 1.01 | ||
| Df(2 L)BSC233 | Enh/Leth | - | - | |||
| Df(3 L)BSC451 | Enh/Leth | - | - | |||
| Df(3R)BSC469 | Enh/Leth | - | - | CJ10 | ||
| Df(3R)BSC491 | Enh/Leth | - | - | |||
| Df(3R)BSC819 | Enh/Leth | - | - | ATPalpha | All Enh/Leth |
Table 4.
Confirmed ATPalpha CJ10 interacting deficiencies
| Df Name | Enh/Sup | Mean +/− SEM | Total N | RI | Hits in region | Coincidence |
|---|---|---|---|---|---|---|
| CJ10 | Control | 50.3 +/− 7.1 | 17 | - | - | - |
| Df(3R)BSC486 | Enh | 168.5 +/− 38.8 | 6 | 4.92 | DTS1 | |
| Df(3 L)Exel6112 | Enh | 144.6 +/− 15.4 | 18 | 4.20 | CJ5 | |
| Df(2 L)BSC180 | Enh | 151.7 +/− 34.5 | 9 | 2.93 | Rbp9 | DTS1 |
| Df(2 L)TW161 | Enh | 103.1 +/− 18.2 | 12 | 2.89 | ||
| Df(3R)BSC469 | Enh | 96.5 +/− 22.9 | 11 | 2.59 | CJ5 | |
| Df(3R)BSC681 | Enh | 98.7 +/− 49.4 | 6 | 2.32 | ||
| Df(3R)A113 | Enh | 92.4 +/− 8.8 | 14 | 2.16 | ||
| Df(3R)BSC501 | Enh | 91.8 +/− 7.8 | 14 | 2.10 | CG14508 | |
| Df(3R)ED5495 | Enh | 139.6 +/− 34.5 | 7 | 1.98 | ||
| Df(3 L)Exel6092 | Enh | 142.8 +/− 31.5 | 20 | 1.85 | spz5, scramb2, FMRFaR, aly | DTS1 |
| Df(2R)BSC664 | Enh | 60.2 +/− 14.1 | 11 | 1.77 | ||
| Df(3R)Exel6196 | Enh | 109.1 +/− 28.5 | 11 | 1.74 | ||
| Df(3 L)BSC410 | Enh | 85.3 +/− 11.1 | 12 | 1.54 | ||
| Df(3 L)ED4475 | Sup | 8.0 +/− 1.6 | 7 | 1.48 | CJ5 | |
| Df(3 L)BSC23 | Sup | 8.0 +/− 3.0 | 18 | 1.43 | rasp, spz5, scramb2, aly | DTS1, CJ5 |
| Df(2 L)BSC240 | Enh | 91.8 +/− 10.9 | 24 | 1.43 | Nckx30C, ppk11, nAChR-alpha6, FKBP59 | |
| Df(2 L)J39 | Sup | 7.9 +/− 2.2 | 25 | 1.43 | FKBP59 | CJ5 |
| Df(2R)BSC361 | Enh | 114.0 +/− 26.3 | 8 | 1.29 | Stj | DTS1 |
| Df(2R)BSC661 | Enh | 78.0 +/− 10.9 | 23 | 1.25 | ||
| Df(3R)ED5577 | Sup | 14.0 +/− 2.0 | 13 | 1.20 | ||
| Df(2 L)ED489 | Sup | 12.1 +/− 2.6 | 25 | 1.19 | Ndae1 | CJ5 |
| Df(3 L)ED230 | Sup | 13.7 +/− 3.7 | 10 | 1.17 | ||
| Df(4)ED6380 | Sup | 12.6 +/− 3.6 | 25 | 1.14 | ||
| Df(3 L)BSC113 | Sup | 14.3 +/− 1.7 | 15 | 1.13 | aay | |
| Df(2 L)ED793 | Sup | 16.2 +/− 4.1 | 25 | 1.09 | Dyrk2, NimB5, nAChRα5 | |
| Df(2 L)BSC149 | Sup | 16.1 +/− 3.3 | 14 | 1.09 | ||
| Df(3R)ED7665 | Sup | 16.5 +/− 5.1 | 21 | 1.06 | DTS1 | |
| Df(3 L)BSC442 | Enh | 79.1 +/− 10.4 | 15 | 1.02 | ||
| Df(3R)BSC467 | Enh/Leth | - | - | DTS1 | ||
| Df(3R)BSC819 | Enh/Leth | - | - | ATPalpha | All Enh/Leth |
Single gene identification and testing
After the verification of critical intervals, genes contained within these intervals were selected for testing. Where practical large intervals were narrowed using smaller Dfs. We obtained classical alleles for integral genes from Bloomington, when possible. Each single gene mutant was mated to the ATPalpha allele it putatively modified and to w1118. All single gene mutants displayed no BS or TS phenotype as heterozygotes (data not shown). Heterozygous double mutants were again assayed for TS or BS with age matched controls. Significant interacting single gene mutants were also tested with the other ATPalpha alleles (Figure 2). Twenty single gene interactions were found using classical mutants for ATPalphaDTS1 including 19 single gene enhancers and one single gene suppressor. Ten single gene suppressors were found for ATPalphaCJ5. Twenty-four single gene interactions were found with ATPalphaCJ10, all but one of which showed suppression of the mutant phenotypes. In total, 35 single gene interactions were found and, importantly, 14 different genes had effects with more than one ATPalpha allele (Table 5).
Table 5.
Single gene effects confirmed for ATPalpha alleles using classical mutants
| Cytological region | Gene | Genotype | Putative function # | ATPα Allele | Nature of interaction | Significance |
|---|---|---|---|---|---|---|
| 10B3 | l(1)10Bb | E04588 | Spliceosome component [44] | CJ10 | Suppressor | * |
| 21B1-21B1 | Galectin | DG25505 | Cell surface protein, galactoside binding [45] | DTS1 | Enhancer | *** |
| 23C9-23C9 | Rbp9 | ∆1 | RNA binding [46] | DTS1 | Enhancer | * |
| 23C9-23C9 | Rbp9 | ∆1 | " | CJ5 | Suppressor | **** |
| 27E-28B1 | Ndae1 | MB05294 | Sodium driven anion exchanger [47] | CJ5 | Suppressor | * |
| 27E-28B1 | Ndae1 | MB05294 | " | DTS1 | Enhancer | * |
| 27E-28B1 | Ndae1 | MB05294 | " | CJ10 | Suppressor | * |
| 29B4-29E4 | Sema-1a | K13702 | Axon guidance signal and receptor [48,49] | DTS1 | Enhancer | * |
| 29B4-29E4 | Glt | EY22126 | Cell surface glycoprotein [50] | DTS1 | Enhancer | **** |
| 29B4-29E4 | Glt | EY22126 | “ | CJ10 | Suppressor | * |
| 30C7-30 F2 | Nckx30C | E00401 | Sodium/Calcium/Potassium exchanger [51] | CJ10 | Enhancer | * |
| 30C7-30 F2 | Ppk11 | MB02012 | Excitatory sodium channel [52] | CJ10 | Suppressor | *** |
| 30C7-30 F2 | Ppk11 | MB02012 | “ | DTS1 | Suppressor | **** |
| 30C7-30 F2 | Ppk11 | MB02012 | “ | CJ5 | Suppressor | **** |
| 30C7-30 F2 | nAChRα6 | MB06675 | ACh receptor subunit | CJ10 | Suppressor | * |
| 30C7-30 F2 | nAChRα6 | MB06675 | “ | CJ5 | Suppressor | **** |
| 31C-32E | FKBP59 | EY03538 | Calcium channel regulator [53] | DTS1 | Enhancer | * |
| 31C-32E | FKBP59 | EY03538 | " | CJ5 | Suppressor | *** |
| 31C-32E | FKBP59 | EY03538 | “ | CJ10 | Suppressor | *** |
| 33A8-33B1 | Pde1c | C04487 | cAMP/cGMP phosphodiesterase [54] | CJ5 | Suppressor | **** |
| 34E4-35B4 | Dyrk2 | 1 | Serine/Threonine kinase [55] | DTS1 | Enhancer | *** |
| 34E4-35B4 | Dyrk2 | 1 | " | CJ10 | Suppressor | **** |
| 34E4-35B4 | Nimb5 | MI01793 | Bacterial defense | CJ10 | Suppressor | ** |
| 34E4-35B4 | nAChRα5 | MB11647 | ACh Receptor subunit | CJ10 | Suppressor | * |
| 35 F1-36A1 | Cact | 7 | Inhibitor of NF-κB [56] | CJ10 | Suppressor | **** |
| 36A8-36 F1 | Beat-Ia & Fas3 | 3/E25 | Neuronal immunoglobulin-like proteins | CJ5 | Suppressor | * |
| 25 F1-36A1 | CG5888 | MB00188 | Toll 3 like receptor | DTS1 | Enhancer | **** |
| 25 F1-36A1 | CG5888 | MB00188 | " | CJ5 | Suppressor | **** |
| 46 F1-47A9 | CG42732 | MB04544 | Predicted potassium channel | DTS1 | Enhancer | **** |
| 46 F1-47A9 | Rpl41/NaCP60E | EP348 | Ribosomal protein; voltage-gated Na+ channel [57] | CJ10 | Suppressor | * |
| 46 F1-47A9 | CG42732 | MB04544 | Predicted potassium channel | CJ5 | Suppressor | ** |
| 46 F1-47A9 | Gαo | MI00833 | Heterotrimeric G-protein subunit | CJ10 | Suppressor | **** |
| 46 F1-47A9 | CYP49A1 & Gαo | MB04922 | Cytochrome P450 & heterotrimeric G-protein subunit | DTS1 | Enhancer | **** |
| 50B1 | CG33156 | MB05931 | Predicted NAD+ kinase | DTS1 | Enhancer | **** |
| 57C5-57 F6 | Pu | r1 | GTP cyclohydrolase [58] | CJ5 | Suppressor | ** |
| 57C5-57 F6 | Pu | r1 | “ | CJ10 | Suppressor | **** |
| 60E6-60E11 | Pain | EP2451 | TRP calcium channel [59] | DTS1 | Enhancer | ** |
| 60E6-60E11 | Pain | EP2451 | “ | CJ10 | Suppressor | **** |
| 60E6-60E11 | Rpl19 | K03704 | Ribosomal component [60] | DTS1 | Enhancer | **** |
| 62E8-63B6 | Spz5 | E03444 | Neurotrophin [61,62] | DTS1 | Enhancer | ** |
| 62E8-63B6 | Spz5 | E03444 | “ | CJ10 | Suppressor | **** |
| 62E8-63B6 | Aly | 1 | Regulator of transcription [63,64] | DTS1 | Enhancer | **** |
| 62E8-63B6 | Rasp | m47 | Palmitoyl transferase [65,66] | DTS1 | Enhancer | ** |
| 62E8-63B6 | Rasp | m47 | “ | CJ10 | Suppressor | **** |
| 63A3-63A3 | Scramb2 | EY01180 | Predicted phosphatidyl serine scramblase | DTS1 | Enhancer | **** |
| 63A3-63A3 | Scramb2 | EY01180 | " | CJ10 | Suppressor | **** |
| 67A2-67D13 | Aay | S042314 | Predicted Phosphoserine phosphatase | CJ10 | Suppressor | ** |
| 93B9-93D4 | Slmb | 295 | Ubiquitin ligase [67,68] | DTS1 | Enhancer | ** |
| 93B9-93D4 | Slmb | 295 | “ | CJ10 | Suppressor | **** |
| 93B9-93D4 | Sec15 | 2 | Protein trafficking [69,70] | DTS1 | Enhancer | ** |
| 93B9-93D4 | Sec15 | 2 | “ | CJ10 | Suppressor | **** |
| 93B9-93D4 | RhoGAP93B | EY07136 | Rac1 GAP [71] | DTS1 | Enhancer | * |
| 98 F10-99B9 | CG14508 | G9163 | Predicted cytochrome C | DTS1 | Enhancer | *** |
| 98 F10-99B9 | CG14508 | G9163 | Predicted cytochrome C | CJ10 | Suppressor | **** |
| 99E1-3Rt | Sro | 1 | Ecdysone biosynthetic pathway | CJ10 | Suppressor | * |
Many genes had an interaction with more than one allele, although some appear to be allele specific. Double mutants were compared to ATPalpha * /+ and heterozygous classical mutant controls. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
#Function per flybase.org and / or listed citation.
Gal4 driven RNAi strains result in a loss-of-function phenotype and are well-suited to confirm the hypomorphic effect of a heterozygous Df. RNAi knockdown was driven with da-Gal4 in ATPalpha mutant backgrounds. Daughterless transcripts are stably expressed throughout the life of a fly and are detectable in every tissue by the FlyAtlas affymetrix array analysis [72,73]. We used this driver to ubiquitously express the RNAi constructs and mimic the effect observed with the Df. RNAi mediated knockdown of candidate genes was compared to age matched controls lacking the UAS-RNAi construct. Twenty-five different genes showed interactions using this method, including 10 genes already identified in the classical mutant screen. Fourteen interactions were identified with ATPalphaDTS1, with nine enhancers and five suppressors. Seventeen interactions, with two enhancers and 15 suppressors, were identified for ATPalphaCJ5. Thirteen interactions, all suppressors, were confirmed with ATPalphaCJ10. In total 15 different genes showed a genetic interaction with two or more ATPalpha alleles (Table 6). In total we have identified 50 genes that interact with ATPalpha, 25 of which were confirmed to interact with at least two independent alleles.
Table 6.
Single gene effects confirmed for ATPalpha alleles using RNAi
| Cytological region | Gene | Putative function # | ATPα Allele | Nature of interaction | Significance |
|---|---|---|---|---|---|
| 21A1-21B1 | Galectin | Galactoside binding [45] | CJ10 | Suppressor | ** |
| 21A1-21B1 | Galectin | “ | CJ5 | Suppressor | *** |
| 22 F4-22 F4 | CG3528 | Unknown | DTS1 | Enhancer | * |
| 22 F4-22 F4 | CG3528 | CJ10 | Suppressor | * | |
| 22 F4-22 F4 | CG3528 | CJ5 | Suppressor | * | |
| 27E-28B1 | Ndae1 | Na + driven anion exchanger [47] | CJ5 | Enhancer | * |
| 29B4-29E4 | Glt | Cell surface glycoprotein [50] | CJ10 | Suppressor | * |
| 30C8-30C9 | Ppk11 | Sodium channel [52] | CJ5 | Suppressor | ** |
| 31C-32E | FKBP59 | Calcium channel regulator [53] | CJ5 | Suppressor | *** |
| 31C-32E | FKBP59 | “ | DTS1 | Enhancer | * |
| 33A1-33A1 | Vha100-5 | ATPase, proton transport | DTS1 | Enhancer | * |
| 33A2-33A2 | Esc | Histone methyltransferase component [74] | DTS1 | Enhancer | *** |
| 33A2-33A2 | Esc | “ | CJ10 | Suppressor | ** |
| 34E4-35B4 | Dyrk2 | Serine/Threonine kinase [55] | DTS1 | Enhancer | * |
| 34E4-35B4 | Dyrk2 | “ | CJ5 | Suppressor | *** |
| 37A2-37A4 | Ham | Transcription factor [75] | DTS1 | Suppressor | * |
| 37A2-37A4 | Ham | “ | CJ5 | Suppressor | ** |
| 37C1-37C1 | Ddc | Amino acid decarboxylase [76] | CJ5 | Suppressor | *** |
| 25 F1-36A1 | CG5888 | Toll 3 like Receptor | CJ10 | Suppressor | ** |
| 25 F1-36A1 | CG5888 | “ | CJ5 | Suppressor | * |
| 50C5-50C6 | Stj | Voltage-gated calcium channel regulatory subunit [77,78] | DTS1 | Enhancer | **** |
| 50C5-50C6 | Stj | “ | CJ5 | Enhancer | ** |
| 51D1-51D1 | Cyp6a19 | Cytochrome P450 | CJ10 | Suppressor | * |
| 62E8-63B6 | Spz5 | Neurotrophin [61,62] | DTS1 | Suppressor | ** |
| 62E8-63B6 | Spz5 | “ | CJ10 | Suppressor | * |
| 62E8-63B6 | Rasp | Palmitoyl transferase [65,66] | CJ5 | Suppressor | *** |
| 63A3-63A3 | FMRFaR | Neuropeptide receptor [79] | DTS1 | Enhancer | ** |
| 63A3-63A3 | FMRFaR | “ | CJ10 | Suppressor | ** |
| 63A3-63A3 | FMRFaR | “ | CJ5 | Suppressor | **** |
| 64C2-64C5 | Con | Homophilic cell adhesion [80] | DTS1 | Suppressor | * |
| 64C2-64C5 | Con | “ | CJ5 | Suppressor | ** |
| 67A2-67D13 | Aay | Predicted phosphoserine phosphatase | CJ10 | Suppressor | * |
| 67A2-67D13 | Aay | “ | CJ5 | Suppressor | ** |
| 67B9-67B9 | Uch-L5 | 26S Proteasome component [81] | DTS1 | Enhancer | * |
| 67D11-67D11 | Scramb1 | Phosphatidyl serine scramblase | CJ10 | Suppressor | ** |
| 99B5-99B6 | Dop1R2 | Dopamine 1-like receptor [82,83] | CJ5 | Suppressor | *** |
| 99B6-99B6 | Ppk21 | Sodium channel | DTS1 | Suppressor | ** |
| 99B6-99B6 | Ppk21 | “ | CJ10 | Suppressor | * |
| 100B9-100B9 | Ppk24 | Sodium channel | DTS1 | Suppressor | ** |
| 100B9-100B9 | Ppk24 | “ | CJ5 | Suppressor | * |
| 100B9-100B9 | Ppk24 | “ | CJ10 | Suppressor | ** |
| 100C1-100C1 | CG11340 | Predicted chloride channel | DTS1 | Suppressor | * |
| 100C1-100C1 | CG11340 | “ | CJ5 | Suppressor | *** |
| 100C1-100C1 | CG11340 | “ | CJ10 | Suppressor | * |
Many genes had an interaction with more than one allele, although some appear to be allele specific. RNAi knockdowns were compared with ATPalpha*, daGal4/+ controls. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
#Function per flybase.org and/or listed citation.
Discussion
The Na+/K+ATPase is central to maintaining cytosolic ion homeostasis suggesting that many of the genes identified in our screen would encode proteins that affect cytosolic ion concentrations and, indeed, this was the case (Figure 3A). Nearly 25% of the genes we identified encode proteins with a known function in ion transport. In our search for single gene modifiers we selected genes known to be expressed in the nervous system. Unsurprisingly, ~50% of our hits are known to cause some neuronal defect when knocked out (Figure 3B). For example, most of the cell adhesion and paracrine signaling molecules we found, such as Galectin (Tables 5 and 6, Figure 4), Glt (Tables 5 and 6), and Sema-1a (Table 5) were previously known to cause malformed or improperly targeted synapses [45,48-50]. However, about half of our genes were not previously linked to neuronal function. Additionally, many genes we identified encode proteins implicated in signaling pathways. In particular we found proteins involved in developmental signaling pathways, such as Wingless and Hedgehog (rasp (Tables 5 and 6) and slmb (Table 5)), and neuronal growth and survival pathways (spz5 (Tables 5 and 6)).
Figure 3.
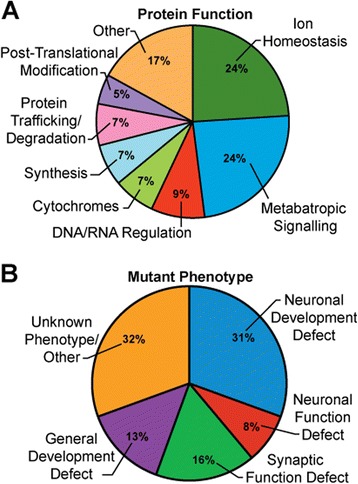
Distribution of validated genetic modifiers. A. Protein function of modifiers, as annotated on flybase.org, grouped into major categories. Stj, rasp, slmb, Rpl41/NaCP60E, and punch were included in two categories. B. Modifier loci categorized according to mutant phenotypes (when available). FKBP59, Cact, Scramb1, and Stj were associated with two phenotypic categories.
Figure 4.
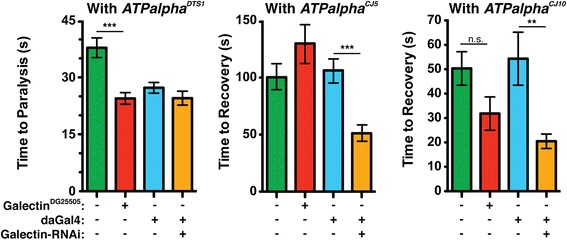
Genetic interaction between Galectin and ATPalpha . Galectin; ATPalpha double mutants and ATPalpha*, Galectin RNAi flies for each ATPalpha mutant were assayed and compared to ATPalpha* heterozygous controls. The RNAi knockdown was driven ubiquitously with daughterless-Gal4 (daGAL4). The genotypes in each graph are: ATPalpha*/+ (green), Galectin DG25505 /+;ATPalpha*/+ (red), daGal4,ATPalpha*/+ (blue), and Galectin-RNAi/+;daGal4,ATPalpha*/+ (orange). Galectin mutants significantly enhanced the ATPalpha DTS1 phenotype while galectin-RNAi significantly suppress ATPalpha CJ5 and ATPalpha CJ10 phenotypes. *p < 0.05, **p < 0.01, ***p < 0.001.
Spz5 (Figure 5) is especially interesting because it has recently been identified as a Drosophila neurotrophin that signals through a Toll receptor [61,62]. Both Slmb and Cact (Table 5) were also identified by our screen and both may function downstream of Spz5. In mammals and flies, Toll signaling activates NF-κB transcription factors, typically through the degradation of an inhibitor of NF-κB (I-κB), such as Cact. Phosphorylated I-κB is targeted for degradation, allowing NF-κB-like transcription factors to translocate to the nucleus. Slmb and its mammalian homolog β-TrCP regulate phospho-I-κB. β-TrCP, and likely Slmb, target an E3 ubiquitin ligase complex to phospho-I-κB and mediate its degradation via ubiquitin proteasome system [68]. Interestingly, we have also identified Uch-L5 (Table 6) in our screen, a member of the 26S regulatory complex which is likely responsible for the deubiquitylation of proteins as they enter the 26S proteasome [81].
Figure 5.
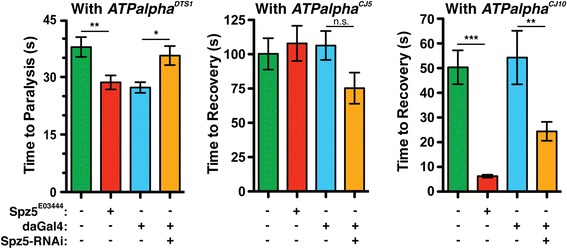
Genetic interaction between Spz5 and ATPalpha . ATPalpha/Spz5 double mutants and ATPalpha*, Spz5 RNAi flies for each ATPalpha mutant were assayed and compared to ATPalpha* heterozygous controls. The RNAi knockdown was driven with da-Gal4. The genotypes in each graph are: ATPalpha*/+ (green), Spz5 E03444 /ATPalpha* (red), daGal4,ATPalpha*/+ (blue), and Spz5-RNAi/daGal4,ATPalpha* (orange). Spz5 mutants significantly enhanced the ATPalpha DTS1 phenotype but Spz5 RNAI significantly suppresses the ATPalpha DTS1 phenotype. The ATPalpha CJ10 phenotype is suppressed in both the Spz5 mutant and RNAi. The ATPalpha CJ5 phenotype was not significantly affected by loss of Spz5. *p < 0.05, **p < 0.01, ***p < 0.001.
Previously published studies of Slmb, and Spz5 show that they play an important role in neural development. Slmb is involved in pruning dendrites and axons during pupation [84] and Spz5 is a neurotrophic signal and axon guidance cue in the embryonic nervous system [61]. Interestingly, animals heterozygous for a loss of function allele of either gene displayed no phenotype in neurons [61,85]. In contrast, our screen examined heterozygous double mutants and found large effects, suggesting ATPalpha mutants are sensitive to otherwise inconsequential changes in neuronal development or another unappreciated function of these proteins. Furthermore, a seemingly insignificant disruption of neuronal survival signals early in development may have dramatic phenotypic effects for ATPalpha mutants since heterozygosity of Slmb, or Spz5 suppressed the loss-of-function ATPalpha phenotype. Additionally, numerous developmental genes were identified implying that neurodevelopmental changes may profoundly affect Na+/K+ ATPase function or this is a general and potent mechanism to modulate locomotor function.
Another interesting possibility is that loss-of-function ATPalpha mutations are disrupting neuronal development through alterations in NF-κB signaling. It has been shown that sub-inhibitory concentrations of ouabain activate NF-κB via an Na+/K+ ATPase dependent mechanism in rat kidney cells. The effect is mediated by slow, inositol triphosphate-dependent, calcium oscillations likely caused by shifting electrochemical gradients [86]. More recently, agrin, a protein involved in synapse formation at NMJs and in the CNS, has been shown to bind to and inhibit the mammalian Na+/K+ ATPase α3 isoform. Furthermore, agrin seems to bind at the same site as ouabain because a protein fragment can prevent ouabain inhibition of the Na+/K+ ATPase [87]. Thus it is possible that agrin exerts its effects through NF-κB. If a similar pathway exists in flies it would likely be constitutively active in our loss-of-function mutants and its dysregulation could cause developmental changes, which might increase seizure susceptibility. This is consistent with our finding that knockdown of proteins required for NF-κB activation suppresses seizures in our loss-of-function mutants. NF-κB activation may be caused by calcium oscillations [86], making it possible that some of the calcium channels we found also play a role in this pathway. FKBP59 (Figure 6) is particularly interesting because it inhibits an inositol triphosphate sensitive, non-specific calcium channel, TrpL [53]. Inhibition of calcium channels would likely be required in calcium oscillations. The preponderance of hits related to the NF-κB pathway suggests a possible role for this pathway in seizure pathogenesis.
Figure 6.
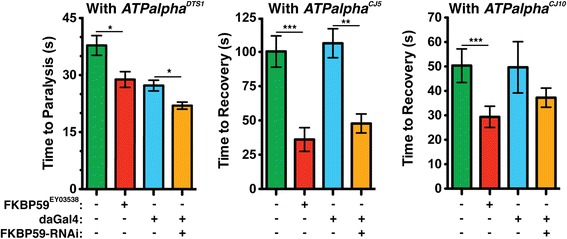
Genetic interactions between FKBP59 and ATPalpha . FKBP59; ATPalpha double mutants and ATPalpha*, FKBP59 RNAi flies were assayed and compared to ATPalpha* heterozygous controls. The RNAi knockdown was driven with da-Gal4. The genotypes in each graph are: ATPalpha*/+ (green), FKBP59 E03444 /+; ATPalpha*/+ (red), daGal4,ATPalpha*/+ (blue), and FKBP59-RNAi/+;daGal4,ATPalpha*/+ (orange). FKBP59 mutants significantly enhanced the ATPalpha DTS1 phenotype. The ATPalpha CJ5 phenotype is suppressed by both the FKBP59 mutant and RNAi. *p < 0.05, **p < 0.01, ***p < 0.001.
In most cases the ATPalphaCJ5 and ATPalphaCJ10 mutant phenotypes were modified in the same direction (enhancement or suppression) and they never had opposite phenotypes in our screen. This is consistent with the finding that both exhibit loss-of-function characteristics. The ATPalphaDTS1 phenotype, however, usually contrasted with the phenotypes of ATPalphaCJ5 and ATPalphaCJ10. This is intriguing as ATPalphaDTS1 is a gain-of-function mutation that can be reverted by a second site mutation to give the characteristic ATPalpha loss-of-function phenotype [23]. In accord with this fact the vast majority (~ 80%) of the single gene interactions with ATPalphaDTS1 modified the loss-of-function alleles in the opposite direction or not at all. Reduction of Ppk11, Ppk21, and Ppk24 function all suppressed the phenotypes of ATPalphaDTS1 and another allele. All three are predicted epithelial sodium channels (DEG/eNaCs) that function in nociception, mechanosensation, gustation and other sensory functions (Reviewed in [88] and [89]). Thus, it is possible that altered sensory function may underlie the ATPalphaDTS1 paralysis phenotype and that a reduction in the ability of the double mutant animals to sense the elevated temperature is sufficient to suppress the TS paralysis. This possibility is consistent with the kinetics of recovery after animals are returned to the permissive temperature. This is also intriguing as the locomotor dysfunction resulting in hemiparalysis in FHM patients has been reported to be associated with sensory dysfunction and FHM patients report having prolonged visual auras [90-92].
Conclusions
FHM, RDP, and AHC are complex human neurological diseases associated with mutations affecting the catalytic alpha subunit of the Na+/K+ ATPase [4-6]. Currently, there is no cure or effective treatment for these diseases. Using three Drosophila strains with different missense mutations in ATPalpha we have performed a large-scale deficiency screen to identify novel genes that interact with the gene encoding the Na+/K+ATPase alpha subunit. In total, we have identified 50 genes that interact with ATPalpha, 25 of which were demonstrated to interact with at least two independent alleles. We have also implicated 50 critical intervals/deficiency regions for which we have yet to identify individual genes that interact with ATPalpha (Tables 2, 3 and 4). Modifier loci that encode proteins expressed in the adult, especially those that phenotypically suppress ATPalpha dysfunction, provide proteins/pathways that could be viable targets for the development of new migraine or anti-epileptic drugs. Additionally, studies of these loci and how they modify ATPalpha dysfunction will help us understand epilepsy, hemiplegia and migraine disease pathogenesis in animals.
Materials and methods
Drosophila strains
Flies were maintained on standard cornmeal-molasses agar medium at 21-22°C. Chromosomal deficiencies were obtained from the Bloomington Deficiency Kit from the Bloomington Stock Center (order date January 2010). The Df Kit we received contained 467 stocks with deletions spanning 97.8% of the Drosophila genome. Three Na+/K+ ATPase alpha subunit mutants were used: ATPalphaDTS1 [23], ATPalphaCJ5 and ATPalphaCJ10 [36]. The other Drosophila strains used were obtained from the Vienna Drosophila RNAi Center (VDRC) or Bloomington Stock Center.
Locomotor assays
F1 offspring heterozygous for an ATPalpha allele and each individual Df were collected upon eclosion (day 0) and aged at 25°C on cornmeal-molasses medium. Temperature sensitivity (TS) was assayed on day 1 and bang sensitivity (BS) was assayed on day 15 as described previously [23]. Aged flies were moved to an empty vial in groups of 5 or fewer using an aspirator. For TS, the vial was submerged in a water bath at 38°C such that the flies were restricted to space in the vial below the waterline. A timer was started when the vial was submerged and time to paralysis was recorded for each fly. For BS, the vial was mechanically shaken using a standard lab Vortex Genie 2 (Daigger, IL) on the highest setting for 20 seconds. Time to recovery for each fly was recorded. Both conditional locomotor assays were stopped after 300 seconds.
Df Interaction screen
Initial Screens
Males with autosomal deficiencies were mated to ATPalphaDTS1, ATPalphaCJ5, and ATPalphaCJ10 virgin females, and X-linked deficiency virgin females were mated with ATPalphaDTS1, ATPalphaCJ5, and ATPalphaCJ10 males. F1 progeny representing a total of 386 deficiency interactions were tested with ATPalphaDTS1 animals (83% of Df kit), 393 were tested with ATPalphaCJ5 (84% of Df kit), and 358 were tested with ATPalphaCJ10 animals (77% of Df kit). Each of the 467 Dfs we received was tested with at least one ATPalpha allele, the vast majority were tested with multiple alleles and >55% were tested with all three alleles. Assays were performed as described above.
Verification screen
Putative modifier Df strains identified in the initial screen were retested in an independent experiment to verify the findings and reduce the rate of false positives. In selecting Df stains to test again, we favored Dfs that suppressed ATPalpha mutant phenotypes and/or interacted with more than one ATPalpha allele. During the verification screen all three ATPalpha alleles were investigated.
Single gene identification
We developed an analysis called the Reproducibility Index (RI) in order to guide our search for single gene modifiers of the ATPalpha alleles. The goal of this index was to rank the most promising Df intervals based on the magnitude and reproducibility with which they modified an ATPalpha allele phenotype. To this end, we first calculated the number of standard deviations of the Df, ATPalpha* double mutant mean from the total mean of the primary screen of each ATPalpha mutant using:
where StdDevtotal is the standard deviation of all deficiencies in the primary screen, Meantotal is the mean of all deficiencies in the primary screen, and MeanDf is the mean response of a Df/ATPalpha double mutant. Num.Std.Dev (#SD) was calculated for the mean response of a Df double mutant in the primary (#SDprim) and verification (#SDveri) screen. We reasoned that these values provide a normalized metric of how much a Df modified an ATPalpha phenotype in each trial. We used these values to calculate the RI:
where
The RI increases for Dfs that were further from the total mean and decreases for Dfs that varied more across the two trials. Thus, a high RI suggests that a region is more likely to contain a gene that interacts with and modifies an ATPalpha allele in a reproducible manner. In some intervals we were able to use small Dfs to narrow the interval further. We, again, prioritized strongly suppressing intervals over enhancing intervals and intervals that interacted with multiple alleles. Single genes were selected from critical intervals using the G-Browse feature (an annotated genome) of flybase.org. In some very small intervals all genes in the region were tested. In large intervals we necessarily focused on genes with described expression within the nervous and or muscular systems, introducing a noted bias. Many of the alleles chosen were P-element or classical mutations reported to knockout the genes of interest. The stocks of interest were ordered from the Bloomington Stock Center.
RNAi analysis
When classical mutants were unavailable for certain loci or to confirm an interaction found using a classical mutant, RNAi analysis was used to examine the gene in question. RNAi stocks were ordered from the VDRC. The RNAi transgenes were driven using daughterless Gal4 strains (daGal4) in each ATPalpha mutant background. RNAi male flies were mated to ATPalpha, daGal4 virgin females. Progeny were raised at 25°C, and TS and BS tests were performed as described previously.
Data collection and statistics
Data were collected and organized using Microsoft Excel (Redmond, WA). Data were analyzed in GraphPad Prism 5 (San Diego, CA). We used ANOVA to compare the ATPalpha mutant heterozygotes, the classical mutant heterozygotes, and flies heterozygous for both alleles. Tukey’s multiple comparison test was performed to determine if the double mutants were significantly different from the ATPalpha mutant heterozygote and the classical mutant heterozygote. Adjusted p-values are reported in Table 5. The effect of RNAi transgenes was analyzed using a Student’s t-test to determine if single gene knockdowns significantly modified the phenotype of ATPalpha*, daGal4 controls. Significant interactions are reported in Table 6.
Acknowledgements
We thank the Bloomington stock center for the Df kit and other fly strains and Troy Novak, Ellis Herman, Nick Brown, Dan Lesky, Dan Wei, John Ries, and James Repko for assistance with the genetic screens. This work could not have been completed without funding from NIH R01AG025046 (MJP) and NIH R01AG027453 (MJP).
Additional file
Data from the primary and verification Df screens.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ADT, JFC, AL and EDW performed the experiments. ADT, JFC, EDW and MJP analyzed the data. ADT, JFC, EDW and MJP wrote the manuscript. MJP designed and coordinated the study. ADT, JFC, AL, EDW and MJP reviewed, edited and approved the manuscript.
Contributor Information
Aaron D Talsma, Email: adt40@pitt.edu.
John F Chaves, Email: john.chaves@jefferson.edu.
Alexandra LaMonaca, Email: ALL118@pitt.edu.
Emily D Wieczorek, Email: wieczorek.emily@medstudent.pitt.edu.
Michael J Palladino, Email: mjp44@pitt.edu.
References
- 1.Lingrel JB. Na, K-ATPase: isoform structure, function, and expression. J Bioenerg Biomembr. 1992;24:263–270. doi: 10.1007/BF00768847. [DOI] [PubMed] [Google Scholar]
- 2.Lopina OD. Na+, K + −ATPase: structure, mechanism, and regulation. Membr Cell Biol. 2000;13:721–744. [PubMed] [Google Scholar]
- 3.Skou JC, Esmann M. The Na, K-ATPase. J Bioenerg Biomembr. 1992;24:249–261. doi: 10.1007/BF00768846. [DOI] [PubMed] [Google Scholar]
- 4.Heinzen EL, Swoboda KJ, Hitomi Y, Gurrieri F, Nicole S, de Vries B, Tiziano FD, Fontaine B, Walley NM, Heavin S, Panagiotakaki E, European Alternating Hemiplegia of Childhood (AHC) Genetics Consortium; Biobanca e Registro Clinico per l'Emiplegia Alternante (I.B.AHC) Consortium; European Network for Research on Alternating Hemiplegia (ENRAH) for Small and Medium-sized Enterpriese (SMEs) Consortium, Fiori S, Abiusi E, Di Pietro L, Sweney MT, Newcomb TM, Viollet L, Huff C, Jorde LB, Reyna SP, Murphy KJ, Shianna KV, Gumbs CE, Little L, Silver K, Ptáček LJ, Haan J: De novo mutations in ATP1A3 cause alternating hemiplegia of childhood.Nat Genetᅟ, 44:1030–1034. [DOI] [PMC free article] [PubMed]
- 5.Kramer PL, Mineta M, Klein C, Schilling K, de Leon D, Farlow MR, Breakefield XO, Bressman SB, Dobyns WB, Ozelius LJ, Brashear A. Rapid-onset dystonia-parkinsonism: linkage to chromosome 19q13. Ann Neurol. 1999;46:176–182. doi: 10.1002/1531-8249(199908)46:2<176::AID-ANA6>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 6.Russell MB, Ducros A. Sporadic and familial hemiplegic migraine: pathophysiological mechanisms, clinical characteristics, diagnosis, and management. Lancet Neurol. 2011;10:457–470. doi: 10.1016/S1474-4422(11)70048-5. [DOI] [PubMed] [Google Scholar]
- 7.Sweney MT, Silver K, Gerard-Blanluet M, Pedespan JM, Renault F, Arzimanoglou A, Schlesinger-Massart M, Lewelt AJ, Reyna SP, Swoboda KJ. Alternating hemiplegia of childhood: early characteristics and evolution of a neurodevelopmental syndrome. Pediatrics. 2009;123:e534–e541. doi: 10.1542/peds.2008-2027. [DOI] [PubMed] [Google Scholar]
- 8.Sasaki M, Ishii A, Saito Y, Hirose S. Intermediate form between alternating hemiplegia of childhood and rapid-onset dystonia-parkinsonism. Mov Disord. 2014;29:153–154. doi: 10.1002/mds.25659. [DOI] [PubMed] [Google Scholar]
- 9.Atkinson NS, Robertson GA, Ganetzky B. A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science. 1991;253:551–555. doi: 10.1126/science.1857984. [DOI] [PubMed] [Google Scholar]
- 10.Littleton JT, Serano TL, Rubin GM, Ganetzky B, Chapman ER. Synaptic function modulated by changes in the ratio of synaptotagmin I and IV. Nature. 1999;400:757–760. doi: 10.1038/23462. [DOI] [PubMed] [Google Scholar]
- 11.Loughney K, Kreber R, Ganetzky B. Molecular analysis of the para locus, a sodium channel gene in Drosophila. Cell. 1989;58:1143–1154. doi: 10.1016/0092-8674(89)90512-6. [DOI] [PubMed] [Google Scholar]
- 12.Pallanck L, Ordway RW, Ganetzky B. A Drosophila NSF mutant. Nature. 1995;376:25. doi: 10.1038/376025a0. [DOI] [PubMed] [Google Scholar]
- 13.Pallanck L, Ordway RW, Ramaswami M, Chi WY, Krishnan KS, Ganetzky B. Distinct roles for N-ethylmaleimide-sensitive fusion protein (NSF) suggested by the identification of a second Drosophila NSF homolog. J Biol Chem. 1995;270:18742–18744. doi: 10.1074/jbc.270.32.18742. [DOI] [PubMed] [Google Scholar]
- 14.Titus SA, Warmke JW, Ganetzky B. The Drosophila erg K+ channel polypeptide is encoded by the seizure locus. J Neurosci. 1997;17:875–881. doi: 10.1523/JNEUROSCI.17-03-00875.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fergestad T, Sale H, Bostwick B, Schaffer A, Ho L, Robertson GA, Ganetzky B. A Drosophila behavioral mutant, down and out (dao), is defective in an essential regulator of Erg potassium channels. Proc Natl Acad Sci U S A. 2010;107:5617–5621. doi: 10.1073/pnas.1001494107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikeda K, Ozawa S, Hagiwara S. Synaptic transmission reversibly conditioned by single-gene mutation in Drosophila melanogaster. Nature. 1976;259:489–491. doi: 10.1038/259489a0. [DOI] [PubMed] [Google Scholar]
- 17.Peixoto AA, Hall JC. Analysis of temperature-sensitive mutants reveals new genes involved in the courtship song of Drosophila. Genetics. 1998;148:827–838. doi: 10.1093/genetics/148.2.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siddiqi O, Benzer S. Neurophysiological defects in temperature-sensitive paralytic mutants of Drosophila melanogaster. Proc Natl Acad Sci U S A. 1976;73:3253–3257. doi: 10.1073/pnas.73.9.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu CF, Ganetzky B, Jan LY, Jan YN, Benzer S. A Drosophila mutant with a temperature-sensitive block in nerve conduction. Proc Natl Acad Sci U S A. 1978;75:4047–4051. doi: 10.1073/pnas.75.8.4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dellinger B, Felling R, Ordway RW. Genetic modifiers of the Drosophila NSF mutant, comatose, include a temperature-sensitive paralytic allele of the calcium channel alpha1-subunit gene, cacophony. Genetics. 2000;155:203–211. doi: 10.1093/genetics/155.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Littleton JT, Chapman ER, Kreber R, Garment MB, Carlson SD, Ganetzky B. Temperature-sensitive paralytic mutations demonstrate that synaptic exocytosis requires SNARE complex assembly and disassembly. Neuron. 1998;21:401–413. doi: 10.1016/S0896-6273(00)80549-8. [DOI] [PubMed] [Google Scholar]
- 22.Reenan RA, Hanrahan CJ, Ganetzky B. The mle(napts) RNA helicase mutation in drosophila results in a splicing catastrophe of the para Na + channel transcript in a region of RNA editing. Neuron. 2000;25:139–149. doi: 10.1016/S0896-6273(00)80878-8. [DOI] [PubMed] [Google Scholar]
- 23.Palladino MJ, Bower JE, Kreber R, Ganetzky B. Neural dysfunction and neurodegeneration in Drosophila Na+/K+ ATPase alpha subunit mutants. J Neurosci. 2003;23:1276–1286. doi: 10.1523/JNEUROSCI.23-04-01276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palladino MJ, Hadley TJ, Ganetzky B. Temperature-sensitive paralytic mutants are enriched for those causing neurodegeneration in Drosophila. Genetics. 2002;161:1197–1208. doi: 10.1093/genetics/161.3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parker L, Padilla M, Du Y, Dong K, Tanouye MA. Drosophila as a model for epilepsy: bss is a gain-of-function mutation in the para sodium channel gene that leads to seizures. Genetics. 2011;187:523–534. doi: 10.1534/genetics.110.123299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Celotto AM, Frank AC, Seigle JL, Palladino MJ. Drosophila model of human inherited triosephosphate isomerase deficiency glycolytic enzymopathy. Genetics. 2006;174:1237–1246. doi: 10.1534/genetics.106.063206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Celotto AM, Liu Z, Vandemark AP, Palladino MJ. A novel Drosophila SOD2 mutant demonstrates a role for mitochondrial ROS in neurodevelopment and disease. Brain Behav. 2012;2:424–434. doi: 10.1002/brb3.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duttaroy A, Paul A, Kundu M, Belton A. A Sod2 null mutation confers severely reduced adult life span in Drosophila. Genetics. 2003;165:2295–2299. doi: 10.1093/genetics/165.4.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fergestad T, Bostwick B, Ganetzky B. Metabolic disruption in Drosophila bang-sensitive seizure mutants. Genetics. 2006;173:1357–1364. doi: 10.1534/genetics.106.057463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pavlidis P, Ramaswami M, Tanouye MA. The Drosophila easily shocked gene: a mutation in a phospholipid synthetic pathway causes seizure, neuronal failure, and paralysis. Cell. 1994;79:23–33. doi: 10.1016/0092-8674(94)90397-2. [DOI] [PubMed] [Google Scholar]
- 31.Celotto AM, Chiu WK, Van Voorhies W, Palladino MJ. Modes of metabolic compensation during mitochondrial disease using the Drosophila model of ATP6 dysfunction. PLoS One. 2011;6:e25823. doi: 10.1371/journal.pone.0025823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hekmat-Scafe DS, Lundy MY, Ranga R, Tanouye MA. Mutations in the K+/Cl- cotransporter gene kazachoc (kcc) increase seizure susceptibility in Drosophila. J Neurosci. 2006;26:8943–8954. doi: 10.1523/JNEUROSCI.4998-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glasscock E, Singhania A, Tanouye MA. The mei-P26 gene encodes a RING finger B-box coiled-coil-NHL protein that regulates seizure susceptibility in Drosophilia. Genetics. 2005;170:1677–1689. doi: 10.1534/genetics.105.043174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glasscock E, Tanouye MA. Drosophila couch potato mutants exhibit complex neurological abnormalities including epilepsy phenotypes. Genetics. 2005;169:2137–2149. doi: 10.1534/genetics.104.028357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song J, Hu J, Tanouye M. Seizure suppression by top1 mutations in Drosophila. J Neurosci. 2007;27:2927–2937. doi: 10.1523/JNEUROSCI.3944-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ashmore LJ, Hrizo SL, Paul SM, Van Voorhies WA, Beitel GJ, Palladino MJ. Novel mutations affecting the Na, K ATPase alpha model complex neurological diseases and implicate the sodium pump in increased longevity. Hum Genet. 2009;126:431–447. doi: 10.1007/s00439-009-0673-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng Y, Huynh L, Takeyasu K, Fambrough DM. The Drosophila Na, K-ATPase alpha-subunit gene: gene structure, promoter function and analysis of a cold-sensitive recessive-lethal mutation. Genes Funct. 1997;1:99–117. doi: 10.1046/j.1365-4624.1997.00006.x. [DOI] [PubMed] [Google Scholar]
- 38.Fergestad T, Ganetzky B, Palladino MJ. Neuropathology in Drosophila membrane excitability mutants. Genetics. 2006;172:1031–1042. doi: 10.1534/genetics.105.050625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schubiger M, Feng Y, Fambrough DM, Palka J. A mutation of the Drosophila sodium pump alpha subunit gene results in bang-sensitive paralysis. Neuron. 1994;12:373–381. doi: 10.1016/0896-6273(94)90278-X. [DOI] [PubMed] [Google Scholar]
- 40.Sun B, Xu P, Wang W, Salvaterra PM. In vivo modification of Na(+), K(+)-ATPase activity in Drosophila. Comp Biochem Physiol B Biochem Mol Biol. 2001;130:521–536. doi: 10.1016/S1096-4959(01)00470-5. [DOI] [PubMed] [Google Scholar]
- 41.Lee LA, Elfring LK, Bosco G, Orr-Weaver TL. A genetic screen for suppressors and enhancers of the Drosophila PAN GU cell cycle kinase identifies cyclin B as a target. Genetics. 2001;158:1545–1556. doi: 10.1093/genetics/158.4.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raftery LA, Twombly V, Wharton K, Gelbart WM. Genetic screens to identify elements of the decapentaplegic signaling pathway in Drosophila. Genetics. 1995;139:241–254. doi: 10.1093/genetics/139.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szuplewski S, Kugler JM, Lim SF, Verma P, Chen YW, Cohen SM. MicroRNA transgene overexpression complements deficiency-based modifier screens in Drosophila. Genetics. 2012;190:617–626. doi: 10.1534/genetics.111.136689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herold N, Will CL, Wolf E, Kastner B, Urlaub H, Luhrmann R. Conservation of the protein composition and electron microscopy structure of Drosophila melanogaster and human spliceosomal complexes. Mol Cell Biol. 2009;29:281–301. doi: 10.1128/MCB.01415-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pace KE, Lebestky T, Hummel T, Arnoux P, Kwan K, Baum LG. Characterization of a novel Drosophila melanogaster galectin. expression in developing immune, neural, and muscle tissues. J Biol Chem. 2002;277:13091–13098. doi: 10.1074/jbc.M112105200. [DOI] [PubMed] [Google Scholar]
- 46.Kim J, Kim YJ, Kim-Ha J. Blood–brain barrier defects associated with Rbp9 mutation. Mol Cells. 2010;29:93–98. doi: 10.1007/s10059-010-0040-0. [DOI] [PubMed] [Google Scholar]
- 47.Romero MF, Henry D, Nelson S, Harte PJ, Dillon AK, Sciortino CM. Cloning and characterization of a Na + −driven anion exchanger (NDAE1). a new bicarbonate transporter. J Biol Chem. 2000;275:24552–24559. doi: 10.1074/jbc.M003476200. [DOI] [PubMed] [Google Scholar]
- 48.Godenschwege TA, Hu H, Shan-Crofts X, Goodman CS, Murphey RK. Bi-directional signaling by Semaphorin 1a during central synapse formation in Drosophila. Nat Neurosci. 2002;5:1294–1301. doi: 10.1038/nn976. [DOI] [PubMed] [Google Scholar]
- 49.Yu L, Zhou Y, Cheng S, Rao Y. Plexin a-semaphorin-1a reverse signaling regulates photoreceptor axon guidance in Drosophila. J Neurosci. 2010;30:12151–12156. doi: 10.1523/JNEUROSCI.1494-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olson PF, Fessler LI, Nelson RE, Sterne RE, Campbell AG, Fessler JH. Glutactin, a novel Drosophila basement membrane-related glycoprotein with sequence similarity to serine esterases. EMBO J. 1990;9:1219–1227. doi: 10.1002/j.1460-2075.1990.tb08229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haug-Collet K, Pearson B, Webel R, Szerencsei RT, Winkfein RJ, Schnetkamp PP, Colley NJ. Cloning and characterization of a potassium-dependent sodium/calcium exchanger in Drosophila. J Cell Biol. 1999;147:659–670. doi: 10.1083/jcb.147.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu L, Leonard AS, Motto DG, Feller MA, Price MP, Johnson WA, Welsh MJ. Contribution of Drosophila DEG/ENaC genes to salt taste. Neuron. 2003;39:133–146. doi: 10.1016/S0896-6273(03)00394-5. [DOI] [PubMed] [Google Scholar]
- 53.Goel M, Garcia R, Estacion M, Schilling WP. Regulation of Drosophila TRPL channels by immunophilin FKBP59. J Biol Chem. 2001;276:38762–38773. doi: 10.1074/jbc.M104125200. [DOI] [PubMed] [Google Scholar]
- 54.Day JP, Dow JA, Houslay MD, Davies SA. Cyclic nucleotide phosphodiesterases in Drosophila melanogaster. Biochem J. 2005;388:333–342. doi: 10.1042/BJ20050057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lochhead PA, Sibbet G, Kinstrie R, Cleghon T, Rylatt M, Morrison DK, Cleghon V. dDYRK2: a novel dual-specificity tyrosine-phosphorylation-regulated kinase in Drosophila. Biochem J. 2003;374:381–391. doi: 10.1042/BJ20030500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bergmann A, Stein D, Geisler R, Hagenmaier S, Schmid B, Fernandez N, Schnell B, Nusslein-Volhard C. A gradient of cytoplasmic Cactus degradation establishes the nuclear localization gradient of the dorsal morphogen in Drosophila. Mech Dev. 1996;60:109–123. doi: 10.1016/S0925-4773(96)00607-7. [DOI] [PubMed] [Google Scholar]
- 57.Zhang T, Liu Z, Song W, Du Y, Dong K. Molecular characterization and functional expression of the DSC1 channel. Insect Biochem Mol Biol. 2011;41:451–458. doi: 10.1016/j.ibmb.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Funderburk CD, Bowling KM, Xu D, Huang Z, O’Donnell JM. A typical N-terminal extensions confer novel regulatory properties on GTP cyclohydrolase isoforms in Drosophila melanogaster. J Biol Chem. 2006;281:33302–33312. doi: 10.1074/jbc.M602196200. [DOI] [PubMed] [Google Scholar]
- 59.Sokabe T, Tsujiuchi S, Kadowaki T, Tominaga M. Drosophila painless is a Ca2 + −requiring channel activated by noxious heat. J Neurosci. 2008;28:9929–9938. doi: 10.1523/JNEUROSCI.2757-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alonso J, Santaren JF. Characterization of the Drosophila melanogaster ribosomal proteome. J Proteome Res. 2006;5:2025–2032. doi: 10.1021/pr0601483. [DOI] [PubMed] [Google Scholar]
- 61.Zhu B, Pennack JA, McQuilton P, Forero MG, Mizuguchi K, Sutcliffe B, Gu CJ, Fenton JC, Hidalgo A. Drosophila neurotrophins reveal a common mechanism for nervous system formation. PLoS Biol. 2008;6:e284. doi: 10.1371/journal.pbio.0060284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McIlroy G, Foldi I, Aurikko J, Wentzell JS, Lim MA, Fenton JC, Gay NJ, Hidalgo A. Toll-6 and Toll-7 function as neurotrophin receptors in the Drosophila melanogaster CNS. Nat Neurosci. 2013;16:1248–1256. doi: 10.1038/nn.3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.White-Cooper H, Schafer MA, Alphey LS, Fuller MT. Transcriptional and post-transcriptional control mechanisms coordinate the onset of spermatid differentiation with meiosis I in Drosophila. Development. 1998;125:125–134. doi: 10.1242/dev.125.1.125. [DOI] [PubMed] [Google Scholar]
- 64.Jiang J, Benson E, Bausek N, Doggett K, White-Cooper H. Tombola, a tesmin/TSO1-family protein, regulates transcriptional activation in the Drosophila male germline and physically interacts with always early. Development. 2007;134:1549–1559. doi: 10.1242/dev.000521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee JD, Treisman JE. Sightless has homology to transmembrane acyltransferases and is required to generate active Hedgehog protein. Curr Biol. 2001;11:1147–1152. doi: 10.1016/S0960-9822(01)00323-2. [DOI] [PubMed] [Google Scholar]
- 66.Micchelli CA, The I, Selva E, Mogila V, Perrimon N. Rasp, a putative transmembrane acyltransferase, is required for Hedgehog signaling. Development. 2002;129:843–851. doi: 10.1242/dev.129.4.843. [DOI] [PubMed] [Google Scholar]
- 67.Jiang J, Struhl G. Regulation of the Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat protein Slimb. Nature. 1998;391:493–496. doi: 10.1038/35154. [DOI] [PubMed] [Google Scholar]
- 68.Spencer E, Jiang J, Chen ZJ. Signal-induced ubiquitination of IkappaBalpha by the F-box protein Slimb/beta-TrCP. Genes Dev. 1999;13:284–294. doi: 10.1101/gad.13.3.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Langevin J, Morgan MJ, Sibarita JB, Aresta S, Murthy M, Schwarz T, Camonis J, Bellaiche Y. Drosophila exocyst components Sec5, Sec6, and Sec15 regulate DE-Cadherin trafficking from recycling endosomes to the plasma membrane. Dev Cell. 2005;9:365–376. doi: 10.1016/j.devcel.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 70.Mehta SQ, Hiesinger PR, Beronja S, Zhai RG, Schulze KL, Verstreken P, Cao Y, Zhou Y, Tepass U, Crair MC, Bellen HJ. Mutations in Drosophila sec15 reveal a function in neuronal targeting for a subset of exocyst components. Neuron. 2005;46:219–232. doi: 10.1016/j.neuron.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 71.Lundstrom A, Gallio M, Englund C, Steneberg P, Hemphala J, Aspenstrom P, Keleman K, Falileeva L, Dickson BJ, Samakovlis C. Vilse, a conserved Rac/Cdc42 GAP mediating Robo repulsion in tracheal cells and axons. Genes Dev. 2004;18:2161–2171. doi: 10.1101/gad.310204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chintapalli VR, Wang J, Dow JA. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet. 2007;39:715–720. doi: 10.1038/ng2049. [DOI] [PubMed] [Google Scholar]
- 73.Cronmiller C, Schedl P, Cline TW. Molecular characterization of daughterless, a Drosophila sex determination gene with multiple roles in development. Genes Dev. 1988;2:1666–1676. doi: 10.1101/gad.2.12a.1666. [DOI] [PubMed] [Google Scholar]
- 74.Ketel CS, Andersen EF, Vargas ML, Suh J, Strome S, Simon JA. Subunit contributions to histone methyltransferase activities of fly and worm polycomb group complexes. Mol Cell Biol. 2005;25:6857–6868. doi: 10.1128/MCB.25.16.6857-6868.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moore AW, Roegiers F, Jan LY, Jan YN. Conversion of neurons and glia to external-cell fates in the external sensory organs of Drosophila hamlet mutants by a cousin-cousin cell-type respecification. Genes Dev. 2004;18:623–628. doi: 10.1101/gad.1170904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Livingstone MS, Tempel BL. Genetic dissection of monoamine neurotransmitter synthesis in Drosophila. Nature. 1983;303:67–70. doi: 10.1038/303067a0. [DOI] [PubMed] [Google Scholar]
- 77.Dickman DK, Kurshan PT, Schwarz TL. Mutations in a Drosophila alpha2delta voltage-gated calcium channel subunit reveal a crucial synaptic function. J Neurosci. 2008;28:31–38. doi: 10.1523/JNEUROSCI.4498-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kurshan PT, Oztan A, Schwarz TL. Presynaptic alpha2delta-3 is required for synaptic morphogenesis independent of its Ca2 + −channel functions. Nat Neurosci. 2009;12:1415–1423. doi: 10.1038/nn.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cazzamali G, Grimmelikhuijzen CJ. Molecular cloning and functional expression of the first insect FMRFamide receptor. Proc Natl Acad Sci U S A. 2002;99:12073–12078. doi: 10.1073/pnas.192442799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nose A, Mahajan VB, Goodman CS. Connectin: a homophilic cell adhesion molecule expressed on a subset of muscles and the motoneurons that innervate them in Drosophila. Cell. 1992;70:553–567. doi: 10.1016/0092-8674(92)90426-D. [DOI] [PubMed] [Google Scholar]
- 81.Holzl H, Kapelari B, Kellermann J, Seemuller E, Sumegi M, Udvardy A, Medalia O, Sperling J, Muller SA, Engel A, Baumeister W. The regulatory complex of Drosophila melanogaster 26S proteasomes. subunit composition and localization of a deubiquitylating enzyme. J Cell Biol. 2000;150:119–130. doi: 10.1083/jcb.150.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Feng G, Hannan F, Reale V, Hon YY, Kousky CT, Evans PD, Hall LM. Cloning and functional characterization of a novel dopamine receptor from Drosophila melanogaster. J Neurosci. 1996;16:3925–3933. doi: 10.1523/JNEUROSCI.16-12-03925.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Han KA, Millar NS, Grotewiel MS, Davis RL. DAMB, a novel dopamine receptor expressed specifically in Drosophila mushroom bodies. Neuron. 1996;16:1127–1135. doi: 10.1016/S0896-6273(00)80139-7. [DOI] [PubMed] [Google Scholar]
- 84.Wong JJ, Li S, Lim EK, Wang Y, Wang C, Zhang H, Kirilly D, Wu C, Liou YC, Wang H, Yu F. A Cullin1-based SCF E3 ubiquitin ligase targets the InR/PI3K/TOR pathway to regulate neuronal pruning. PLoS Biol. 2013;11:e1001657. doi: 10.1371/journal.pbio.1001657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Watts RJ, Hoopfer ED, Luo L. Axon pruning during Drosophila metamorphosis: evidence for local degeneration and requirement of the ubiquitin-proteasome system. Neuron. 2003;38:871–885. doi: 10.1016/S0896-6273(03)00295-2. [DOI] [PubMed] [Google Scholar]
- 86.Aizman O, Uhlen P, Lal M, Brismar H, Aperia A. Ouabain, a steroid hormone that signals with slow calcium oscillations. Proc Natl Acad Sci U S A. 2001;98:13420–13424. doi: 10.1073/pnas.221315298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hilgenberg LG, Su H, Gu H, O’Dowd DK, Smith MA. Alpha3Na+/K + −ATPase is a neuronal receptor for agrin. Cell. 2006;125:359–369. doi: 10.1016/j.cell.2006.01.052. [DOI] [PubMed] [Google Scholar]
- 88.Bianchi L, Driscoll M. Protons at the gate: DEG/ENaC ion channels help us feel and remember. Neuron. 2002;34:337–340. doi: 10.1016/S0896-6273(02)00687-6. [DOI] [PubMed] [Google Scholar]
- 89.Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev. 2002;82:735–767. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- 90.Guedj E, Belenotti P, Serratrice J, Ene N, Pineau S, Donnet A, Mundler O, Weiller PJ. Partially reversible cortical metabolic dysfunction in familial hemiplegic migraine with prolonged aura. Headache. 2010;50:872–877. doi: 10.1111/j.1526-4610.2010.01634.x. [DOI] [PubMed] [Google Scholar]
- 91.Thomsen LL, Eriksen MK, Roemer SF, Andersen I, Olesen J, Russell MB. A population-based study of familial hemiplegic migraine suggests revised diagnostic criteria. Brain. 2002;125:1379–1391. doi: 10.1093/brain/awf132. [DOI] [PubMed] [Google Scholar]
- 92.Thomsen LL, Ostergaard E, Olesen J, Russell MB. Evidence for a separate type of migraine with aura: sporadic hemiplegic migraine. Neurology. 2003;60:595–601. doi: 10.1212/01.WNL.0000046524.25369.7D. [DOI] [PubMed] [Google Scholar]