

Abstract
Damage from spinal cord injury occurs in two phases – the trauma of the initial mechanical insult and a secondary injury to nervous tissue spared by the primary insult. Apart from damage sustained as a result of direct trauma to the spinal cord, the post-traumatic inflammatory response contributes significantly to functional motor deficits exacerbated by the secondary injury. Attenuating the detrimental aspects of the inflammatory response is a promising strategy to potentially ameliorate the secondary injury, and promote significant functional recovery. This review details how the inflammatory component of secondary injury to the spinal cord can be treated currently and in the foreseeable future.
Keywords: acute spinal cord injury, inflammation, treatment, secondary injury, neuroprotective, hypothermia, erythropoietin, estrogen, etanercept, rolipram
Research Highlights
-
(1)
The molecular and cellular events underlying the robust inflammatory response in acute spinal cord injury are key targets for potential pharmacological therapies.
-
(2)
Appropriate modulation of post-traumatic inflammation, to augment its beneficial aspects and attenuate its detrimental aspects, may promote functional recovery.
-
(3)
Anti-inflammatory mechanisms of systemic hypothermia, and minocycline and erythropoietin therapies, suggest the promise of clinical interventions for spinal cord injury, without the risks associated with high dose steroid therapy.
-
(4)
Preclinical therapeutic agents studied in experimental models should continue to be pursued to establish more effective treatments for inflammation after acute spinal cord injury.
Abbreviations
CNS, central nervous system; TNF-α, tumor necrosis factor α; IL-1β, interleukin-1β; IL-6, interleukin-6; MP, methylprednisolone; EPO, erythropoietin; cAMP, cyclic adenosine monophosphate; PDE, phosphodiesterase
INTRODUCTION
Spinal cord injury affects an estimated 250 000 persons in the United States with an incidence of approximately 12 000 new cases per year[1]. Most of these injuries are produced by trauma which accounts for about 10 000 of the new cases annually[2]. The life expectancy for spinal cord injured persons remains below that for uninjured persons though it continues to increase with advances in spinal cord injury emergency care and rehabilitation. The mortality rate for injured persons is 750 to 1 000 deaths per year. Septicemia, pneumonia, and other respiratory conditions have replaced renal failure as the leading causes of death among spinal cord injured persons[1]. The prognosis for recovery after spinal cord injury remains challenged by severe motor and sensory deficits and the onset of secondary co-morbidities. The robust inflammatory response elicited by traumatic injury to the spinal cord has a central and complex role in the onset and persistence of these conditions. Inflammation contributes to a milieu in the injured spinal cord which promotes cell death and tissue destruction, and is largely inhibitory to neural repair and regeneration. Hence, molecular and cellular mediators of the immune system have been considered as possible therapeutic targets for spinal cord injury. Despite the advancement of multiple neuroprotective agents into clinical trials, the therapeutic efficacy of these agents has been largely inadequate. There are no current treatments for spinal cord injury which effectively alleviate the associated pathology, such that only limited regeneration and functional recovery can be achieved.
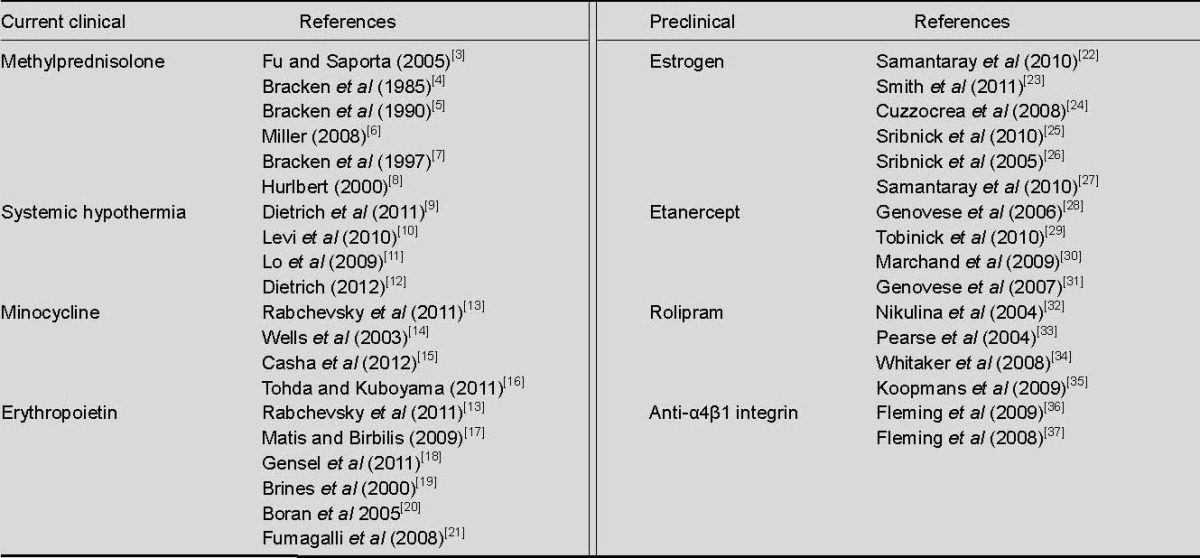
In this review, the contribution of the post-traumatic inflammatory response to secondary degeneration in spinal cord injury is highlighted through the discussion of established and prospective therapies intended to attenuate the detrimental aspects, and augment the beneficial properties of inflammation. Established clinical treatments, therapies evaluated in clinical trials, and preclinical treatments presently under investigation, are examined and discussed (Table 1).
Table 1.
Most significant treatments for inflammation in current clinical practice and preclinical investigations
PATHOPHYSIOLOGY
Spinal cord trauma can be caused by a number of injuries to the spine. Traumatic injury typically results from vertebral fractures or dislocations, which produce bony fragments that may compress or contuse the spinal cord. The direct insult causes glial and nerve cell damage, and disruption of neural tracks, forming a primary lesion characterized by vascular damage, hemorrhaging, tissue disruption, and ischemia[38]. The primary injury typically occurs within the first 20 minutes of spinal cord trauma and is essentially untreatable.
Subsequent damage to healthy tissue, spared by the initial insult, is described as the secondary injury, which is potentially treatable. The mechanisms which underlie the onset and progression of secondary degeneration include blood-spinal cord barrier breakdown, ischemia, free radical formation, oxidative stress, excitotoxicity, cellular dysfunction, and inflammatory and immune changes[13,38,39]. Ultimately resulting in tissue necrosis, and the progressive loss of neurons and oligodendrocytes, these vascular, biochemical and cellular events are largely initiated and orchestrated by the immune response to spinal cord injury[39].
Ischemic conditions at the primary lesion generate oxygen free radicals, resulting in considerable damage to cell structures or oxidative stress. Free radicals induce progressive lipid peroxidation in cell membranes, which significantly contributes to neural tissue damage observed in spinal cord injury[40]. Injury stimulates biochemical pathways that increase the release of excitatory neurotransmitters by neurons and glia into the extracellular space, and also impairs the ability of astrocytes to clear and metabolize them[38,41]. Consequently, neurotransmitters accumulate to cytotoxic levels.
Most notably, glutamate release causes excessive stimulation and prolonged excitation of neurons and oligodendrocytes by allowing a high influx of calcium ions (Ca2+) into the cell. Elevated intracellular Ca2+ activates proteases which damage and degrade cell structures. The result is neuronal and oligodendroglial cell death by excitotoxicity[38].
Vascular endothelial cells of spinal cord blood vessels, damaged by the mechanical injury, secrete vasoactive substances, such as nitric oxide, that produce extravasation of plasma proteins to cause spinal cord tissue edema. The combined effect of increased vascular permeability and the upregulation of adhesion molecules by endothelial cells, permit the infiltration of peripheral immune cells into the spinal cord, a hallmark of the onset of inflammation in acute spinal cord injury.
As resident central nervous system (CNS) macrophages, microglia are first-responders in the cellular line of host defense against tissue damage sustained in spinal cord trauma[42]. The primary injury triggers microglial activation and production of pro-inflammatory cytokines, specifically, tumor necrosis factor alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6)[38]. These cytokines exacerbate the initial damage to the spinal cord, contribute to several aspects of secondary degeneration, and most importantly, play a central role in orchestrating the inflammatory response immediately after injury. Collectively, they promote synthesis of adhesion molecules and chemotactic factors by endothelial cells responsible for mobilizing monocytes and neutrophils from the blood to the primary lesion. Peripheral immune cells also produce pro-inflammatory cytokines, further increasing the extent of the inflammatory response.
Prolonged secretion of IL-1β and TNF-α is implicated in the damage or loss of myelin sheaths, resulting in oligodendroglial and neuronal cell death[39,41].
Furthermore, these potent cytokines stimulate reactive astrogliosis and formation of a glial scar. Astrogliosis has been shown to have both beneficial and detrimental effects on repair after spinal cord injury[41]. Although glial scarring serves to contain the spread of tissue damage, it presents challenges to neurogenesis by also serving as a physical and chemical barrier to axonal regeneration.
The specific role of each type of immune cell in the milieu of the injured spinal cord still requires further investigation. In general, the removal of necrotic tissue and cellular debris at the lesion has been attributed to immune cells[39]. Contrary to reports that microglia and peripheral macrophages play a role in wound healing[43], they have also been implicated in damage to healthy tissue initially spared by the mechanical injury[39]. Phagocytosis by peripheral macrophages and neutrophils generates free radicals which increases the magnitude of secondary damage in the spinal cord due to oxidation of proteins, DNA, and lipids[39]. For example, reactive oxygen intermediates and NO generated during phagocytosis, induce glial and neuronal apoptosis. Peripheral macrophages, neutrophils, and microglia also promote vascular permeability, leukocyte infiltration, and secondary damage in the injured spinal cord through the secretion of matrix metalloproteinases[44].
CLINICAL INFLAMMATORY TREATMENTS
Methylprednisolone (MP)
The neuroprotective potential of corticosteroids in traumatic spinal cord injury has been investigated largely in animal studies on the glucocorticosteroid MP. MP inhibits free oxygen radical-induced lipid peroxidation which is essential to the degenerative events that comprise the secondary injury[45,46,47]. High dose MP is reported to protect against secondary neuronal degeneration[47], decrease accumulation of intracellular calcium, reduce tissue necrosis[45], limit progression of spinal cord ischemia[47], and improve neurological recovery in animal models of spinal cord injury[45,46]. MP is believed to intercalate into cell membranes where it inhibits the propagation of peroxidation reactions locally[47]. Furthermore, high dose MP inhibits the release of interleukins known to exacerbate post-traumatic inflammation[3]. Given the above, MP therapy for acute spinal cord injury was evaluated in three successive multi-center clinical trials on human subjects.
The first trial, the National Acute Spinal Cord Injury Study (NASCIS I), compared the efficacy of low and high dose MP on neurological recovery after acute spinal cord injury[48]. The results failed to show a difference in the degree of neurological improvement produced by the low and high dose regimen at 6 weeks and 6 months after injury[48]. Patients given either treatment regimen also had almost identical rates of neurological recovery at the 1-year follow-up[4]. Notably, NASCIS I did not compare either treatment regimen to a placebo, providing additional reason to study MP further in subsequent clinical trials. The second trial, NASCIS II, examined a higher dose of MP (30 mg/kg) than in the high dose regimen of NASCIS I (14.3 mg/kg), which was believed to be closer to the theoretical threshold for any therapeutic effect[5]. NASCIS II also examined naloxone, an opiate receptor blocker, as a potential therapy, and was placebo-controlled. For all measures of functional motor and sensory recovery, the MP group consistently outperformed the naloxone group at 6 weeks and 6 months after injury; however, this result was only concluded for patients who were administered treatment within 8 hours of injury.
Consequently, the results of the NASCIS trials led to MP as the first drug therapy to improve neurological recovery of patients after spinal cord injury[5]. Advocated by such organizations as the National Institute of Neurological Disorders and Stroke (NINDS) and the National Institute of Health (NIH)[6,49], the NASCIS II protocol was widely disseminated and became the standard for administration of the steroid in a large proportion of hospitals in the United States[6,49]. The recommended treatment regimen is an initial bolus of MP (30 mg/kg over 15 minutes), followed by a 24-hour infusion at 5.4 mg/kg per hour within 8 hours of injury for optimal outcomes[5]. In the subsequent NASCIS III trial, which compared the effects of 24-hour and 48-hour infusions of MP on neurological recovery, a 48-hour infusion was reported to improve recovery for a subgroup of patients at 6 weeks and 6 months after injury[7], as well as 1 year after injury[50]. Notably, this subgroup received treatment between 3 and 8 hours after injury. However, no difference in recovery of patients was reported if MP therapy was initiated within 3 hours of injury, regardless of the length of infusion[50].
Despite support for the benefits of early steroid treatment after acute spinal cord injury, the efficacy of MP remains moderate at best. An analysis by the AANS/CNS Joint Section on Disorders of the Spine and Peripheral Nerves on the clinical care of spinal cord injuries recommends usage of corticosteroids as an option, but with the understanding that the risks of such treatment outweighs any benefits suggested by clinical evidence[51]. Furthermore, MP has not yet received approval as a treatment for spinal cord injury by the Food and Drug Administration (FDA)[49,52]. Several complications, perhaps associated with the high dosage regimen, were reported in NASCIS II and III: increased incidences of wound infections, pulmonary emboli, gastrointestinal hemorrhages, severe sepsis, and severe pneumonias[5,7]. The reported respiratory complications associated with the 48-hour infusion therapy resulted in a six-fold higher mortality rate in the 48-hour group as compared to the 24-hour group. In his independent evaluation of the NASCIS II and III results, Hurlbert asserts this high incidence of mortality is cause to consider a 48-hour infusion of MP after spinal cord injury as potentially harmful and possibly lethal to patients[8].
More importantly, Hurlbert's review revealed a lack of compelling data, inappropriate statistical methods employed to determine the significance of the results, exclusion of measurable outcomes important to patients, and the absence of reproducible results validated by peer review. In short, these inadequacies undermined the credibility of NASCIS II and III, which in fact, failed to demonstrate improvement in primary outcome measures due to MP therapy[8]. Similar criticisms are made in several other reviews of the NASCIS trials[6,49,53]. Subsequently, steroid therapy was relegated to experimental use in clinical spinal cord injury, and not recommended as a standard of care[8]. The reported shortcomings of the NASCIS trials have certainly disputed the reliability and promise of high dose MP as an effective therapy, and resulted in its use largely at the discretion of the practicing physician and medical institution. As a result of a risk-benefit analysis determined at our institution, some of our physicians do not use a high dose steroid regimen in the clinical management of spinal cord injury.
Systemic hypothermia
Historically, systemic hypothermia has been implemented to protect nervous tissue from cerebral ischemia and trauma[9]. Preclinical research demonstrates lowering core body temperature to 32–34°C is protective against neurologic injury without any deleterious systemic effects of cooling[9,10]. The safety and efficacy of moderate, systemic hypothermia has been suggested in experimental models of stroke, cardiac arrest, and brain and spinal cord injury[9]. In addition to improving locomotor function in rats after spinal cord injury[11,54,55], early initiation of hypothermia reduces the area of tissue damage in the spinal cord[54], and preserves gray and white matter structures through sparing of neurons and axonal connections[11]. Hypothermic treatment after spinal cord injury reduces microglial activation[55], decreases accumulation of microglia and granular leukocytes[10], decreases swelling of axons and spinal cord parenchyma, reduces tissue hemorrhage, and decreases apoptosis, oxidative stress, and glutamate release, in the spinal cord. Thus, by modulating events of secondary degeneration, hypothermia is both neuroprotective and anti-inflammatory.
Local hypothermia has already been used to reduce spinal cord temperatures during laminectomy or decompression surgery in clinical trials[9,10]. However, the small cohort of patients, lack of randomization and absence of placebo groups in these trials detracted from the statistical power of the findings. Local hypothermia was also initiated as part of a combinatorial treatment, making it difficult to consider any of the clinical outcomes as an effect of spinal cord cooling specifically[10,56]. Other limiters to more widespread use of local hypothermia in spinal cord injury management include technical difficulties in administration, risk of local infection, and maintenance of cooling for extended periods of time[11]. Hence, there has been increasing focus on systemic hypothermia therapy in clinical spinal cord injury. Although preliminary guidelines for its application in spinal cord injury have been developed at some institutions[10], these have yet to be peer-reviewed and published[9]. The American Association of Neurological Surgeons/Congress of Neurological Surgeons (AANS/CNS) Joint Section on Disorders of the Spine and Peripheral Nerves, and the AANS/CNS Joint Section on Trauma, requires controlled clinical trials in order to establish systemic hypothermia as a treatment option for spinal cord injury[9]. To date, the safety and efficacy of systemic hypothermia has only been characterized in small population of patients with cervical spinal cord injuries[9].
Recently, Levi et al[56] conducted a phase I clinical trial in 14 patients presenting with acute, complete ASIA A C4-6 spinal injuries. Moderate hypothermia was administered to patients through endovascular techniques for 48 hours, in the absence of any pharmacological agent. This duration of cooling parallels the critical window of time in which the degenerative events of the secondary injury are induced, if hypothermia is initiated within hours after injury[11]. Furthermore, all patients received spinal decompression surgery during cooling, or at the target body temperature of 33°C. On average, hypothermia was initiated within 9 hours of injury. At the time of follow-up, 1 year post-injury, six patients in the hypothermia group experienced some recovery of motor function to improve by one or more ASIA grade[56]. No further deterioration in injury was observed in the remaining patients of the hypothermia group. Although more patients improved in ASIA grade after hypothermia treatment compared to control patients, the improvement in the final ASIA grade was not reported to be significant.
Respiratory complications were commonly reported including atelectasis, pneumonia, pleural effusions, iatrogenic or traumatic pneumothorax, pulmonary edema, and acute respiratory distress syndrome[56]. Urinary tract infections, anemia, and acid-base, water, and electrolyte imbalances, were also prevalent. Nevertheless, the incidence and extent of these complications in the control group was comparable. Although some measure of functional recovery is known to occur spontaneously after spinal cord injury, the improvements reported in this study, as indicated by ASIA grade conversions, exceed improvements typically observed without treatment. Given the small patient population, the study lacked the statistical power to adequately evaluate the efficacy of hypothermia, but is sufficient to support the design of phase II and III multicenter trials. Accordingly, efforts are currently being made to implement a larger, randomized, multi-center trial to better define the therapeutic value of systemic hypothermia in acute spinal cord injury[12].
TREATMENTS IN CLINICAL TRIALS
Minocycline
Minocycline is a synthetic derivative of a tetracycline antibiotic which modulates several mechanisms of secondary degeneration in spinal cord injury through anti-apoptotic and anti-inflammatory effects[13,14,15]. Minocycline inhibits the expression and activity of inflammatory cytokines, free radicals, and matrix metalloproteinases, all of which are mediators of tissue injury[14]. Alterations, in these mechanisms may account for attenuated secondary degeneration and improved functional recovery reported in various animal models of spinal cord injury after minocycline therapy[16]. In one study, minocycline-treated animals had improved recovery of hindlimb function, compared with vehicle-treated animals, as early as 3 days post-injury. While the minocycline group achieved weight-supported plantar stepping on average at 4-week post-injury, the vehicle group achieved only slight movements of the hip, knee, and ankle joints, on average[14]. Furthermore, superior behavioral recovery after spinal cord injury was attributed to minocycline as compared to MP. Its ability to ameliorate inflammation is mediated in part by inhibition of microglial activation during spinal cord injury[16]. Its neuroprotective effects are reported in animal models of other CNS trauma and pathologies. Minocycline inhibits inflammatory induced apoptotic effects in stroke, Huntington's disease, Parkinson's disease, amyotrophic lateral sclerosis, and multiple sclerosis[13,16]. Sufficient evidence led to a single-center, randomized, placebo-controlled, phase I/II clinical trial on acute spinal cord injury patients with complete and incomplete spinal cord lesions[15]. Preliminary data suggests intravenous minocycline administered 7 days after traumatic spinal cord injury produces moderate improvement in the motor function of patients. These findings have encouraged further study of minocycline in a multi-center, phase III trial in order to evaluate the efficacy of minocycline in treating inflammation in spinal cord injury.
Erythropoietin (EPO)
The role of the endogenous growth factor EPO is to stimulate proliferation and differentiation of erythroid precursor cells; however, EPO exhibits non-hematopoietic effects in the CNS[13]. Studies show endogenous expression of EPO occurs in the spinal cord after injury as part of a physiological response to hypoxia[17]. EPO attenuates secondary spinal cord injury by reducing inflammation and apoptosis, promoting tissue sparing, and restoring vascular integrity[17,18]. The anti-inflammatory effect is mediated by reduced infiltration of leukocytes into the injured spinal cord and protection of neurons against apoptosis[17,19]. In one study, exposure of neurons to an apoptosis-inducing toxin elicited glial cell production of TNF; however, treatment of neurons with EPO inhibited production of the pro-inflammatory cytokine[17]. Neurological and locomotor recovery is also enhanced after experimental spinal cord injury with exogenous EPO therapy[13,20]. In a model of spinal cord injury caused by contusion and compression, recombinant human EPO (rhEPO) produced superior functional motor recovery within 12 hours and 4 days after injury, respectively[57]. Furthermore, reduced detection of inflammatory cells in stained sections of contused spinal cord also suggested secondary inflammation was attenuated with rhEPO therapy.
The neuroprotective properties of EPO, and availability of non-hematopoietic analogues, encouraged its evaluation in clinical trials for acute spinal cord injury[17]. rhEPO has already been assessed in clinical trials as a potential therapy for stroke patients with non-hemorrhagic infarcts of the middle cerebral artery. rhEPO improved outcomes after stroke when administered within 8 hours of the onset of symptoms[17]. Currently underway is a phase III clinical trial evaluating the safety and efficacy of acute EPO administration (compared to MP) on clinical outcome, sensory and motor function, neurogenic pain, and spasticity in spinal cord injury patients; however, preliminary data is not yet available[13,18,58]. Nonetheless, the therapeutic potential of EPO has been rarely reported. Studies have published results contrary to benefits reported for EPO or its derivatives[18]. Chronic EPO treatment may exacerbate CNS injury through increased red blood cell volume, thus an EPO analogue lacking hematopoietic effects may avert these side effects[21].
PRECLINICAL INFLAMMATORY TREATMENTS
Estrogen
The hormone estrogen is currently considered to be a powerful, multi-active neuroprotective agent[22]. Several studies demonstrate its therapeutic potential in neurodegenerative disorders including stroke, multiple sclerosis, and Parkinson's disease[23]. Estrogen classically binds signals through a nuclear receptor to alter mRNA transcription and protein expression. Two types of receptors exist, estrogen receptor alpha, and estrogen receptor beta, of which only estrogen receptor beta has a significant role in the CNS[22]. Growing scientific evidence from animal models supports estrogen as a potential therapy for acute spinal cord injury. Notably, most of these studies have utilized 17β- estradiol[24], an endogenous form of the hormone, which activates the estrogen receptor beta[22,23,25,26,27].
High dose estrogen administered immediately after acute spinal cord injury reduced spinal cord tissue edema, infiltration of inflammatory cells, and myelin loss at the lesion site as well as at penumbral areas of the lesion, increasing preservation of spinal cord architecture[26]. These outcomes were correlated with increased nuclear factor-kappa B, a transcriptional regulator involved in inflammatory responses[26]. Although some studies have utilized high dose estrogen in animal models of spinal cord injury with beneficial outcomes[26], estrogen administered at a low dosage is believed to minimize any undesirable effects of hormone therapy that may occur[27]. Samantaray et al[27] demonstrated low dose estrogen promotes survival of sensory and motor neurons in spinal cord injured rats, suggesting recovery of the neuronal population in the injured cord is important for reversing functional deficits. No side effects were reported for the low dose regimen in the study. Thus, estrogen is believed to mediate neuroprotection through genomic, receptor-dependent transcriptional regulation[22], as well as anti-oxidative, anti-apoptotic, and anti-inflammatory mechanisms[22,23]. Its anti-inflammatory effects are well documented[23,24,25,26]. Recruitment, adhesion, and infiltration of leukocytes into tissues are mediated by chemokines, cytokines, and selectins, whose production can be blocked by estrogen[26]. Smith et al[23] demonstrated this in microglial cultures where estrogen prevented cell death by attenuating the release of pro-inflammatory cytokines, TNF-α, IL-1β, and IL-1α, after lipopolysaccharide toxicity.
In another study, administration of estrogen prior to spinal cord injury also decreased the degree of tissue damage, tissue edema, and myelin loss in the spinal cord[24]. In addition to decreased leukocyte infiltration, reduced expression of potent pro-inflammatory mediators, TNF-α, IL-1β, and IL-6; cytokine-inducible nitric oxide synthase; and cyclooxygenase-2 was detected in spinal cords of injured mice. Pre-treatment with estrogen also ameliorated functional motor deficits in these mice, which is a more clinically relevant outcome. Moreover, estrogen therapy also exhibits anti-inflammatory effects in chronic spinal cord injury, attenuating microglia/macrophage activation and cyclooxygenase-2 activity, and improving motor function[25]. Its efficacy in reducing post-traumatic inflammation, mitigating secondary degeneration in the spinal cord, and relative safety when administered at low dose, suggests estrogen therapy is a strong candidate for application and evaluation in clinical trials.
Etanercept
Etanercept, like other TNF-α inhibitors, has demonstrated anti-inflammatory effects[28]. TNF-α is a major therapeutic target in medicine due to its role as a mediator in inflammatory neurological diseases such as Alzheimer's disease, sciatica, and traumatic brain and spinal cord injury. This was first elucidated after an association between excess TNF-α in the cerebral spinal fluid of patients with rapid progression of Alzheimer's disease was established[29]. After spinal cord injury, increased levels of TNF-α and IL-1β at the primary lesion[28] promote the onset and persistence of inflammation as well as oligodendroglial cell death. Moreover, studies have revealed a link between increased expressions of TNF-α or the TNF-α receptor, with development of forms of neuropathic pain, including spinal cord injury-related allodynia or hyperalgesia[29,30].
Etanercept may antagonize these effects by competitively binding to TNF-α, and favorably modulating the post-traumatic inflammatory response[28]. The goal of drug therapy is to reduce excess TNF-α and not deplete it entirely, in order to restore the cytokine to homeostatic levels[29]. Indeed, reduced TNF-α and IL-1β in the injured spinal cord was detected in an animal model after treatment with etanercept[29]. Genovese et al[31] found etanercept, administered in combination with the glucocorticoid dexamethasone, significantly reduced the degree of spinal cord inflammation after injury. Furthermore, attenuation of inflammation by etanercept has yielded enhanced functional motor recovery in several experimental spinal cord injury models[25,28,29]. Through the regulation of cytokine secretion, etanercept mediates beneficial effects in the injured spinal cord aside from anti-inflammation, including reduced tissue injury, and cell apoptosis[28,30,31].
As a FDA-approved drug, the use of etanercept as a neurological therapeutic for spinal cord injury is promising. Clinical studies on etanercept as treatment for Alzheimer's disease, sciatica, and intervertebral disc-related pain have optimized a method for drug delivery. Perispinal administration via the vertebral venous plexus, instead of conventional epidural and intrathecal routes, is believed to enhance the therapeutic efficacy of etanercept[29]. This approach is less invasive, avoids the risk of dural puncture, and drug distribution is facilitated by the vertebral venous system. This system lacks valves, so blood flow is bidirectional. Thus, etanercept can freely and rapidly distribute in venous blood to the spinal cord. This is believed to explain the increasing pain relief patients with disc-related pain experienced within 2–3 minutes of receiving treatment[29]. The clinical feasibility of etanercept is further strengthened by the localization of its effects to specific spinal structures, such as the spinal cord or spinal nerve roots, which is possible with administration via the vertebral venous plexus.
Rolipram
Rolipram functions in maintaining levels of cyclic adenosine monophosphate (cAMP), an intracellular second messenger, through inhibition of phosphodiesterase 4 (PDE4)[32]. Studies have shown cAMP is a promising target for modulation of the inhibitory environment at the lesion produced by spinal cord trauma[33]. Inhibitory molecules that are upregulated in myelin and the glial scar after injury present challenges for re-growth of axons across the lesion area, and re-establishment of continuous neural pathways in the spinal cord. Elevated intracellular cAMP in neurons, prior to spinal cord injury, can promote regeneration of damaged axons by overcoming inhibitors of regeneration. Nikulina et al[32] reported that rolipram promotes axon regeneration, attenuates the formation of the glial scar, and significantly enhances functional recovery in a hemisection model of spinal cord injury. Rolipram-treated animals experienced reduced astrogliosis and staining density of astrocyte marker glial fibrillary acidic protein, by one-third less than in vehicle-treated animals. In addition to facilitating axonal regeneration, maintaining elevated cAMP with rolipram may also be a mechanism for increased oligodendrocyte survival after spinal cord injury[34].
Elevated cAMP has potent anti-inflammatory effects mediated through inhibition of TNF-α and IL-1β production, and prevention of immune cell activation[33]. However, the dosage of rolipram known to produce such effects is three to six-fold higher than the optimal dose administered in the study by Nikulina et al[32]. In another study, increased oligodendrocyte survival in rolipram-treated injured rats was attributed to decreased production of TNF-α, which exacerbates excitotoxicity and inflammation in spinal cord injury[34]. Rolipram, in concert with thalidomide, also reduced spinal cord lesion size by improving white matter sparing, suggesting attenuated TNF-α and IL-1β expression as the mechanism. Together, modulation of cytokine expression and increased white matter sparing resulted in improved locomotor recovery[35]. Experimental models of ischemia, traumatic brain injury, and spinal cord injury have also demonstrated benefits of lowered TNF-α and IL-1β production or signalling by rolipram. Again, reduced secondary degeneration and improved functional outcomes were attributed to changes in cytokine expression[35].
Since it readily crosses the blood-brain barrier[32], rolipram is a strong candidate as an effective therapy for spinal cord injury. Furthermore, rolipram is already an FDA-approved drug[35]. It can be administered orally or subcutaneously, strengthening its clinical applicability. Its inhibitory target, the enzyme PDE4 subtype, accounts for approximately 70–80% of PDEs present in nervous tissue, thus minimizing the likelihood of side effects on other tissues where the prevalence of PDE4 is lower[32]. However, side effects such as nausea, vomiting and sedation have been reported with its use[35].
Anti-α4β1 Integrin
The integrin family of adhesion molecules plays a key role in the migration, activation, proliferation, and survival of leukocytes. Integrin α4β1 and integrin α4β2 in particular, mediate adhesion and extravasation of leukocytes, facilitating their migration from blood vessels into tissue[36]. Leukocyte activation and migration into the spinal cord tissue is a hallmark of post-traumatic inflammation. Through secretion of pro-inflammatory cytokines and free radicals, leukocytes contribute to ischemia, as well as glial and neuronal cell death. Blocking interactions between integrin molecules and cell-surface receptors can reduce inflammation after acute spinal cord injury[36]. Thus, integrin α4β1 expressed on neutrophils and monocytes/macrophages, is an ideal therapeutic target. Administration of anti-α4β1 integrin (Natalizumab) as treatment for relapsing-remitting multiple sclerosis, has been clinically effective in reducing relapses in patients[59]. However, several cases of progressive multifocal leukoencephalopathy, characterized by myelin damage and other adverse events, have been documented with its repetitive usage[59,60].
Fleming et al[37] reported decreased intra-spinal inflammation in a clip-compression model of spinal cord injury, after antagonizing α4β1 integrin with a monoclonal antibody treatment administered intravenously. Detection of neutrophils and monocytes/macrophages was significantly less in spinal cords treated 2 hours after spinal cord injury compared to untreated spinal cords. Early inhibition of leukocyte infiltration is optimal, as beneficial roles in wound healing and repair have been ascribed to neutrophils and macrophage/monocyte activity at later phases of inflammation[36]. The treatment regimen in the study by Fleming et al[36], limits the effects of the anti-integrin α4β1 antibody at approximately 3 days after treatment is administered. Since treatment was administered immediately after spinal cord injury, it is concomitant with the duration of peak neutrophil infiltration into the spinal cord after injury. Anti-integrin α4β1-treated animals significantly outperformed control animals in motor function[36]. This group also achieved weight-supported stepping earlier than the control group, a clinically relevant change which could delay onset of paraplegia in patients, or better, enable greater stabilization during standing or walking[37]. Other benefits of treatment include increased tissue sparing at the lesion, reduced mechanical allodynia, indicative of decreased neuropathic pain, as well as improved autonomic function. Considering that progressive multifocal leukoencephalopathy remains the greatest risk associated with anti-α4β1 integrin therapy[59], any potential application in clinical spinal cord injury should be with caution and knowledge of any additional reported complications.
CONCLUSION
In the hours following traumatic injury to the spinal cord, activation of the immune system contributes to a complex pathophysiology which exacerbates neurological deficits. Modulating the post-traumatic inflammatory response to mitigate its harmful aspects is one approach under widespread experimentation. The ideal therapeutic agent modulates inflammation to curtail the extent of secondary injury to spinal cord tissue, while promoting reparative mechanisms intrinsic to the CNS. Once purported as a standard of care for acute spinal cord injury, MP has now fallen out of favor after detailed analyses of the NASCIS trials as well as reevaluation of the safety and efficacy of the glucocorticoid steroid. Despite its neuroprotective and anti-inflammatory effects, the associated high risk of mortality and harmful complications, as reported in the NASCIS trials, has increasingly discouraged its use. While steroid therapy may still be implemented at some medical institutions, it is not a recommendation of this review for clinical management of spinal cord injury.
Promisingly, alternative therapies under investigation in preclinical studies and human trials for acute spinal cord injury exhibit anti-inflammatory effects. The prospective therapeutic agents highlighted in this review target the biochemical, molecular or cellular cascade of events underlying inflammation at the spinal cord lesion site. A subset of these therapies is multi-active, affecting multiple levels of the inflammatory response, which further strengthens the potential for their application in clinical spinal cord injury. Systemic hypothermia and etanercept both act on molecular and cellular levels of the inflammatory response, while estrogen exerts effects at the biochemical and molecular levels. Nonetheless, the immune-modulatory properties demonstrated by the remaining therapies, and associated effects on histological, physiological, and functional measures of recovery after spinal cord injury, are encouraging. Ultimately, the progression of more therapeutic agents studied in experimental models of spinal cord injury, which produce the most meaningful outcomes for patients, into clinical trials, will increase prospects for establishing additional beneficial treatments for acute spinal cord injury patients.
Footnotes
Conflicts of interest: None declared.
(Edited by Zhang HL/Jin Y/Song LP)
REFERENCES
- [1].Birmingham: The University of Alabama at Birmingham; [Accessed June 15, 2011]. National Spinal Cord Injury Statistical Center. Spinal Cord Injury Facts and Figures at a Glance. https://www.nscisc.uab.edu/public_content/pdf/Facts%202011%20Feb%20Final.pdf . [Google Scholar]
- [2].Dawodu ST. Spinal Cord Injury-Definition, Epidemiology, Pathophysiology. Medscape Reference. [Accessed June 6, 2011]. http://emedicine.medscape.com/article/322480-overview .
- [3].Fu ES, Saporta S. Methylprednisolone inhibits production of interleukin-1beta and interleukin-6 in the spinal cord following compression injury in rats. J Neurosurg Anesthesiol. 2005;17(2):82–85. doi: 10.1097/01.ana.0000163199.10365.38. [DOI] [PubMed] [Google Scholar]
- [4].Bracken MB, Shepard MJ, Hellenbrand KG, et al. Methylprednisolone and neurological function 1 year after spinal cord injury. Results of the National Acute Spinal Cord Injury Study. J Neurosurg. 1985;63(5):704–713. doi: 10.3171/jns.1985.63.5.0704. [DOI] [PubMed] [Google Scholar]
- [5].Bracken MB, Shepard MJ, Collins WF, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the Second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322(20):1405–1411. doi: 10.1056/NEJM199005173222001. [DOI] [PubMed] [Google Scholar]
- [6].Miller SM. Methylprednisolone in acute spinal cord injury: a tarnished standard. J Neurosurg Anesthesiol. 2008;20(2):140–142. doi: 10.1097/01.ana.0000314442.40952.0d. [DOI] [PubMed] [Google Scholar]
- [7].Bracken MB, Shepard MJ, Holford TR, et al. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the Third National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. JAMA. 1997;277(20):1597–1604. [PubMed] [Google Scholar]
- [8].Hurlbert RJ. Methylprednisolone for acute spinal cord injury: an inappropriate standard of care. J Neurosurg. 2000;93(1 Suppl):1–7. doi: 10.3171/spi.2000.93.1.0001. [DOI] [PubMed] [Google Scholar]
- [9].Dietrich WD, Cappuccino A, Cappuccino H. Systemic hypothermia for the treatment of acute cervical spinal cord injury in sports. Curr Sports Med Rep. 2011;10(1):50–54. doi: 10.1249/JSR.0b013e318205e0b3. [DOI] [PubMed] [Google Scholar]
- [10].Levi AD, Casella G, Green BA, et al. Clinical outcomes using modest intravascular hypothermia after acute cervical spinal cord injury. Neurosurgery. 2010;66(4):670–677. doi: 10.1227/01.NEU.0000367557.77973.5F. [DOI] [PubMed] [Google Scholar]
- [11].Lo TP, Jr, Cho KS, Garg MS, et al. Systemic hypothermia improves histological and functional outcome after cervical spinal cord contusion in rats. J Comp Neurol. 2009;514(5):433–448. doi: 10.1002/cne.22014. [DOI] [PubMed] [Google Scholar]
- [12].Dietrich WD. Therapeutic hypothermia for acute severe spinal cord injury: ready to start large clinical trials? Crit Care Med. 2012;40(2):691–692. doi: 10.1097/CCM.0b013e318236eacb. [DOI] [PubMed] [Google Scholar]
- [13].Rabchevsky AG, Patel SP, Springer JE. Pharmacological interventions for spinal cord injury: Where do we stand?How might we step forward? Pharmacol Ther. 2011;132(1):15–29. doi: 10.1016/j.pharmthera.2011.05.001. [DOI] [PubMed] [Google Scholar]
- [14].Wells JE, Hurlbert RJ, Fehlings MG, et al. Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain. 2003;126(Pt 7):1628–1637. doi: 10.1093/brain/awg178. [DOI] [PubMed] [Google Scholar]
- [15].Casha S, Zygun D, McGowan MD, et al. Results of a phase II placebo-controlled randomized trial of minocycline in acute spinal cord injury. Brain. 2012;135(4):1224–1236. doi: 10.1093/brain/aws072. [DOI] [PubMed] [Google Scholar]
- [16].Tohda C, Kuboyama T. Current and future therapeutic strategies for functional repair of spinal cord injury. Pharmacol Ther. 2011;132(1):57–71. doi: 10.1016/j.pharmthera.2011.05.006. [DOI] [PubMed] [Google Scholar]
- [17].Matis GK, Birbilis TA. Erythropoietin in spinal cord injury. Eur Spine J. 2009;18(3):314–323. doi: 10.1007/s00586-008-0829-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gensel JC, Donnelly DJ, Popovich PG. Spinal cord injury therapies in humans: an overview of current clinical trials and their potential effects on intrinsic CNS macrophages. Expert Opin Ther Targets. 2011;15(4):505–518. doi: 10.1517/14728222.2011.553605. [DOI] [PubMed] [Google Scholar]
- [19].Brines ML, Ghezzi P, Keenan S, et al. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci U S A. 2000;97(19):10526–10531. doi: 10.1073/pnas.97.19.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Boran BO, Colak A, Kutlay M. Erythropoietin enhances neurological recovery after experimental spinal cord injury. Restor Neurol Neurosci. 2005;23(5-6):341–345. [PubMed] [Google Scholar]
- [21].Fumagalli F, Madaschi L, Brenna P, et al. Single exposure to erythropoietin modulates Nerve Growth Factor expression in the spinal cord following traumatic injury: comparison with methylprednisolone. Eur J Pharmacol. 2008;578(1):19–27. doi: 10.1016/j.ejphar.2007.09.021. [DOI] [PubMed] [Google Scholar]
- [22].Samantaray S, Sribnick EA, Das A, et al. Neuroprotective efficacy of estrogen in experimental spinal cord injury in rats. Ann N Y Acad Sci. 2010;1199:90–94. doi: 10.1111/j.1749-6632.2009.05357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Smith JA, Das A, Butler JT, et al. Estrogen or estrogen receptor agonist inhibits lipopolysaccharide induced microglial activation and death. Neurochem Res. 2011;36(9):1587–1593. doi: 10.1007/s11064-010-0336-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cuzzocrea S, Genovese T, Mazzon E, et al. Effect of 17beta-estradiol on signal transduction pathways and secondary damage in experimental spinal cord trauma. Shock. 2008;29(3):362–371. doi: 10.1097/shk.0b013e31814545dc. [DOI] [PubMed] [Google Scholar]
- [25].Sribnick EA, Samantaray S, Das A, et al. Postinjury estrogen treatment of chronic spinal cord injury improves locomotor function in rats. J Neurosci Res. 2010;88(8):1738–1750. doi: 10.1002/jnr.22337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sribnick EA, Wingrave JM, Matzelle DD, et al. Estrogen attenuated markers of inflammation and decreased lesion volume in acute spinal cord injury in rats. J Neurosci Res. 2005;82(2):283–293. doi: 10.1002/jnr.20622. [DOI] [PubMed] [Google Scholar]
- [27].Samantaray S, Matzelle DD, Ray SK, et al. Physiological low dose of estrogen-protected neurons in experimental spinal cord injury. Ann N Y Acad Sci. 2010;1199:86–89. doi: 10.1111/j.1749-6632.2009.05360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Genovese T, Mazzon E, Crisafulli C, et al. Immunomodulatory effects of etanercept in an experimental model of spinal cord injury. J Pharmacol Exp Ther. 2006;316(3):1006–1016. doi: 10.1124/jpet.105.097188. [DOI] [PubMed] [Google Scholar]
- [29].Tobinick E. Perispinal etanercept: a new therapeutic paradigm in neurology. Expert Rev Neurother. 2010;10(6):985–1002. doi: 10.1586/ern.10.52. [DOI] [PubMed] [Google Scholar]
- [30].Marchand F, Tsantoulas C, Singh D, et al. Effects of Etanercept and Minocycline in a rat model of spinal cord injury. Eur J Pain. 2009;13(7):673–681. doi: 10.1016/j.ejpain.2008.08.001. [DOI] [PubMed] [Google Scholar]
- [31].Genovese T, Mazzon E, Crisafulli C, et al. Combination of dexamethasone and etanercept reduces secondary damage in experimental spinal cord trauma. Neuroscience. 2007;150(1):168–181. doi: 10.1016/j.neuroscience.2007.06.059. [DOI] [PubMed] [Google Scholar]
- [32].Nikulina E, Tidwell JL, Dai HN, et al. The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc Natl Acad Sci U S A. 2004;101(23):8786–8790. doi: 10.1073/pnas.0402595101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pearse DD, Pereira FC, Marcillo AE, et al. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med. 2004;10(6):610–616. doi: 10.1038/nm1056. [DOI] [PubMed] [Google Scholar]
- [34].Whitaker CM, Beaumont E, Wells MJ, et al. Rolipram attenuates acute oligodendrocyte death in the adult rat ventrolateral funiculus following contusive cervical spinal cord injury. Neurosci Lett. 2008;438(2):200–204. doi: 10.1016/j.neulet.2008.03.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Koopmans GC, Deumens R, Buss A, et al. Acute rolipram/thalidomide treatment improves tissue sparing and locomotion after experimental spinal cord injury. Exp Neurol. 2009;216(2):490–498. doi: 10.1016/j.expneurol.2009.01.005. [DOI] [PubMed] [Google Scholar]
- [36].Fleming JC, Bao F, Chen Y, et al. Timing and duration of anti-alpha4beta1 integrin treatment after spinal cord injury: effect on therapeutic efficacy. J Neurosurg Spine. 2009;11(5):575–587. doi: 10.3171/2009.6.SPINE08915. [DOI] [PubMed] [Google Scholar]
- [37].Fleming JC, Bao F, Chen Y, et al. Alpha4beta1 integrin blockade after spinal cord injury decreases damage and improves neurological function. Exp Neurol. 2008;214(2):147–159. doi: 10.1016/j.expneurol.2008.04.024. [DOI] [PubMed] [Google Scholar]
- [38].Hausmann ON. Post-traumatic inflammation following spinal cord injury. Spinal Cord. 2003;41(7):369–378. doi: 10.1038/sj.sc.3101483. [DOI] [PubMed] [Google Scholar]
- [39].Jones TB, McDaniel EE, Popovich PG. Inflammatory- mediated injury and repair in the traumatically injured spinal cord. Curr Pharm Des. 2005;11(10):1223–1236. doi: 10.2174/1381612053507468. [DOI] [PubMed] [Google Scholar]
- [40].Hall ED, Braughler JM. Role of lipid peroxidation in post-traumatic spinal cord degeneration: a review. Cent Nerv Syst Trauma. 1986;3(4):281–294. doi: 10.1089/cns.1986.3.281. [DOI] [PubMed] [Google Scholar]
- [41].Donnelly DJ, Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol. 2008;209(2):378–388. doi: 10.1016/j.expneurol.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schwartz M. Macrophages and microglia in central nervous system injury: are they helpful or harmful? J Cereb Blood Flow Metab. 2003;23(4):385–394. doi: 10.1097/01.WCB.0000061881.75234.5E. [DOI] [PubMed] [Google Scholar]
- [43].Elkabes S, DiCicco-Bloom EM, Black IB. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J Neurosci. 1996;16(8):2508–2521. doi: 10.1523/JNEUROSCI.16-08-02508.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fleming JC, Norenberg MD, Ramsay DA, et al. The cellular inflammatory response in human spinal cords after injury. Brain. 2006;129(Pt 12):3249–3269. doi: 10.1093/brain/awl296. [DOI] [PubMed] [Google Scholar]
- [45].Anderson DK, Saunders RD, Demediuk P, et al. Lipid hydrolysis and peroxidation in injured spinal cord: partial protection with methylprednisolone or vitamin E and selenium. Cent Nerv Syst Trauma. 1985;2(4):257–267. doi: 10.1089/cns.1985.2.257. [DOI] [PubMed] [Google Scholar]
- [46].Braughler JM, Hall ED, Means ED, et al. Evaluation of an intensive methylprednisolone sodium succinate dosing regimen in experimental spinal cord injury. J Neurosurg. 1987;67(1):102–105. doi: 10.3171/jns.1987.67.1.0102. [DOI] [PubMed] [Google Scholar]
- [47].Hall ED. Neuroprotective actions of glucocorticoid and nonglucocorticoid steroids in acute neuronal injury. Cell Mol Neurobiol. 1993;13(4):415–432. doi: 10.1007/BF00711581. [DOI] [PubMed] [Google Scholar]
- [48].Bracken MB, Collins WF, Freeman DF, et al. Efficacy of methylprednisolone in acute spinal cord injury. JAMA. 1984;251(1):45–52. [PubMed] [Google Scholar]
- [49].Coleman WP, Benzel D, Cahill DW, et al. A critical appraisal of the reporting of the National Acute Spinal Cord Injury Studies (II and III) of methylprednisolone in acute spinal cord injury. J Spinal Disord. 2000;13(3):185–199. doi: 10.1097/00002517-200006000-00001. [DOI] [PubMed] [Google Scholar]
- [50].Bracken MB, Shepard MJ, Holford TR, et al. Methylprednisolone or tirilazad mesylate administration after acute spinal cord injury: 1-year follow up. Results of the third National Acute Spinal Cord Injury randomized controlled trial. J Neurosurg. 1998;89(5):699–706. doi: 10.3171/jns.1998.89.5.0699. [DOI] [PubMed] [Google Scholar]
- [51].Hadley MN, Walters BC, Grabb PA, et al. Guidelines for the management of acute cervical spine and spinal cord injuries. Clin Neurosurg. 2002;49:407–498. [PubMed] [Google Scholar]
- [52].Rozet I. Methylprednisolone in acute spinal cord injury: is there any other ethical choice? J Neurosurg Anesthesiol. 2008;20(2):137–139. doi: 10.1097/01.ana.0000314441.63823.b0. [DOI] [PubMed] [Google Scholar]
- [53].Nesathurai S. Steroids and spinal cord injury: revisiting the NASCIS 2 and NASCIS 3 trials. J Trauma. 1998;45(6):1088–1093. doi: 10.1097/00005373-199812000-00021. [DOI] [PubMed] [Google Scholar]
- [54].Yu CG, Jimenez O, Marcillo AE, et al. Beneficial effects of modest systemic hypothermia on locomotor function and histopathological damage following contusion-induced spinal cord injury in rats. J Neurosurg. 2000;93(1 Suppl):85–93. doi: 10.3171/spi.2000.93.1.0085. [DOI] [PubMed] [Google Scholar]
- [55].Ha KY, Kim YH. Neuroprotective effect of moderate epidural hypothermia after spinal cord injury in rats. Spine (Phila Pa 1976) 2008;33(19):2059–2065. doi: 10.1097/BRS.0b013e31818018f6. [DOI] [PubMed] [Google Scholar]
- [56].Levi AD, Green BA, Wang MY, et al. Clinical application of modest hypothermia after spinal cord injury. J Neurotrauma. 2009;26(3):407–415. doi: 10.1089/neu.2008.0745. [DOI] [PubMed] [Google Scholar]
- [57].Gorio A, Gokmen N, Erbayraktar S, et al. Recombinant human erythropoietin counteracts secondary injury and markedly enhances neurological recovery from experimental spinal cord trauma. Proc Natl Acad Sci U S A. 2002;99(14):9450–9455. doi: 10.1073/pnas.142287899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].ClinicalTrials. gov. Evaluation of tolerability and efficacy of erythropoietin (EPO) treatment in spinal shock: comparative study versus methylprednisolone (MP) http://clinicaltrials.gov/ct2/show/NCT00561067 .
- [59].Schowinsky J, Corboy J, Vollmer T, et al. Natalizumab- associated complication. First case of peripheral T cell lymphoma? Acta Neuropathol. 2012;123(5):751–752. doi: 10.1007/s00401-012-0967-7. [DOI] [PubMed] [Google Scholar]
- [60].Midaglia L, Rodriguez Ruiz M, Muñoz-García D. Severe haematological complications during treatment with natalizumab. Mult Scler. doi: 10.1177/1352458512442262. in press. [DOI] [PubMed] [Google Scholar]