

Abstract
Induced pluripotent stem (iPS) cells can be generated from somatic cells by coexpression of four transcription factors: Sox2, Oct4, Klf4, and c-Myc. However, the low efficiency in generating iPS cells and the tendency of tumorigenesis hinder the therapeutic applications for iPS cells in treatment of human diseases. To this end, it remains largely unknown how the iPS process is subjected to regulation by upstream signaling pathway(s). Here, we report that Akt regulates the iPS process by modulating posttranslational modifications of these iPS factors in both direct and indirect manners. Specifically, Akt directly phosphorylates Oct4 to modulate the Oct4/Sox2 heterodimer formation. Furthermore, Akt either facilitates the p300-mediated acetylation of Oct4, Sox2, and Klf4, or stabilizes Klf4 by inactivating GSK3, thus indirectly modulating stemness. As tumorigenesis shares possible common features and mechanisms with iPS, our study suggests that Akt inhibition might serve as a cancer therapeutic approach to target cancer stem cells.
Keywords: Akt, iPS cell, Klf4, Oct4, p300, Sox2
Introduction
Embryonic stem (ES) cells are derived from the inner cell mass of mammalian blastocysts and have been demonstrated to maintain pluripotency 1,2. The pluripotent potential of human ES cells has been proposed to be beneficial in the treatment of various human diseases including Parkinson's disease, spinal cord injury, and diabetes 3,4. However, the therapeutic use of ES cells requires a comprehensive understanding of the molecular mechanisms that control the proliferation and differentiation of ES cells 5,6. More importantly, ethical controversy regarding the use of human embryos also prevents its therapeutic application. To overcome this issue, coexpression of Oct4, Sox2, Klf4, and Nanog or c-Myc in normal mouse or human fibroblasts has been shown to actively reprogram these differentiated cells into an induced pluripotent stem (iPS) cell state 2,7–9. It has been further demonstrated that pluripotency can be induced without c-Myc, however, with lower efficiency 10,11. This provides a manageable approach to generate large quantities of human iPS cells for therapeutic purposes 12. However, the low efficiency of this iPS-generating process hinders the development of iPS technology into clinical applications. Although multiple methods have been attempted to enhance the iPS process, such as adding SV40 large T antigen 13, co-overexpressing MyoD 14, enhancing RA signaling 15, or depleting Mdb3 that is a core component of the NuRD complex 16, it remains largely unknown how the iPS process is regulated by upstream signaling pathways in vivo 17.
Another critical issue that limits the potential application of iPS technology is that all four transcription factors (Sox2, Oct4, Klf4, and c-Myc) are found to be frequently overexpressed in various cancers 6. Thus, it is not surprising that a high percentage of mice derived from iPS cells developed tumors 2. Moreover, it is noteworthy that the iPS process, which requires the coexpression of four transcriptional factors, mirrors the transformation of primary human cells, in which overexpression of the Ha-Ras and hTERT oncogenes and inactivation of both the p53 and Rb tumor suppressor pathways by SV40 LT antigen are required 18,19. More interestingly, tumorigenesis and embryonic development share many similarities. For example, both of them are immortalized and could form tumors when implanted subcutaneously in mice. There is also a growing body of evidence suggesting that many tumors arise by acquiring genetic mutations in only a small population of transformed cells termed cancer stem cells, or cancer initiating cells 20–22. As cancer stem cells are more resistant to common chemotherapeutic interventions, it is critical to understand their biological features in order to develop better anticancer regimens 23,24. To this end, it has been proposed that cancer stem cells were derived from either normal stem cells through acquiring genetic mutations or terminally differentiated somatic cells by activating a subset of genes typically overexpressed in stem cells to acquire a stem cell-like phenotype 24. In this scenario, cellular transformation correlates with the dedifferentiation process. Nonetheless, in both cases genetic mutations are a driving force to cancer stem cell formation. Thus, acquiring mutations that promote tumorigenesis might also partially convert somatic cells into a stem cell-like phenotype through pathways partially resembling the iPS process. In support of this idea, recently it has been shown that inactivation of the p53 tumor suppressor pathway greatly enhanced iPS efficiency 25–29, suggesting that key signaling pathway(s), frequently altered in human cancers, might also be involved in stem cell maintenance 30.
In addition to p53, the PTEN/PI3K/Akt pathway is found commonly hyperactivated in various human carcinomas through various means of genetic alterations and is considered as a hallmark of cancer 31,32. In support of a critical role for Akt in stem cell regulation, constitutive activation of Akt was shown to be capable of substituting for basic fibroblast growth factor 33 or leukemia inhibitory factor to maintain stemness. Consistently, loss of PTEN is found to affect the hematopoietic stem cell renewal process 34,35. However, further in-depth evaluations revealed that PTEN negatively regulated the mTORC2/Akt signaling only in adult, but not neonatal hematopoietic stem cells 34. This not only highlighted a development stage-dependent role for PTEN in maintaining stemness but also suggested a potential temporal regulation difference between stem cell self-renewal and tumorigenesis. However, even though Akt has been characterized as a driving oncogene to facilitate tumorigenesis, it remains largely elusive how Akt participates in stem cell fate regulation and whether similar to its oncogenic role, Akt could enhance the efficiency of the iPS process.
Methods
Plasmids
CMV-Flag-Sox2, CMV-Flag-Oct4, CMV-Flag-Klf4, and CMV-Flag-Nanog were obtained from Addgene (Cambridge, MA). pcDNA3-HA-p300, Myc-p300, and pcDNA3-HA-CBP were obtained from Dr. James DeCaprio (Dana-Farber Cancer Institute, Boston, MA). pcDNA3-HA-Myr-Akt1 construct was obtained from Dr. Alex Toker (Beth Israel Deaconess Medical Center, Boston, MA) and described previously 36. ERK1, p38-mitogen-activated protein kinase (MAPK), GSK3, and HA-Fbw7 expression plasmids were described previously 37. Various mutation constructs of Flag-Klf4, Flag-Oct4, and Flag-Sox2 were generated using the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions.
siRNAs
Scramble, luciferase, β-TRCP1+2, Fbw7, Skp2, and Cdh1 siRNA oligos and siRNA transfection methods have been described previously 36.
Antibodies
Anti-β-catenin, Skp2, cyclin E, and polyclonal anti-HA antibodies were purchased from Santa Cruz (Dallas, TX). Anti-phospho-Akt substrate (RxRxxpS/T), acetylated-Lys, and Klf4 antibodies were purchased from Cell Signaling Technology (Danvers, MA). Anti-Tubulin and Vinculin antibodies, polyclonal and monoclonal anti-Flag antibodies, anti-Flag agarose beads, anti-HA agarose beads, and anti-mouse and rabbit horseradish peroxidase-conjugated secondary antibodies were purchased from Sigma (St. Louis, MO). Monoclonal anti-HA antibody was purchased from Covance (Princeton, NJ). Anti-GFP and Cdh1 antibodies were purchased from Invitrogen (Carlsbad, CA).
Immunoblots and immunoprecipitation
Cells were harvested with EBC buffer (50 mmol/L Tris pH 7.5, 120 mmol/L NaCl, 0.5% NP-40) containing protease inhibitors (Roche, Indianapolis, IN) and phosphatase inhibitors (Calbiochem, Billerica, MA). Whole cell lysates were subjected to immunoblot analyses with indicated antibodies. Immunoprecipitations were carried out by incubating 1 mg of whole cell lysates with 8 μL of HA or Flag slurry beads (Sigma) for 3–4 h at 4°C. Immunoprecipitants were washed five times with NETN buffer (20 mmol/L Tris, pH 8.0, 100 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid, and 0.5% NP-40) and resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) for immunoblot analyses.
Cell culture and transfection
Cell culture and transfection procedures have been described previously 36,38. For cell transfection, cells were transfected using Lipofectamine (Life Technologies, Woburn, MA) in OptiMEM medium (Life Technologies) according to the manufacturer's instructions. Forty-eight hours posttransfection, transfected cells were further subjected to immunoblot analysis.
Results
Oct4 and Klf4 are phosphorylated by Akt in vivo
Consistent with previous reports 39,40, we observed that a constitutive active Akt (N-terminal tagged with a myristoylation tag) could phosphorylate Oct4 in cells. Furthermore, Klf4, in addition to Oct4, but not Sox2 or Nanog, was also found phosphorylated by exogenous Akt1 (Fig.1A). By scanning the Oct4 protein sequence, we identified an AGC kinase consensus motif “RxRxxpS/pT” 41 located at the Thr235 residue (Fig.1B) and mutation of this site to a nonphosphorylatable residue, Ala, almost completely abolished Akt1-mediated Oct4 phosphorylation (Fig.1C). The critical function of Oct4 in stem cell regulation is mainly attributed to its role as a transcriptional regulator, which is achieved by direct binding of Oct4 to its canonical octamer motif through its DNA-binding domains 42,43. Interestingly, Oct4 contains two distinct DNA-binding domains and it can form either a homodimer with itself, or a heterodimer with other transcription factors, depending on the octamer half-sites present in the enhancer region of the target gene. Thus, it is interesting to investigate whether phosphorylation of Oct4 potentially affects its ability to form homodimers or heterodimers. To this end, we did not observe a significant effect of Akt-mediated Oct4 phosphorylation on Oct4 homodimer formation (Fig.1D). As generally Sox proteins require partners such as other transcription factors for activation and previous studies has demonstrated that Oct4 could form a complex with Sox2 on DNA to control the expression of embryonic development-related genes 44–47, next we examined whether Oct4 phosphorylation on T235 affects its interaction with Sox2. Interestingly, phosphorylation of Oct4 significantly reduced Oct4 interaction with Sox2 (Fig.1D). Consistent with this result, ectopic expression of Akt1 led to dissociation of Sox2 from wild-type Oct4 but not T235A Oct4 (Fig.1E). These data suggest that Akt-mediated phosphorylation of Oct4 on T235 might regulate cellular stemness in a signal dependent manner through modulating heterodimer formation with Sox2. Notably, a recent study illustrated that phosphorylation of Oct4 on T235 led to enhanced binding of Oct4 to Sox2 to differentially regulate transcription of stemness genes 39. The discrepancy between their observation and our study might stem from different cell lines examined, which might suggest that Akt-mediated phosphorylation of Oct4 at T235 might regulate transcription of stemness genes through modulating Oct4/Sox2 complex formation in a cellular context-dependent manner, and warrants further investigation. In addition, phosphorylation of Oct4 at S229 and Y327 has also been observed to have differential effects on its transcriptional activity toward multiple targets 48. Furthermore, Sox2 phosphorylation at T118 by Akt has also been reported to enhance the transcriptional ability of Sox2 in ESCs by unknown mechanisms 49. Similarly, phosphorylation of Nanog was observed in cell as well but has not been connected with any characterized function 50.
Figure 1.

Oct4 and Klf4 are phosphorylated by Akt in vivo. (A) Immunoblot (IB) analysis of whole cell lysates (WCLs) and immunoprecipitates (IPs) derived from 293T cells transfected with HA-tagged Myr-Akt1 and indicated Flag-tagged constructs. Akt-mediated phosphorylation was recognized by an Akt substrate-motif phosphorylation-specific antibody (RxRxxpS/pT). (B) Sequence alignment of the Thr235 putative Akt phosphorylation site in Oct4 among different species. (C) Akt specifically phosphorylated Oct4 at Thr235. (D) Phosphomimetic mutation at Thr235 of Oct4 decreased Oct4 interaction with Sox2. IB analysis and Flag-IP derived from 293T cells transfected with indicated constructs. (E) Phosphorylation of Oct4 on Thr235 led to attenuated Oct4 interaction with Sox2. WCLs of 293T cells transfected with indicated constructs were subjected to immunoprecipitation with Flag antibody. The Flag-IPs and WCLs were immunoblotted with indicated antibodies. (F) Akt phosphorylated Klf4 at Thr399. WCLs of 293T cells transfected with indicated constructs were subjected to immunoprecipitation with Flag antibody. The Flag-IPs and WCLs were immunoblotted with indicated antibodies. (G) Sequence alignment of the Thr429 putative Akt phosphorylation site in Klf4 among different species.
Furthermore, we observed that in addition to Oct4, Klf4 phosphorylation was also increased when co-overexpressed with Myr-Akt1 in cells (Fig.1F). By truncating Klf4 we narrowed down the Akt-mediated phosphorylation site(s) within amino acids 340–483, which contain an evolutionarily conserved AGC consensus motif located at T429. More importantly, mutation of Thr429 to Ala completely abolished the phosphorylation of the C-terminal portion of Klf4 by Akt (Fig.1F and G). As Klf4 could directly interact with the Oct4/Sox2 complex to facilitate somatic cell reprogramming 51, it is plausible that phosphorylation of Klf4 at T429, which resides in its zinc finger motif, might participate in modulating cellular stemness by affecting its interaction with the Oc4/Sox2 complex. Interesting, recent work has clearly demonstrated the critical role for Oct4 in pluripotency regulation, while Klf4 could be substituted by other factors 8,52, therefore we hypothesize that Akt controls the induced pluripotency process in large part by phosphorylation of Oct4, or by shifting Oct4-binding partners. Taken together, these results indicate that Akt-mediated phosphorylation might be an upstream regulatory mechanism responsible for the formation of iPS cells.
Sox2, Oct4, and Klf4 are acetylated by p300 in vivo
In addition to phosphorylation, various posttranslational modifications have been demonstrated as regulatory mechanisms in controlling the transcriptional activities of Oct4, Sox2, Klf4, and Nanog. For example, SUMOylation of Oct4 or Sox2 has been observed but with opposite effects on protein function. Specifically, Oct4 SUMOylation led to enhanced stability and DNA-binding ability 53, while Sox2 SUMOylation resulted in attenuated DNA-binding ability 54. As Akt has been demonstrated to modulate protein acetylation process by direct phosphorylation of the acetyl-transferases p300 55 or CBP 56, next we examined whether any of the four iPS factors, Oct4, Sox2, Klf4, or Nanog, were subjected to acetylation-mediated regulation. From our initial screening by coexpression of an iPS factor with either p300 or CBP in cells, we observed various acetylation patterns among these iPS factors. First, p300- or CBP-dependent acetylation of Nanog was not detected in our experimental condition by an Ac-K antibody (Fig.2A). Second, Sox2 displayed a high level of basal acetylation and a slight increase in acetylation in the presence of ectopic p300 or CBP (Fig.2A). Third, acetylation of both Oct4 and Klf4 was induced by p300-WT but not with a p300-acetyltransferase dead mutant, or CBP (Fig.2A). Interestingly, ectopic expression of Akt1 induced the acetylation of Klf4, but not Sox2, Oct4, or Nanog (Fig.2B), suggesting that the Akt activity directly and/or indirectly regulates acetylation state of iPS factors in a different mechanism. More importantly, p300-mediated acetylation of Oct4/Sox2 led to a dramatically decreased interaction between Sox2 and Oct4 (Fig.2C), suggesting that acetylation of Oct4/Sox2 behaves similarly to Oct4 phosphorylation-mediated impairment of the association between Oct4 and Sox2 (Fig.1D), which subsequently shifts the transcription activity of Oct4 toward a certain subset of genes 46,47.
Figure 2.
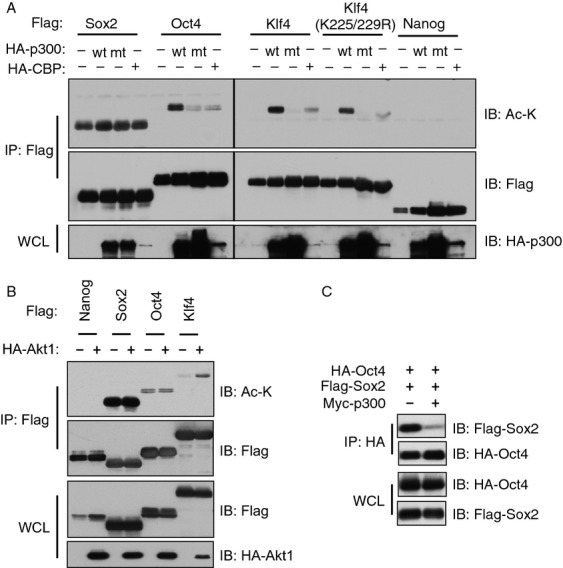
Oct4 and Klf4 are acetylated by p300 in vivo. (A) Whole cell lysates (WCLs) of 293T cells transfected with HA-tagged p300 or CBP with indicated Flag-tagged constructs were subjected to immunoprecipitation (IP) with Flag antibody. The Flag-IPs and WCLs were immunoblotted with indicated antibodies. Acetylation was detected by a Lys-acetylation (Ac-K) antibody. (B) WCLs of 293T cells transfected with indicated constructs were subjected to immunoprecipitation with Flag antibody. The Flag-IPs and WCLs were immunoblotted with indicated antibodies. (C) WCLs of 293T cells transfected with indicated constructs were subjected to immunoprecipitation with HA antibody. The HA-IPs and WCLs were immunoblotted with Flag and HA antibodies.
Sox2 is acetylated by p300 on multiple sites in vivo
To obtain mechanistic insights into how acetylation of Sox2 and Oct4 modulate their complex formation, we tried to pinpoint the acetylation sites on both Sox2 and Oct4. As Sox2 displayed both basal and p300-dependent acetylation events (Fig.2A), we first truncated Sox2 to narrow down the possible acetylation region(s) (Fig.3A). Interestingly, all truncations missing the first 1–48 amino acids showed dramatically reduced acetylation signals (Fig.3B), suggesting that the major basal Sox2 acetylation sites are located within the first 48 amino acids. As the HMG domain is the critical functional module for Sox2 function and there are two lysine residues (K37 and K44) located within this critical region, we further examined whether these two sites were the acetylation targets. Notably, mutation of K37, K44, or both sites to Arg to abolish possible acetylation did not significantly affect the basal acetylation state of Sox2 (Fig.3C), indicating that neither K37 nor K44 is the major acetylation site. In addition to K37 and K44, there is only one lysine, K10, left in the first 48 amino acids, thus it warrants further investigation to pinpoint whether K10 is the major site for Sox2 basal acetylation. Furthermore, for p300-dependent Sox2 acetylation, multiple sites might be involved, as a series of truncations displayed a gradual decrease in Sox2 acetylation (Fig.3D). One of these acetylation events on K75 has been recently reported to be critical to export Sox2 to the cytoplasm to terminate its transcriptional activity in the nucleus 57, which is consistent with our model that p300-mediated acetylation on Sox2/Oct4 impairs their ability to transcribe downstream genes.
Figure 3.

Mapping of Sox2 acetylation sites in vivo. (A) Schematic illustration of a series of generated Sox2 truncation mutations. (B) WCLs of 293T cells transfected with various Sox2 mutants were subjected to immunoprecipitation with Flag antibody. The Flag-IPs and WCLs were immunoblotted with Ac-K and Flag antibodies. (C) WCLs of 293T cells transfected with various Sox2 K-to-R substitution mutants were subjected to immunoprecipitation with Flag antibody. The Flag-IPs and WCLs were immunoblotted with Ac-K and Flag antibodies. (D) WCLs of 293T cells transfected with HA-p300 and various Sox2 truncation mutants were subjected to immunoprecipitation with Flag antibody. The Flag-IPs and WCLs were immunoblotted with Ac-K and Flag antibodies. WCLs, whole cell lysates; IP, immunoprecipitation.
Oct4 is acetylated by p300 on multiple sites in cells
Similarly, we constructed a series of Oct4 truncation mutants to further map the acetylation sites on Oct4 (Fig.4A). In cells we observed that the acetylation sites were mainly located within amino acids 220–233 (Fig.4B), while the major p300 interacting motif was mapped within amino acids 140–163 (Fig.4C). As the POU and Homeodomain serve as a bipartite DNA-binding domain in Sox2 (Fig.4A), and part of the POU and HM domains are located within these two regions, we next generated single or double K-R mutations to examine which lysine was critical for Oct4 acetylation. Notably, all single KR mutation tested exhibited attenuated Oct4 acetylation status (Fig.4D), indicating that p300-mediated Oct4 acetylation occurs on multiple sites 16,58, which has been reported in many of p300 downstream substrates. Nevertheless, Oct4 and Sox2 acetylation led to impaired Oct4/Sox2 heterodimer formation (Fig.2C) and subsequent attenuated transcriptional activity 46,47.
Figure 4.

Mapping of Oct4 acetylation sites in vivo. (A) Schematic illustration of a series of generated Oct4 truncation mutations. (B and C) WCLs of 293T cells transfected with HA-p300 and various Oct4 truncation mutants were subjected to immunoprecipitation with Flag antibody. The Flag-IPs and WCLs were immunoblotted with indicated antibodies. (D) WCLs of 293T cells transfected with HA-p300 and various Oct4 K-to-R substitution mutants were subjected to immunoprecipitation with Flag antibody. The Flag-IPs and WCLs were immunoblotted with indicated antibodies. WCLs, whole cell lysates; IP, immunoprecipitation.
Klf4 is acetylated by p300 on multiple sites in cells
Furthermore, we generated Klf4 truncation mutations to pinpoint its p300-dependent acetylation site(s) (Fig.5A). By this method, we narrowed down the Klf4 acetylation sites to amino acids 1–151 (Fig.5B) and further to 25–151 (Fig.5C). Notably, a previous report indicated that p300 mainly acetylated Klf4 on K225 and K229 to enhance its transcriptional activity 59. However, in our experimental system, mutation of both K225 and K229 to Arg did not significantly reduce Klf4 acetylation status (Fig.5C), suggesting that there might be other sites involved in p300-mediated acetylation of Klf4. Consistent with this notion, we identified two lysine residues within amino acids 24–151, K32 and K52, that were possible acetylation sites (Fig.5D). However, mutating K52 to arginine did not noticeably affect Klf4 acetylation, suggesting that K32 or both K32 and K52 are possible p300 acetylation targets (Fig.5D) that warrant further investigation.
Figure 5.

Mapping of Klf4 acetylation sites in vivo. (A) Schematic illustration of a series of Klf4 truncation mutations generated. (B) WCLs of 293T cells transfected with HA-p300 and various Klf4 truncation mutants were subjected to immunoprecipitation with Flag antibody. The Flag-IPs and WCLs were immunoblotted with indicated antibodies. (C and D) WCLs of 293T cells transfected with HA-p300 and various Klf4 truncation or K-to-R substitution mutants were subjected to immunoprecipitation with Flag antibody. The Flag-IPs and WCLs were immunoblotted with indicated antibodies. WCLs, whole cell lysates; IP, immunoprecipitation.
Fbw7 possibly governs Klf4 stability in a GSK3-dependent manner
During our examination of Klf4 acetylation, consistent with a previous report that Klf4 was subjected to 26S proteasome-mediated degradation 60, we observed that Klf4 was unstable. However, its upstream E3 ligases remain largely unknown. To this end, we screened a panel of E3 ligases for their possible roles in governing Klf4 stability by various siRNAs (Fig.6A). Interestingly, compared to the mock treatment, only depletion of Fbw7, but not other E3s examined, led to an accumulation of Klf4 (Fig.6A). The regulation of Klf4 by Fbw7 is further confirmed by the Fbw7 knockdown experiment with multiple independent shRNAs against Fbw7 (Fig.6B). As it is well characterized that Fbw7 only recognizes substrates with proper posttranslational modifications 61,62, next we attempted to identify its possible upstream modifying kinase(s). To this end, we observed that coexpression with GSK3, but neither ERK1 nor p38-MAPK, resulted in an efficient degradation of Klf4 (Fig.6C), and depletion of GSK3β led to accumulation of Klf4 (Fig.6D), demonstrating that GSK3 is a major upstream kinase responsible for Klf4 turnover mediated by Fbw7. This is consistent with a previous report that activation of the Akt pathway by peroxisome proliferator-activated receptor gamma agonist could stabilize Klf4 by reducing its ubiquitination 63. As phosphorylation of GSK3 by Akt can inactivate its kinase activity 64, which could lead to reduced Klf4 phosphorylation by GSK3, therefore evading Fbw7-mediated proteolysis. Through a close examination of the Klf4 protein sequence, we identified two putative Fbw7 consensus degrons 61 on Klf4 (Fig.6E) that are evolutionarily conserved (Fig.6F), which further supports Klf4 as a possible Fbw7 substrate and warrants further investigations.
Figure 6.
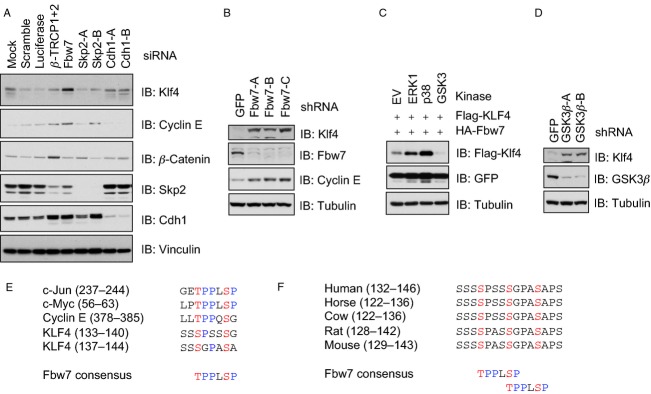
Fbw7 possibly governs Klf4 stability in a GSK3-dependent manner. (A) Fbw7-siRNA treatment in HeLa cells led to increased Klf4 expression. (B) Fbw7-shRNA treatments in HeLa cells led to Klf4 accumulation. (C) Overexpression of Fbw7 and GSK3 led to the destruction of Klf4. 293T cells were cotransfected with HA-Fbw7, Flag-Klf4 and indicated kinases and Klf4 abundance was measured by immunoblots with anti-Flag antibody. GFP was included as an internal transfection control and tubulin served as a loading control. (D) GSK3β-shRNA treatments in HeLa cells led to elevated level of Klf4. (E and F) Sequence alignment of the putative Fbw7 degrons in Klf4 (E) among different species (F).
Discussion
Recent scientific advances have demonstrated that tumors arise in a step-wise fashion through gain-of-function mechanisms from certain oncogenes, concomitantly with the loss of expression of key tumor suppressor proteins 65,66. In addition to the p53 tumor suppressor pathway that is inactivated in about 50% of all human cancers, hyperactivation of the PTEN/PI3K/Akt pathway was observed in over 40% of human carcinomas 67,68. Recent work indicates that PTEN is directly linked to the hematopoietic stem cell renewal process 69,70. Furthermore, it is well established that mouse stem cells are committed to differentiation after withdrawal of the LIF ligand, indicating that a yet unknown downstream signal transduction pathway triggered by LIF is critical to maintain the stem cell state. Surprisingly, constitutive activation of Akt was shown to adequately substitute the function of LIF 71. However, it remains unclear how constitutive activation of Akt signaling is sufficient to maintain pluripotency.
Oct4, Sox2, and Klf4 co-occupy a substantial portion of their downstream target genes to build up a regulatory circuit consisting of multiple autoregulatory and feed-forward loops. In this scenario, it creates consistent activity above a threshold level to maintain the pluripotent state, while the whole system is unstable, and might quickly shut down when one critical regulator becomes negative 72,73. To this end, we observed that Akt directly phosphorylated key transcriptional factors including Oct4 and Klf4 to restrict their activities below this threshold. These results also indicate that Akt-mediated phosphorylation might be an upstream regulatory mechanism responsible for the formation of iPS cells (Fig.7). Therefore, it will be intriguing to examine whether coexpression of Myr-Akt (an active form of Akt) together with Sox2, Oct4, and Klf4 will greatly enhance the efficiency of iPS formation. Furthermore, whether point mutations of all the potential Akt sites in Oct4 or Klf4 would attenuate this phenotype.
Figure 7.
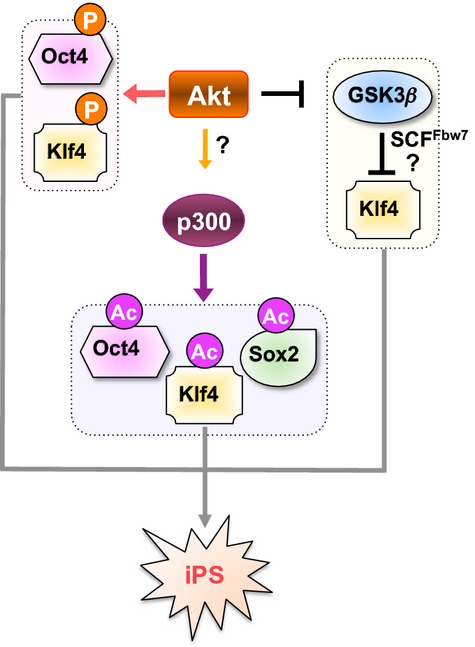
A schematic model for how Akt mediates the induced pluripotent stem (iPS) process through direct or indirect regulation of posttranslational modifications of iPS-inducing factors. Akt directly phosphorylates Oct4 to modulate its interaction with Sox2, leading to a shift of Oct4/Sox2-mediated transcription events. Akt may also activate p300 to promote acetylation of Oct4, Sox2, and Klf4 at multiple sites to change their transcription activity. Furthermore, Akt phosphorylates GSK3, resulting in reduced Klf4 phosphorylation by GSK3 that could possibly trigger Fbw7-mediated degradation of Klf4.
In addition to a direct role of Akt-mediated phosphorylation of iPS factors in regulating the iPS process, Akt could also indirectly affect stemness by modulating other posttranslational modifications of iPS factors. To this end, we have identified that Akt could either facilitate p300-mediated acetylation of Oct4, Sox2, and Klf4 by directly activating p300, or stabilizing Klf4 by directly phosphorylating and inactivating GSK3 to evade Fbw7-mediated degradation of Klf4 (Fig.7). Consistently, multilayer regulations of iPS factors have been identified to cooperatively regulate the pluripotency. For example, a chemical agent ATRA has been shown to increase the interaction of Klf4 with p300 by inducing Klf4 phosphorylation via activation of c-Jun N-terminal kinase and p38 MAPK signaling, and Klf4 acetylation by p300 increased its activity to transactivate the Mfn-2 promoter 74. Since Oct4, Sox2, and Klf4 were found to be key regulators of stem cells, it is possible that Akt could directly or indirectly modulate their transcriptional activities, thus influencing the maintenance of pluripotency.
As cellular reprogramming and the carcinogenic process share many similar features and mechanisms 75,76, it is not surprising that these iPS markers might play critical roles in tumorigenesis as well. To this end, overexpression of Sox2 77–79 or Oct4 80–82 has been observed in multiple cancer types. Similarly, Klf4 overexpression was observed to promote malignant transformation through downregulation of the Cdk inhibitor p21 83. Klf4 belongs to the family of Kruppel-like transcription factors, whose functions have been implicated in regulation of tissue-specific development 84. Elevated Klf4 overexpression is frequently observed in many types of cancers 85 and overexpressing Klf4 in mice led to squamous cell cancer 86, while the molecular mechanisms remain unclear. However, on the other hand, recent studies demonstrated that rather than an oncogene, Klf4 serves as an inhibitor for tumor cell growth and migration 85,87,88. Interestingly, loss of Fbw7 is frequently found in T-cell acute leukemia (T-ALL), a disease caused by the blockage of proper differentiation from progenitor cells to mature T cells. In this study, we found that Fbw7 could possibly degrade Klf4 in a GSK3-dependent manner. As a result, loss of Fbw7 could cause accumulation of the Klf4 transcription factor, which might subsequently block the proper differentiation process, leading to the development of leukemia. Taken together, our study provided insight into the critical role of the Akt oncogenic pathway in regulating stem cell reprogramming and impact on the cancer stem cells. Thus, it will provide the rationale, therefore opening new avenues for developing Akt-specific inhibitors as efficient anticancer drugs.
Acknowledgments
This work was supported by grants from the National Institute of Health (H. I., AG041218). P. L. is supported by 5T32HL007893.
Conflict of Interest
None declared.
References
- 1.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 3.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 4.Muller LU, Daley GQ, Williams DA. Upping the ante: recent advances in direct reprogramming. Mol. Ther. 2009;17:947–953. doi: 10.1038/mt.2009.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Malley J, Woltjen K, Kaji K. New strategies to generate induced pluripotent stem cells. Curr. Opin. Biotechnol. 2009;20:516–521. doi: 10.1016/j.copbio.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 6.Zhao R, Daley GQ. From fibroblasts to iPS cells: induced pluripotency by defined factors. J. Cell. Biochem. 2008;105:949–955. doi: 10.1002/jcb.21871. [DOI] [PubMed] [Google Scholar]
- 7.Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 8.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 9.Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- 10.Wernig M, Meissner A, Cassady JP, Jaenisch R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell. 2008;2:10–12. doi: 10.1016/j.stem.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- 12.Nishikawa S, Goldstein RA, Nierras CR. The promise of human induced pluripotent stem cells for research and therapy. Nat. Rev. Mol. Cell Biol. 2008;9:725–729. doi: 10.1038/nrm2466. [DOI] [PubMed] [Google Scholar]
- 13.Mali P, Ye Z, Hommond HH, Yu X, Lin J, Chen G, et al. Improved efficiency and pace of generating induced pluripotent stem cells from human adult and fetal fibroblasts. Stem Cells. 2008;26:1998–2005. doi: 10.1634/stemcells.2008-0346. [DOI] [PubMed] [Google Scholar]
- 14.Hirai H, Katoku-Kikyo N, Karian P, Firpo M, Kikyo N. Efficient iPS cell production with the MyoD transactivation domain in serum-free culture. PLoS One. 2012;7:e34149. doi: 10.1371/journal.pone.0034149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W, Yang J, Liu H, Lu D, Chen X, Zenonos Z, et al. Rapid and efficient reprogramming of somatic cells to induced pluripotent stem cells by retinoic acid receptor gamma and liver receptor homolog 1. Proc. Natl. Acad. Sci. USA. 2011;108:18283–18288. doi: 10.1073/pnas.1100893108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rais Y, Zviran A, Geula S, Gafni O, Chomsky E, Viukov S, et al. Deterministic direct reprogramming of somatic cells to pluripotency. Nature. 2013;502:65–70. doi: 10.1038/nature12587. [DOI] [PubMed] [Google Scholar]
- 17.Loh YH, Ng JH, Ng HH. Molecular framework underlying pluripotency. Cell Cycle. 2008;7:885–891. doi: 10.4161/cc.7.7.5636. [DOI] [PubMed] [Google Scholar]
- 18.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 19.Wei W, Jobling WA, Chen W, Hahn WC, Sedivy JM. Abolition of cyclin-dependent kinase inhibitor p16Ink4a and p21Cip1/Waf1 functions permits Ras-induced anchorage-independent growth in telomerase-immortalized human fibroblasts. Mol. Cell. Biol. 2003;23:2859–2870. doi: 10.1128/MCB.23.8.2859-2870.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polyak K, Hahn WC. Roots and stems: stem cells in cancer. Nat. Med. 2006;12:296–300. doi: 10.1038/nm1379. [DOI] [PubMed] [Google Scholar]
- 21.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat. Med. 2009;15:1010–1012. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 22.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 23.Marotta LL, Polyak K. Cancer stem cells: a model in the making. Curr. Opin. Genet. Dev. 2009;19:44–50. doi: 10.1016/j.gde.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 24.Shipitsin M, Polyak K. The cancer stem cell hypothesis: in search of definitions, markers, and relevance. Lab. Invest. 2008;88:459–463. doi: 10.1038/labinvest.2008.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132–1135. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140–1144. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Collado M, Villasante A, Strati K, Ortega S, Cañamero M, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136–1139. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marion RM, Strati K, Li H, Murga M, Blanco R, Ortega S, et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149–1153. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, et al. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009;460:1145–1148. doi: 10.1038/nature08285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krizhanovsky V, Lowe SW. Stem cells: the promises and perils of p53. Nature. 2009;460:1085–1086. doi: 10.1038/4601085a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Testa JR, Tsichlis PN. AKT signaling in normal and malignant cells. Oncogene. 2005;24:7391–7393. doi: 10.1038/sj.onc.1209100. [DOI] [PubMed] [Google Scholar]
- 32.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv. Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 33.Kimura T, Tomooka M, Yamano N, Murayama K, Matoba S, Umehara H, et al. AKT signaling promotes derivation of embryonic germ cells from primordial germ cells. Development. 2008;135:869–879. doi: 10.1242/dev.013474. [DOI] [PubMed] [Google Scholar]
- 34.Magee JA, Ikenoue T, Nakada D, Lee JY, Guan KL, Morrison SJ. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell. 2012;11:415–428. doi: 10.1016/j.stem.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tesio M, Oser GM, Baccelli I, Blanco-Bose W, Wu H, Göthert JR, et al. Pten loss in the bone marrow leads to G-CSF-mediated HSC mobilization. J. Exp. Med. 2013;210:2337–2349. doi: 10.1084/jem.20122768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao D, Inuzuka H, Tseng A, Chin RY, Toker A, Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat. Cell Biol. 2009;11:397–408. doi: 10.1038/ncb1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104–109. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG., Jr Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428:194–198. doi: 10.1038/nature02381. [DOI] [PubMed] [Google Scholar]
- 39.Lin Y, Yang Y, Li W, Chen Q, Li J, Pan X, et al. Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol. Cell. 2012;48:627–640. doi: 10.1016/j.molcel.2012.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swaney DL, Wenger CD, Thomson JA, Coon JJ. Human embryonic stem cell phosphoproteome revealed by electron transfer dissociation tandem mass spectrometry. Proc. Natl. Acad. Sci. USA. 2009;106:995–1000. doi: 10.1073/pnas.0811964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 42.Chambers I, Tomlinson SR. The transcriptional foundation of pluripotency. Development. 2009;136:2311–2322. doi: 10.1242/dev.024398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lam CS, Mistri TK, Foo YH, Sudhaharan T, Gan HT, Rodda D, et al. DNA-dependent Oct4-Sox2 interaction and diffusion properties characteristic of the pluripotent cell state revealed by fluorescence spectroscopy. Biochem. J. 2012;448:21–33. doi: 10.1042/BJ20120725. [DOI] [PubMed] [Google Scholar]
- 44.Wilson M, Koopman P. Matching SOX: partner proteins and co-factors of the SOX family of transcriptional regulators. Curr. Opin. Genet. Dev. 2002;12:441–446. doi: 10.1016/s0959-437x(02)00323-4. [DOI] [PubMed] [Google Scholar]
- 45.Wissmuller S, Kosian T, Wolf M, Finzsch M, Wegner M. The high-mobility-group domain of Sox proteins interacts with DNA-binding domains of many transcription factors. Nucleic Acids Res. 2006;34:1735–1744. doi: 10.1093/nar/gkl105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuroda T, Tada M, Kubota H, Kimura H, Hatano SY, Suemori H, et al. Octamer and Sox elements are required for transcriptional cis regulation of Nanog gene expression. Mol. Cell. Biol. 2005;25:2475–2485. doi: 10.1128/MCB.25.6.2475-2485.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodda DJ, Chew JL, Lim LH, Loh YH, Wang B, Ng HH, et al. Transcriptional regulation of nanog by OCT4 and SOX2. J. Biol. Chem. 2005;280:24731–24737. doi: 10.1074/jbc.M502573200. [DOI] [PubMed] [Google Scholar]
- 48.Saxe JP, Tomilin A, Schöler HR, Plath K, Huang J. Post-translational regulation of Oct4 transcriptional activity. PLoS One. 2009;4:e4467. doi: 10.1371/journal.pone.0004467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeong CH, Cho YY, Kim MO, Kim SH, Cho EJ, Lee SY, et al. Phosphorylation of Sox2 cooperates in reprogramming to pluripotent stem cells. Stem Cells. 2010;28:2141–2150. doi: 10.1002/stem.540. [DOI] [PubMed] [Google Scholar]
- 50.Yates A, Chambers I. The homeodomain protein Nanog and pluripotency in mouse embryonic stem cells. Biochem. Soc. Trans. 2005;33(Pt 6):1518–1521. doi: 10.1042/BST0331518. [DOI] [PubMed] [Google Scholar]
- 51.Wei Z, Yang Y, Zhang P, Andrianakos R, Hasegawa K, Lyu J, et al. Klf4 interacts directly with Oct4 and Sox2 to promote reprogramming. Stem Cells. 2009;27:2969–2978. doi: 10.1002/stem.231. [DOI] [PubMed] [Google Scholar]
- 52.Kim JB, Greber B, Araúzo-Bravo MJ, Meyer J, Park KI, Zaehres H, et al. Direct reprogramming of human neural stem cells by OCT4. Nature. 2009;461:649–653. doi: 10.1038/nature08436. [DOI] [PubMed] [Google Scholar]
- 53.Wei F, Scholer HR, Atchison ML. SUMOylation of Oct4 enhances its stability, DNA binding, and transactivation. J. Biol. Chem. 2007;282:21551–21560. doi: 10.1074/jbc.M611041200. [DOI] [PubMed] [Google Scholar]
- 54.Tsuruzoe S, Ishihara K, Uchimura Y, Watanabe S, Sekita Y, Aoto T, et al. Inhibition of DNA binding of Sox2 by the SUMO conjugation. Biochem. Biophys. Res. Commun. 2006;351:920–926. doi: 10.1016/j.bbrc.2006.10.130. [DOI] [PubMed] [Google Scholar]
- 55.Huang WC, Chen CC. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol. Cell. Biol. 2005;25:6592–6602. doi: 10.1128/MCB.25.15.6592-6602.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, Xing ZB, Zhang JH, Fang Y. Akt kinase targets the association of CBP with histone H3 to regulate the acetylation of lysine K18. FEBS Lett. 2013;587:847–853. doi: 10.1016/j.febslet.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 57.Baltus GA, Kowalski MP, Zhai H, Tutter AV, Quinn D, Wall D, et al. Acetylation of sox2 induces its nuclear export in embryonic stem cells. Stem Cells. 2009;27:2175–2184. doi: 10.1002/stem.168. [DOI] [PubMed] [Google Scholar]
- 58.Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell. Biol. 2009;29:2658–2672. doi: 10.1128/MCB.01639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Evans PM, Zhang W, Chen X, Yang J, Bhakat KK, Liu C. Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J. Biol. Chem. 2007;282:33994–34002. doi: 10.1074/jbc.M701847200. [DOI] [PubMed] [Google Scholar]
- 60.Chen ZY, Wang X, Zhou Y, Offner G, Tseng CC. Destabilization of Kruppel-like factor 4 protein in response to serum stimulation involves the ubiquitin-proteasome pathway. Cancer Res. 2005;65:10394–10400. doi: 10.1158/0008-5472.CAN-05-2059. [DOI] [PubMed] [Google Scholar]
- 61.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 62.Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol. 2013;14:369–381. doi: 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun Y, Zheng B, Zhang XH, He M, Guo ZW, Wen JK. PPAR-gamma agonist stabilizes KLF4 protein via activating Akt signaling and reducing KLF4 ubiquitination. Biochem. Biophys. Res. Commun. 2014;443:383–388. doi: 10.1016/j.bbrc.2013.11.129. [DOI] [PubMed] [Google Scholar]
- 64.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 65.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 66.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 67.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 68.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 70.Guo W, Lasky JL, Chang CJ, Mosessian S, Lewis X, Xiao Y, et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature. 2008;453:529–533. doi: 10.1038/nature06933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Watanabe S, Umehara H, Murayama K, Okabe M, Kimura T, Nakano T. Activation of Akt signaling is sufficient to maintain pluripotency in mouse and primate embryonic stem cells. Oncogene. 2006;25:2697–2707. doi: 10.1038/sj.onc.1209307. [DOI] [PubMed] [Google Scholar]
- 72.Muraro MJ, Kempe H, Verschure PJ. Concise review: the dynamics of induced pluripotency and its behavior captured in gene network motifs. Stem Cells. 2013;31:838–848. doi: 10.1002/stem.1340. [DOI] [PubMed] [Google Scholar]
- 73.Sanchez Alvarado A. Stem cells: time to check our premises. Cell Stem Cell. 2008;3:25–29. doi: 10.1016/j.stem.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 74.Zhang R, Han M, Zheng B, Li YJ, Shu YN, Wen JK. Kruppel-like factor 4 interacts with p300 to activate mitofusin 2 gene expression induced by all-trans retinoic acid in VSMCs. Acta Pharmacol. Sin. 2010;31:1293–1302. doi: 10.1038/aps.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Iacovides D, Michael S, Achilleos C, Strati K. Shared mechanisms in stemness and carcinogenesis: lessons from oncogenic viruses. Front. Cell. Infect. Microbiol. 2013;3:66. doi: 10.3389/fcimb.2013.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kuhn NZ, Tuan RS. Regulation of stemness and stem cell niche of mesenchymal stem cells: implications in tumorigenesis and metastasis. J. Cell. Physiol. 2010;222:268–277. doi: 10.1002/jcp.21940. [DOI] [PubMed] [Google Scholar]
- 77.Chen S, Xu Y, Chen Y, Li X, Mou W, Wang L, et al. SOX2 gene regulates the transcriptional network of oncogenes and affects tumorigenesis of human lung cancer cells. PLoS One. 2012;7:e36326. doi: 10.1371/journal.pone.0036326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jia X, Li X, Xu Y, Zhang S, Mou W, Liu Y, et al. SOX2 promotes tumorigenesis and increases the anti-apoptotic property of human prostate cancer cell. J. Mol. Cell. Biol. 2011;3:230–238. doi: 10.1093/jmcb/mjr002. [DOI] [PubMed] [Google Scholar]
- 79.Bareiss PM, Paczulla A, Wang H, Schairer R, Wiehr S, Kohlhofer U, et al. SOX2 expression associates with stem cell state in human ovarian carcinoma. Cancer Res. 2013;73:5544–5555. doi: 10.1158/0008-5472.CAN-12-4177. [DOI] [PubMed] [Google Scholar]
- 80.Li XL, Jia LL, Shi MM, Li X, Li ZH, Li HF, et al. Downregulation of KPNA2 in non-small-cell lung cancer is associated with Oct4 expression. J. Transl. Med. 2013;11:232. doi: 10.1186/1479-5876-11-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li C, Yan Y, Ji W, Bao L, Qian H, Chen L, et al. OCT4 positively regulates Survivin expression to promote cancer cell proliferation and leads to poor prognosis in esophageal squamous cell carcinoma. PLoS One. 2012;7:e49693. doi: 10.1371/journal.pone.0049693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang YD, Cai N, Wu XL, Cao HZ, Xie LL, Zheng PS. OCT4 promotes tumorigenesis and inhibits apoptosis of cervical cancer cells by miR-125b/BAK1 pathway. Cell Death Dis. 2013;4:e760. doi: 10.1038/cddis.2013.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rowland BD, Peeper DS. KLF4, p21 and context-dependent opposing forces in cancer. Nat. Rev. Cancer. 2006;6:11–23. doi: 10.1038/nrc1780. [DOI] [PubMed] [Google Scholar]
- 84.Katz JP, Perreault N, Goldstein BG, Actman L, McNally SR, Silberg DG, et al. Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology. 2005;128:935–945. doi: 10.1053/j.gastro.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 85.Yu F, Li J, Chen H, Fu J, Ray S, Huang S, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161–2172. doi: 10.1038/onc.2010.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tetreault MP, Wang ML, Yang Y, Travis J, Yu QC, Klein-Szanto AJ, et al. Klf4 overexpression activates epithelial cytokines and inflammation-mediated esophageal squamous cell cancer in mice. Gastroenterology. 2010;139:2124–2134.e9. doi: 10.1053/j.gastro.2010.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang J, Place RF, Huang V, Wang X, Noonan EJ, Magyar CE, et al. Prognostic value and function of KLF4 in prostate cancer: RNAa and vector-mediated overexpression identify KLF4 as an inhibitor of tumor cell growth and migration. Cancer Res. 2010;70:10182–10191. doi: 10.1158/0008-5472.CAN-10-2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dang DT, Chen X, Feng J, Torbenson M, Dang LH, Yang VW. Overexpression of Kruppel-like factor 4 in the human colon cancer cell line RKO leads to reduced tumorigenecity. Oncogene. 2003;22:3424–3430. doi: 10.1038/sj.onc.1206413. [DOI] [PMC free article] [PubMed] [Google Scholar]