

Abstract
Mutations in p53 occur in over 50% of the human head and neck squamous cell carcinomas (SCCHN). The majority of these mutations result in the expression of mutant forms of p53, rather than deletions in the p53 gene. Some p53 mutants are associated with poor prognosis in SCCHN patients. However, the molecular mechanisms that determine the poor outcome of cancers carrying p53 mutations are unknown. Here, we generated a mouse model for SCCHN and found that activation of the endogenous p53 gain-of-function mutation p53R172H, but not deletion of p53, cooperates with oncogenic K-ras during SCCHN initiation, accelerates oral tumour growth, and promotes progression to carcinoma. Mechanistically, expression profiling of the tumours that developed in these mice and studies using cell lines derived from these tumours determined that mutant p53 induces the expression of genes involved in mitosis, including cyclin B1 and cyclin A, and accelerates entry in mitosis. Additionally, we discovered that this oncogenic function of mutant p53 was dependent on K-ras because the expression of cyclin B1 and cyclin A decreased, and entry in mitosis was delayed, after suppressing K-ras expression in oral tumour cells that express p53R172H. The presence of double-strand breaks in the tumours suggests that oncogene-dependent DNA damage resulting from K-ras activation promotes the oncogenic function of mutant p53. Accordingly, DNA damage induced by doxorubicin also induced increased expression of cyclin B1 and cyclin A in cells that express p53R172H. These findings represent strong in vivo evidence for an oncogenic function of endogenous p53 gain-of-function mutations in SCCHN and provide a mechanistic explanation for the genetic interaction between oncogenic K-ras and mutant p53.
Keywords: mutant p53, head and neck cancer, mouse model, ras, gain-of-function mutations
Introduction
Head and neck cancer is the sixth leading cancer worldwide, with over 500 000 cases diagnosed every year [1]. The vast majority of head and neck cancers (>90%) are squamous cell carcinomas (SCCHN) that arise from the epithelial lining of the upper aerodigestive tract, including the oral cavity, tongue, and lips. Current therapeutic options for patients with advanced SCCHN (>60%) are largely limited to chemotherapy and radiation [2]. However, tumour recurrence is extremely high in patients with advanced SCCHN, and the estimated 5-year survival rates are less than 30%, as recurrent tumours tend to be aggressive and refractory to current therapies [3,4].
Similar to other solid cancers, SCCHN arise through the accumulation of genetic alterations that result in the activation of oncogenes and/or inactivation of tumour suppressor genes [5,6]. The p53 gene is one of the most frequently mutated genes in human cancers, including SCCHN [7–10], and loss of p53 in mice predisposes to cancer development [11,12]. However, most p53 mutations found in human cancers are missense mutations that result in the expression of mutant forms of p53 [13]. In addition to inactivating the tumour suppressor function of p53, some of these mutations may confer gain-of-function properties to mutant p53, including the induction of tumourigenic potential [14–16]. In human cancers, including SCCHN, p53 mutations are associated with poor prognosis. Thus, a decreased survival and poor response to chemotherapy in patients with SCCHN who carry a subset of p53 missense mutations were recently reported [10,17], and accelerated tumour recurrence was also observed in patients with SCCHN bearing p53 mutations [18,19].
Consistent with the clinical observations, several mouse models provided evidence to support a gain-of-function role for certain p53 mutations, as documented by the finding that mice carrying endogenous p53 missense mutations develop a different tumour spectrum with increased metastatic potential compared with p53 knockout mice [20,21]. In addition, tissue-specific activation of p53 gain-of-function mutations accelerates tumour progression and confers metastatic potential to skin and pancreatic cancers, compared with loss of p53 [22,23]. Despite the strong clinical and experimental evidence, the molecular mechanisms involved in the gain-of-function of p53 mutants remain elusive. During the past years, different models have been proposed to explain the gain-of-function properties of mutant p53, including binding and inactivation of the p53 family members p63 and p73, modulation of the activity of the transcription factor NF-Y, inactivation of the DNA damage sensor ATM, and induction of integrin recycling, among others [24–28]. This broad spectrum of mechanistic models suggests that p53 mutants may exert multiple functions that operate in a cell context-dependent manner [29].
Here, we generated a mouse model to determine the role of endogenous p53 gain-of-function mutations during head and neck cancer development. By analysing the oral tumours that developed in these mice and cell lines derived from the tumours, we addressed mechanisms involved in the gain-of-function of p53 mutations in head and neck cancer. These studies provide new evidence to explain the oncogenic function of mutant p53 and may have an impact in the interpretation of the clinical observations outlined above and the design of novel therapeutic interventions to treat patients with head and neck cancer.
Materials and methods
Mouse models
All compound mice used in this study were generated by crossings involving the following mouse lines: K5.Cre*PR1, Neo-p53R172H, floxed-p53, and LSL-K-rasG12D [20,30–32]. Mice were genotyped as previously described [22]. All comparative studies were conducted using littermates with the indicated genotypes. All research involving mice was performed in compliance with the Institutional Animal Care and Use Committee of the MD Anderson Cancer Center.
Histology and immunohistochemistry
Oral tumours were fixed in 10% neutral buffered formalin, paraffin-embedded, sectioned, and stained with haematoxylin and eosin. For immunohistochemistry, tissue sections were deparaffinized and rehydrated using xylene and alcohol series. Antigen retrieval was carried out in 100 mM sodium citrate (pH 6.0) and endogenous peroxidase was blocked with 1% hydrogen peroxide. Tissue sections were then incubated with primary antibodies for p53 (NCL-p53-CM5p; Leica Microsystems, Buffalo Grove, IL, USA), phospho-Histone H3 (Ser10) (06–570; Millipore, Billerica, MA, USA), and γ-H2AX (05–636; Millipore). Signal was detected with biotinylated secondary antibodies using the Elite Vectastain ABC kit and peroxidase substrate DAB kit (Vector Laboratories, Burlingame, CA, USA).
Gene expression analysis
RNA was purified using the RNeasy Mini Kit according to the manufacturer’s instructions (Qiagen, Valencia, CA, USA). RNA quality was confirmed using the Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). Total RNA was used for in vitro transcription, biotin labelling, and hybridization to Affymetrix Genechip Mouse Genome 430 2.0 arrays (Affymetrix, Santa Clara, CA, USA) according to standard protocols in the Microarray Core at Baylor College of Medicine (Houston, TX, USA). Fluorescence intensities were captured with an Affymetrix GeneArray 2500 Scanner, quantified with Affymetrix Microarray Suite 5.0, and analysed following the model-based expression method implemented by dChip [33]. The cut-off for selecting differentially expressed genes was established at 90% confidence lower bound fold change ≥1.2. Gene ontology analysis was performed using DAVID and p values were determined by a modified Fisher exact test implemented in DAVID [34]. Gene interaction networks were generated with IPA (http://www.ingenuity.com/). Microarray data have been deposited in the GEO repository (http://www.ncbi.nlm.nih.gov/geo/) with the accession number GSE28203.
Expression changes were confirmed by real-time RT-PCR. Briefly, total RNA was reverse-transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA, USA). Quantitative RT-PCR was performed in CFX96 Real Time System C1000 Thermal Cycles (Bio-Rad, Hercules, CA, USA) using specific Taqman® Gene Expression Assays (Applied Biosystems) for Ccnb1, Ccna2, Mmp10, and Mmp13. All samples were amplified in triplicate and the relative amount of mRNA was calculated using the comparative 2−ΔΔCt method. 18s RNA was used as an internal control.
Immunofluorescence
Cells were fixed in 100% methanol at −20 °C, washed with PBS, blocked with 1 mg/ml BSA, and incubated with antibodies for K14 [35], p53 (sc-6243; Santa Cruz Biotechnology, Santa Cruz, CA, USA), phospho-Histone H3 (Ser10) (06–570, Millipore), γ-tubulin (T5326; Sigma-Aldrich, St Louis, MO, USA), and anti BrdU-FITC (347 583; BD Biosciences, San Jose, CA, USA). Cells were washed with PBS–0.1% Tween and PBS, and then incubated with 1 mg/ml BSA followed by Alexa 488-labelled secondary antibodies. Slides were washed with PBS–0.1% Tween and PBS, rinsed with dH2O, 100% ethanol, mounted in Vectashield with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Labs), and analysed on a Leica DML fluorescence microscope.
Luciferase reporter assay
Cells growing on 12-well dishes were transfected with different concentrations of a plasmid carrying a murine p53R172H minigene [36], 0.5 μg of cyclin B1 reporter plasmid (521-CycB1-LUC) [37], and 200 ng of a pCMV-LacZ plasmid. Luciferase and β-galactosidase activity was determined 36 h after transfection using the Dual Light Luciferase and β-galactosidase reporter gene assay (Applied Biosystems) according to the manufacturer’s instructions.
Western blot
Whole cell lysates were obtained by solubilizing cells in protein loading buffer and heating at 100 °C for 10 min. An equal amount of proteins from each sample was separated by electrophoresis on SDS-PAGE gels and transferred onto Hybond P membranes (GE Healthcare, Piscataway, NJ, USA). Membranes were incubated with primary antibodies for p53 (NCL-p53-CM5p; Leica Microsystems) and Santa Cruz antibodies for β-actin (sc-1616R), cyclin B1 (sc-245), cyclin A (sc-751), K-ras (sc-30), and phospho-p53 (Ser15) (9284; Cell Signaling, Danvers, MA, USA). Signal was detected with the ECL-Plus detection system (GE Healthcare) and quantified using Scion Image Beta 4.0.3 (Scion Corporation, Frederick, MD, USA).
RNA interference
Cells were transfected with K-ras siRNA (D-043 846-01; Dharmacon, Lafayette, CO, USA) or control siRNA (D-001 210-04-05; Dharmacon) using the TransIT-TKO system (Mirus Corporation, Madison, WI, USA) following the manufacturer’s recommendations. For mitosis labelling, the cells were plated on coverslips and transfected with the siRNAs indicated above or an siRNA for p53 (D-040 642-01). Twenty-four hours later, the cells were pulsed with BrdU for 30 min, fixed, and subjected to double immunofluorescence for BrdU and pHH3.
Statistical analysis
All comparisons between groups were analysed by the two-tail Student’s t-test. Error bars represent the mean ± standard error of the mean.
Results
An endogenous p53R172H gain-of-function mutation promotes SCCHN progression
To determine the role of the p53R172H gain-of-function mutation (equivalent to human p53R175H) during head and neck cancer development, we generated mice in which the endogenous p53R172H mutation was activated exclusively in the oral epithelium of mice carrying a conditional p53R172H allele [20] and the K5.Cre*PR1 transgene that allows expression of an RU486-inducible Cre recombinase [30] (Supporting information, Supplementary Figure 1). In these mice (K-p53R172H/wt mice), oral tumour formation was initiated by activation of oncogenic K-rasG12D [30,32]. In addition, a floxed-p53 allele (p53f) [31] was used to induce deletion of the remaining p53 allele, to rule out potential dominant negative effects of mutant p53 (K-p53R172H/f mice). The potential gain-of-function properties of mutant p53 were analysed by comparing the tumour rates in these mice with mice in which both copies of the p53 gene were deleted in the presence of K-rasG12D (K-p53f/f mice). Control mice expressing K-rasG12D and wild-type (wt) p53 were also generated (K-p53wt/wt mice). The conditional alleles in all of these mice were activated in the oral cavity by topical application of RU486.
We found that K-p53R172H/f and K-p53R172H/wt mice developed twice as many tumours as K-p53f/f or K-p53wt/wt mice, indicating that mutant p53 contributes to early oral squamous tumourigenesis (Figure 1A). Moreover, tumours that developed in the K-p53R172H/f and K-p53R172H/wt mice grew faster than tumours that developed in K-p53f/f mice or mice with wt p53 (Figure 1B). Most of the tumours that developed in these mice were classified as benign papillomas (Figures 2A–B and 2D–E). Notably, progression to carcinoma occurred only in K-p53R172H/f mice, but not in K-p53f/f mice (Figures 2C and 2F). We detected carcinomas in 15% of the K-p53R172H/f mice that survived at least 44 weeks, with a mean latency of 42 weeks after activation of the conditional alleles. However, the penetrance of progression to carcinoma could not be fully determined due to the short lifespan of K-p53R172H/f mice (12–15 months). Nevertheless, K-p53f/f mice had a longer lifespan but never developed carcinomas, indicating that mutant p53, but not loss of p53, predisposed to carcinoma formation. Activation of the conditional alleles in the tumours was verified by PCR using specific primers that allow the identification of activated mutant and wt alleles [22] (Supporting information, Supplementary Figure 1D). The expression of mutant p53 in tumours with an activated p53R172H allele was further confirmed by immunohistochemistry (Figures 2G–2I). Collectively, these observations document a role for the mutant p53R172H in oral cancer initiation, tumour growth, and progression to carcinoma.
Figure 1.
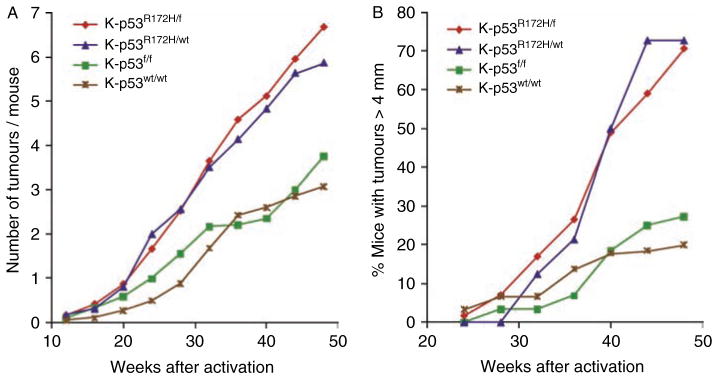
Kinetics of oral tumour formation and growth. (A) Kinetics of oral tumour formation in mice carrying different combinations of p53 alleles: K-p53R172H/f mice (red line, n = 62), K-p53R172H/wt mice (blue line, n = 17), K-p53f/f mice (green line, n = 30), and K-p53wt/wt mice (brown line, n = 18). Tumour initiation was determined by the average number of tumours that developed per mouse after Cre activation induced by topical application of RU486 in the oral cavity. (B) Kinetics of tumour growth in mice described in panel A. Each time point represents the percentage of mice with tumours larger than 4 mm.
Figure 2.

Histopathology of the mouse oral tumours. (A, B) Gross appearance of papillomas (Pap) that developed in the oral cavity of (A) K-p53f/f mice or (B) K-p53R172H/f mice, 8 months after activation of the conditional alleles. (C) Frontal view showing the gross appearance of a carcinoma (SCC) that developed in a K-p53R172H/f mouse. (D–F) Haematoxylin and eosin staining of oral papillomas that developed in (D) K-p53f/f mice or (E) K-p53R172H/f mice, and (F) the carcinoma shown in panel C. (G–I) Immunohistochemistry for p53 in oral papillomas that developed in (G) K-p53f/f mice and (H) papillomas or (I) carcinomas that developed in K-p53R172H/f mice.
Mutant p53 modulates the expression of genes involved in mitosis and the mitotic rates in oral tumours
To identify molecular mechanisms involved in the oncogenic function of mutant p53, we compared the expression profiles of p53-null oral papillomas that developed in K-p53f/f mice (hereafter referred as K-p53−/− tumours) with those of papillomas that expressed endogenous mutant p53R172H (K-p53R172H/− and K-p53R172H/+). Gene expression analysis identified 131 genes that were up-regulated and 70 genes down-regulated in p53R172H tumours, with a false discovery rate less than 5% (Figure 3A and Supporting information, Supplementary Table 1). Analysis of the functional significance of these changes using DAVID [34] revealed enrichment for several gene ontology terms related to mitosis in the gene set with increased expression in p53R172H tumours (Figure 3B). In addition, the gene network with the highest score (p = 1.0E−41) generated with Ingenuity Pathways Analysis (IPA) using genes up-regulated in p53R172H tumours contained cyclin B1 (Ccnb1) and cyclin A (Ccna2) as the highest connected nodes in a network that included 22 genes up-regulated in p53R172H tumours (Figure 3C). The increased expression of cyclin B1, cyclin A, and other genes found up-regulated in K-p53R172H tumours was confirmed by quantitative real-time-PCR (Figure 3D).
Figure 3.

Gene expression analysis of p53 mutant oral tumours. (A) Hierarchical clustering of oral tumour samples using a gene list containing genes with differential expression in oral tumours that expressed p53R172H (K-p53R172H/− and K-p53R172H/+ tumours) compared with tumours that lacked p53 (K-p53−/− tumours). Three tumours per genotype were analysed. (B) Gene ontology terms enriched in genes up-regulated in tumours that expressed p53R172H. Each bar represents a distinct term, indicating in parentheses the gene ontology category: biological process (BP) or cell component (CC). (C) Gene network generated with IPA using genes up-regulated in p53R172H tumours. Icons in red represent genes with increased expression in p53R172H tumours; white icons represent genes with unchanged expression. The highest connected nodes were moved to the centre of the network and other genes were brought to the periphery. Note that cyclin B1 (Ccnb1) and cyclin A (Ccna2) are the most connected genes in this network. (D) Real-time RT-PCR for the indicated genes. Three tumours per genotype were analysed in triplicate. *p = 0.01–0.05; **p < 0.01.
To determine the impact of these changes in gene expression in modulating mitotic rates in the tumours, we identified cells in G2–M in the oral tumours by staining with an antibody specific for phosphorylated histone H3 (pHH3). We found that the number of pHH3-positive cells was slightly higher in K-p53R172H/− tumours than in K-p53−/− tumours (Figure 4A), although this difference did not reach statistical significance. However, a more detailed evaluation of the staining pattern for pHH3 that allows the discrimination of cells in G2 (punctate staining) from cells undergoing mitosis (solid staining) [38] (Figure 4B) revealed that the fraction of cells in mitosis was significantly higher in K-p53R172H/− tumours than in K-p53−/− tumours (Figure 4C). Thus, 70% of the pHH3-positive cells in K-p53−/− tumours were in G2 and only 30% were in mitosis, whereas the K-p53R172H/− tumours contained a similar number of cells in G2 and mitosis, suggesting that entry in mitosis was accelerated in tumours that express mutant p53.
Figure 4.
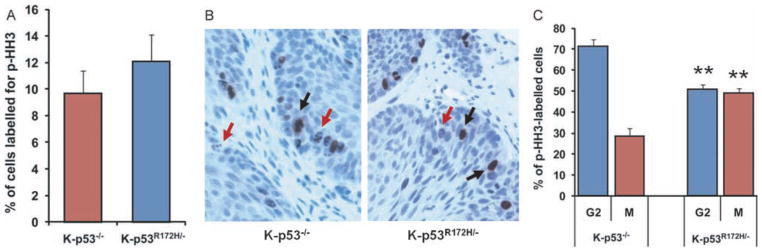
Quantification of mitosis in oral tumours. (A) Percentage of cells labelled with an antibody for pHH3 in tumours with the indicated genotypes. Five tumours per genotype were analysed and over 400 cells per tumour were counted. (B) Immunohistochemistry for pHH3 in K-p53−/− tumours (left panel) and K-p53R172H/− tumours (right panel). Note the distinctive punctate staining (red arrows) and solid staining (black arrows) in the tumours. (C) Cells in G2 (punctate staining for pHH3) and in mitosis (solid staining for pHH3) were counted and are represented as a percentage of all pHH3-positive cells. Five tumours per genotype were analysed. **p < 0.01.
Mutant p53 induces the expression of cyclin B1 and cyclin A in oral tumour cells and accelerates entry in mitosis
To further analyse the oncogenic function of mutant p53, we generated cell lines from K-p53R172H/− and K-p53−/− mouse oral tumours (Figures 5A and 5B). We found that K-p53R172H/− cells tend to accumulate significantly higher number of cells with multiple centrosomes (Figure 5C). In addition, transfection of p53R172H into K-p53−/− cells induced a significant increase in the number of cells that contained more than two centrosomes (Figure 5D), thereby confirming that mutant p53 can induce centrosome amplification in oral tumour cells. Consistent with these findings and the accelerated tumour growth observed in the K-p53R172H primary tumours, we also found that tumours induced upon subcutaneous injection of K-p53R172H/− cells grew faster than tumours induced by K-p53−/− cells (Figure 5E), supporting an increased tumourigenic potential for the K-p53R172H/− oral tumour cells.
Figure 5.

Cyclin B1 expression and accelerated entry in mitosis induced by mutant p53 in cell lines derived from mouse oral tumours. (A) Bright field images illustrating the epithelial morphology of the cells (left panels), and immunofluorescence staining showing the organization of keratin 14 (K14) filaments in K-p53−/− (top panels) and K-p53R172H/− cell lines (bottom panels), and p53 accumulation in the nuclei of K-p53R172H/− cells, but not in K-p53−/− cells. (B) Western blot for p53 in K-p53R172H/− and K-p53−/− cell lines. Two independent cell lines from each genotype are shown. (C) Centrosomes were visualized by immunofluorescence staining with a γ-tubulin antibody. The number of cells with more than two centrosomes is represented. Three independent cell lines per genotype were analysed. (D) Transfection of mutant p53 into K-p53−/− oral tumour cells using the p53R172H minigene (mp53) induces centrosome amplification. Centrosomes were scored as indicated in panel C. (E) Tumour growth induced upon subcutaneous injection of K-p53R172H/− and K-p53−/− cell lines in athymic nude mice. 106 cells per cell line were injected. Three cell lines of each genotype were analysed. (F) Luciferase reporter assay showing that transfection of K-p53−/− oral tumour cells with increasing concentrations of a plasmid carrying a p53R172H minigene results in increased activity of the cyclin B1 promoter. (G) Luciferase reporter assay for the cyclin B1 promoter in K-p53−/− and K-p53R172H/− cell lines. Data are expressed as mean of triplicates. (H) Real-time RT-PCR for Ccnb1 (cyclin B1) and Ccna2 (cyclin A) using RNA purified from K-p53−/− oral tumour cells transfected with a control empty plasmid (Contr) or a plasmid carrying a p53R172H minigene (mp53). (I) Protein expression of cyclin B1 and cyclin A in K-p53R172H/− and K-p53−/− cell lines. Three independent cell lines per genotype were analysed. (J) Percentage of BrdU-labelled mitosis at different times after BrdU labelling. To identify cells in mitosis, cells pulsed with BrdU were stained for pHH3 at different times after chasing BrdU labelling. The rate of entry in mitosis was determined by analysing the ratio of BrdU-labelled mitosis. Two cell lines per genotype were analysed and over 100 cells per time point were counted in each cell line. *p = 0.01–0.05; **p < 0.01.
Using these cells, we found that transfection of mutant p53 into K-p53−/− cells resulted in increased activity of the cyclin B1 promoter in a luciferase reporter assay (Figure 5F). Similarly, the activity of the cyclin B1 promoter was higher in K-p53R172H/− cells than in K-p53−/− cells (Figure 5G). Moreover, mutant p53 also induced the RNA expression of both cyclin B1 and cyclin A (Figure 5H), and the protein levels of cyclin B1 and cyclin A were higher in K-p53R172H/− cells than in K-p53−/− cells (Figure 5I). Consistent with these observations, we detected faster accumulation of BrdU-labelled mitosis in K-p53R172H/− cell lines, compared with K-p53−/− cells, in an experiment in which cells pulsed with BrdU were stained for pHH3 at different times to determine the rates of entry in mitosis (Figure 5J). These results indicate a faster transition from G2 to M in cells that express p53R172H. Collectively, these findings indicate that mutant p53 can induce the expression of genes involved in mitosis, including cyclin B1 and cyclin A, and promote accelerated entry in mitosis as part of its oncogenic function during head and neck cancer development.
Mutant p53 induces cyclin B1 and cyclin A in response DNA damage and K-ras
It was previously shown that mutant p53 can modulate the expression of cyclin B1 and cyclin A in breast and colon cancer cell lines in response to DNA damage [25]. To determine whether DNA damage can also induce cyclin B1 and cyclin A expression in oral tumour cells that express mutant p53, we treated K-p53R172H/− and K-p53−/− cells with doxorubicin for up to 36 h. We observed that exposure to doxorubicin induced a higher increase in the cyclin B1 and cyclin A levels in K-p53R172H/− cells than in K-p53−/− cells (Figures 6A–6D). This treatment also induced a slight increase in the phosphorylation of p53 at serine 15, accompanied by a modest raise in the mutant p53 levels, suggesting that in these tumour cells mutant p53 is only moderately stabilized in response to DNA damage (Supporting information, Supplementary Figure 2E). Together, our observations indicate that p53R172H can induce the expression of cyclin B1 and cyclin A in head and neck tumours, and in response to DNA damage.
Figure 6.

Induction of cyclin B1 and cyclin A in response to DNA damage and K-ras. (A) Western blot analysis of cellular extracts from K-p53R172H/− and K-p53−/− cells treated with 200 ng/ml doxorubicin for up to 36 h, using cyclin B1 and β-actin antibodies. (B) Protein levels in panel A are represented as fold increase relative to time 0, after normalization with β-actin, used as a loading control. (C) Analysis of cyclin A protein levels using the cell lysates described in panel A. (D) Quantification of the cyclin A protein levels shown in panel C, normalized with β-actin. (E) Immunohistochemistry for γ-H2AX in a K-p53wt/wt oral tumour. (F) Suppression of K-ras expression in K-p53R172H/− cells. Control or K-ras siRNAs were transfected into K-p53R172H/− cells, and the expression of cyclin B1, cyclin A, and K-ras was analysed by western blot. Quantification of cyclin B1 and cyclin A levels is shown in the Supporting information, Supplementary Figures 2A and 2B. (G) Suppression of K-ras expression in K-p53−/− cells. Control or K-ras siRNAs were transfected into K-p53−/− cells, and the expression of cyclin B1 and cyclin A was analysed by western blot. Quantification of cyclin B1 and cyclin A levels is shown in the Supporting information, Supplementary Figures 2C and 2D. (H) Suppression of K-ras and mutant p53 expression delays entry in mitosis in cells that express p53R172H. Percentage of labelled mitosis was determined as in Figure 5J in cells transfected with a control siRNA (blue line), K-ras siRNA (green line) or p53 siRNA (red line). Duplicates for each time point are represented and over 100 cells per time point were counted. p < 0.01 for both K-ras siRNA and p53 siRNA compared with control siRNA.
However, the oral tumours that developed in our mice appeared spontaneously upon activation of oncogenic mutations, with no further exposure to carcinogens or any other external source of DNA damage. As oncogenes can also induce DNA damage in tumours [39,40], we asked whether DNA damage occurred in the mouse oral tumours. Indeed, multiple tumour cells were stained with an antibody for γ-H2AX, a marker for double-strand breaks, thus confirming the presence of DNA damage in the tumours (Figure 6E). As we speculated that DNA damage in the oral tumours was a consequence of oncogenic K-ras expression, we suppressed the expression of K-ras in oral tumour cells to determine whether cyclin B1 and cyclin A expression was modulated by oncogenic K-ras. Remarkably, K-ras suppression in K-p53R172H/− cells resulted in decreased levels of cyclin B1 and cyclin A (Figure 6F and Supporting information, Supplementary Figures 2A and 2B). However, the expression of cyclin B1 and cyclin A did not change after K-ras suppression in oral tumour cells that lack p53 (Figure 6G and Supporting information, Supplementary Figures 2C and 2D). Consistent with these findings, suppression of K-ras delayed entry in mitosis in K-p53R172H/− cells, indicating that the accelerated entry in mitosis induced by mutant p53 in these cells was dependent on oncogenic K-ras (Figure 6H). Similarly, entry in mitosis was delayed after suppressing mutant p53 expression, thereby confirming that modulation of mitosis in these cells is a direct effect of mutant p53 (Figure 6H). Together, these observations indicate that K-ras can induce the expression of cyclin B1 and cyclin A only in the presence of mutant p53, and suggest that the increased expression of cyclin B1 and cyclin A and the accelerated entry in mitosis observed in oral tumours that express mutant p53 may respond to oncogenic K-ras activation.
Discussion
We generated mice that developed spontaneous SCCHN through a stepwise mechanism upon activation of endogenous mutations, and demonstrated an oncogenic role for the mutant p53R172H in the early stages of oral tumour formation and during progression to carcinoma. We found that mutant p53 exerts this oncogenic function by inducing the expression of genes involved in mitosis, including cyclin B1 and cyclin A, and promoting accelerated entry in mitosis. The increased expression of cyclin B1 and cyclin A induced by DNA damage in oral tumour cells that express mutant p53, the presence of DNA damage in tumours induced by K-ras activation, the induction of cyclin B1 and cyclin A by K-ras only in oral tumour cells that express mutant p53, and the faster entry in mitosis induced by K-ras in mutant p53 cells suggest that the oncogenic function of mutant p53 in SCCHN may be promoted by oncogene-dependent DNA damage. These findings uncover a novel mechanism that promotes the oncogenic function of mutant p53 and provides a mechanistic explanation for the cooperation between oncogenic Ras and p53-gain-of-function mutations in tumour initiation and progression.
The increased number of oral tumours induced by co-activation of the p53R172H and K-rasG12D mutations compared with activation of K-rasG12D alone clearly indicates that tumours do not arise from every oral epithelial cell expressing oncogenic K-ras. In fact, oncogenes, including Ras genes, can induce a DNA damage response that results in cell cycle arrest or apoptosis, thereby preventing tumour initiation and progression of benign tumours [39,40]. However, expression of mutant p53 in cells with activated K-ras may overcome DNA damage induced by oncogenic K-ras and facilitate the appearance of oral tumours. This contribution of mutant p53 to oral tumour initiation is a consequence of a gain-of-function role for mutant p53 because deletion of p53 did not affect tumour initiation induced by K-ras. Additionally, tumours that expressed mutant p53 grew faster than p53-null tumours, indicating that mutant p53 also promotes accelerated tumour growth.
Analysis of the expression profiles of the mouse oral tumours and functional studies in cell lines derived from the tumours revealed that genes involved in mitosis, including cyclin B1 and cyclin A, may contribute to the oncogenic function of mutant p53. Using these cell lines, we demonstrated that mutant p53 can induce the expression of both cyclin B1 and cyclin A. Therefore, it is unlikely that the observed changes in gene expression are secondary effects resulting from additional mutations that may arise as the tumours progress. Supporting this idea, we found that mutant p53 induces accelerated entry in mitosis both in the tumours and in culture tumour cells. These findings suggest that up-regulation of cyclin B1 and cyclin A and accelerated transition through mitosis contribute to the oncogenic function of mutant p53 during oral tumour development. Remarkably, the induction of mitotic cyclins was modulated by oncogenic K-ras, suggesting that oncogene activation is critical to expose the oncogenic function of mutant p53. In addition, DNA damage induced by exposing cells to doxorubicin resulted in a greater induction of cyclin B1 and cyclin A in cells that express mutant p53, compared with p53-null cells. These observations, together with the induction of cyclins by oncogenic K-ras only in K-p53R172H/− cells, and the presence of DNA damage in the tumours induced by K-ras suggest that mutant p53 may induce the expression of cyclin B1 and cyclin A in response to DNA damage promoted by oncogene activation (K-ras) or chemotherapy (doxorubicin). It is worth noting that cyclin B1 and cyclin A were reported to be over-expressed in human SCCHN, suggesting that these cyclins may also be involved in human SCCHN progression, although the p53 status of these tumours was not assessed [41,42].
In addition to accelerating tumour growth, the forced entry in mitosis promoted by mutant p53 may allow the accumulation of genomic alterations that ultimately facilitates progression to carcinoma. Since carcinomas developed in our mice only upon activation of the p53R172H mutation, we suggest that molecular changes induced by mutant p53 may predispose benign tumours to progress to malignant stages, although further studies will be required to determine whether overexpression of cyclin B1 and/or cyclin A can promote progression to carcinoma. Importantly, carcinomas were induced only in the presence of mutant p53 but not upon deletion of the p53 gene. Similarly, in a previously described mouse model, oral tumours induced by K-ras did not progress to carcinoma after deletion of p53, except when tumours developed in the ventral area of the tongue [43]. We did not detect conversion to carcinoma upon deletion of p53 in K-ras-initiated oral tumours, perhaps because of the limited number of tumours that developed in our mice in the area of the tongue that seems to be more sensitive to progression in the absence of p53, due to the more restrictive nature of Cre activation in our model.
Importantly, the major risk factors for SCCHN are tobacco smoking and heavy alcohol consumption, which may be attributed, at least in part, to the genotoxic effects of metabolites that allow the accumulation of genetic alterations [44,45]. It is tempting to speculate that p53 mutations may also facilitate head and neck cancer formation and progression in individuals exposed to DNA damage induced by alcohol and tobacco. In addition, certain p53 mutations are associated with poor prognosis of SCCHN [10,17,18]. Our mouse models provide genetic evidence to support these clinical observations and propose a role for p53 gain-of-function mutations during head and neck cancer progression. In this context, it is worth mentioning that since the initial discovery of the mutant p53 gain-of-function it was appreciated the distinct tumourigenic potential of different p53 mutants was appreciated [14], an observation that was later confirmed in mice carrying different germline mutations in the p53 gene [20,46]. Given the broad range of p53 mutations found in human cancers, including SSCHN [47], it is expected that different mutations may have a different impact on prognosis. Indeed, a shorter survival was observed in patients with SCCHN containing p53 mutations compared with patients with wt p53 tumours. However, when the p53 mutations were classified as disruptive or non-disruptive, based on the differential polarities of the wt and mutant amino acids and the location in the protein, it was found that disruptive mutations accounted for most of the association with the short survival, indicating that this group may contain most of the transforming p53 mutants [10]. Interestingly, the p53R175H mutation, the human equivalent to mouse p53R172H, falls into the ‘non-disruptive’ category, although it was one of the most transforming mutants in vitro and in animal models, including the one reported here [14,20–22]. By contrast, the human p53R175P mutation, equivalent to mouse p53R172P, is considered a disruptive mutation. However, germline activation of p53R172P in mice showed that tumours in these mice developed at much lower rates compared with p53-null mice because p53R172P can induce cell cycle arrest, although it is deficient in inducing apoptosis [46]. Collectively, these observations indicate that classifying p53 mutations certainly helps to predict SCCHN outcomes, but additional efforts in refining the classification methods will help to improve prediction and design potential therapies to target these mutants.
Supplementary Material
Acknowledgments
We would like to thank Tyler Jacks and David Tuveson for providing LSL-K-rasG12D mice, Anton Berns for the floxed-p53 mice, Lisa White for assistance with microarray experiments, Jeffrey Rosen and Karen Katula for reagents, Guillermina Lozano for the Neo-p53R172H mice and critically reading the manuscript, and Jeffrey Myers for helpful discussions. This work was supported by NIH grants CA105491 to D Roop and C Caulin, and DE015344 to C Caulin.
Footnotes
Author contribution statement
SA and ZL conceived and carried out experiments, and analysed data. OM carried out experiments. DRR provided materials and participated in the initial conception of the study. AE-N analysed tumour pathology. CC designed the study, conceived experiments, analysed data, and wrote the manuscript.
No conflicts of interest were declared.
SUPPORTING INFORMATION ON THE INTERNET
The following supporting information may be found in the online version of this article.
Figure S1. Schematic representation of the conditional alleles and activation in tumors.
Figure S2. Quantification of the western blots shown in Figures 6F–6G; p53 levels in response to DNA damage and control staining for γ-H2AX.
Table S1. Genes differentially expressed in K-p53R172H/− and K-p53R172H/+ oral tumors compared to K-p53−/− tumors.
References
- 1.Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Seiwert TY, Salama JK, Vokes EE. The chemoradiation paradigm in head and neck cancer. Nature Clin Pract Oncol. 2007;4:156–171. doi: 10.1038/ncponc0750. [DOI] [PubMed] [Google Scholar]
- 3.Vokes EE, Weichselbaum RR, Lippman SM, et al. Head and neck cancer. N Engl J Med. 1993;328:184–194. doi: 10.1056/NEJM199301213280306. [DOI] [PubMed] [Google Scholar]
- 4.Culliney B, Birhan A, Young AV, et al. Management of locally advanced or unresectable head and neck cancer. Oncology (Williston Park) 2008;22:1152–1161. [PubMed] [Google Scholar]
- 5.Califano J, van der Riet P, Westra W, et al. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res. 1996;56:2488–2492. [PubMed] [Google Scholar]
- 6.Forastiere A, Koch W, Trotti A, et al. Head and neck cancer. N Engl J Med. 2001;345:1890–1900. doi: 10.1056/NEJMra001375. [DOI] [PubMed] [Google Scholar]
- 7.El-Naggar AK, Lai S, Luna MA, et al. Sequential p53 mutation analysis of pre-invasive and invasive head and neck squamous carcinoma. Int J Cancer. 1995;64:196–201. doi: 10.1002/ijc.2910640309. [DOI] [PubMed] [Google Scholar]
- 8.Olshan AF, Weissler MC, Pei H, et al. p53 mutations in head and neck cancer: new data and evaluation of mutational spectra. Cancer Epidemiol Biomarkers Prev. 1997;6:499–504. [PubMed] [Google Scholar]
- 9.Andrews GA, Xi S, Pomerantz RG, et al. Mutation of p53 in head and neck squamous cell carcinoma correlates with Bcl-2 expression and increased susceptibility to cisplatin-induced apoptosis. Head Neck. 2004;26:870–877. doi: 10.1002/hed.20029. [DOI] [PubMed] [Google Scholar]
- 10.Poeta ML, Manola J, Goldwasser MA, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. 2007;357:2552–2561. doi: 10.1056/NEJMoa073770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 12.Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 13.Hainaut P, Soussi T, Shomer B, et al. Database of p53 gene somatic mutations in human tumors and cell lines: updated compilation and future prospects. Nucleic Acids Res. 1997;25:151–157. doi: 10.1093/nar/25.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dittmer D, Pati S, Zambetti G, et al. Gain of function mutations in p53. Nature Genet. 1993;4:42–46. doi: 10.1038/ng0593-42. [DOI] [PubMed] [Google Scholar]
- 15.Sun Y, Nakamura K, Wendel E, et al. Progression toward tumor cell phenotype is enhanced by overexpression of a mutant p53 tumor-suppressor gene isolated from nasopharyngeal carcinoma. Proc Natl Acad Sci U S A. 1993;90:2827–2831. doi: 10.1073/pnas.90.7.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nature Rev Cancer. 2009;9:701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 17.Perrone F, Bossi P, Cortelazzi B, et al. TP53 mutations and pathologic complete response to neoadjuvant cisplatin and fluorouracil chemotherapy in resected oral cavity squamous cell carcinoma. J Clin Oncol. 2010;28:761–766. doi: 10.1200/JCO.2009.22.4170. [DOI] [PubMed] [Google Scholar]
- 18.Shin DM, Lee JS, Lippman SM, et al. p53 expressions: predicting recurrence and second primary tumors in head and neck squamous cell carcinoma. J Natl Cancer Inst. 1996;88:519–529. doi: 10.1093/jnci/88.8.519. [DOI] [PubMed] [Google Scholar]
- 19.Hassan NM, Tada M, Hamada J, et al. Presence of dominant negative mutation of TP53 is a risk of early recurrence in oral cancer. Cancer Lett. 2008;270:108–119. doi: 10.1016/j.canlet.2008.04.052. [DOI] [PubMed] [Google Scholar]
- 20.Lang GA, Iwakuma T, Suh YA, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li–Fraumeni syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 21.Olive KP, Tuveson DA, Ruhe ZC, et al. Mutant p53 gain of function in two mouse models of Li–Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Caulin C, Nguyen T, Lang GA, et al. An inducible mouse model for skin cancer reveals distinct roles for gain- and loss-of-function p53 mutations. J Clin Invest. 2007;117:1893–1901. doi: 10.1172/JCI31721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morton JP, Timpson P, Karim SA, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A. 2010;107:246–251. doi: 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaiddon C, Lokshin M, Ahn J, et al. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol. 2001;21:1874–1887. doi: 10.1128/MCB.21.5.1874-1887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Di Agostino S, Strano S, Emiliozzi V, et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell. 2006;10:191–202. doi: 10.1016/j.ccr.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 26.Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nature Cell Biol. 2007;9:573–580. doi: 10.1038/ncb1571. [DOI] [PubMed] [Google Scholar]
- 27.Muller PA, Caswell PT, Doyle B, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327–1341. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 28.Goh AM, Coffill CR, Lane DP. The role of mutant p53 in human cancer. J Pathol. 2011;223:116–126. doi: 10.1002/path.2784. [DOI] [PubMed] [Google Scholar]
- 29.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Protoc. 2010;2:a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caulin C, Nguyen T, Longley MA, et al. Inducible activation of oncogenic K-ras results in tumor formation in the oral cavity. Cancer Res. 2004;64:5054–5058. doi: 10.1158/0008-5472.CAN-04-1488. [DOI] [PubMed] [Google Scholar]
- 31.Jonkers J, Meuwissen R, van der Gulden H, et al. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nature Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 32.Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li C, Hung WW. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2:RESEARCH0032. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dennis G, Jr, Sherman BT, Hosack DA, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- 35.Roop DR, Cheng CK, Titterington L, et al. Synthetic peptides corresponding to keratin subunits elicit highly specific antibodies. J Biol Chem. 1984;259:8037–8040. [PubMed] [Google Scholar]
- 36.Li B, Murphy KL, Laucirica R, et al. A transgenic mouse model for mammary carcinogenesis. Oncogene. 1998;16:997–1007. doi: 10.1038/sj.onc.1201621. [DOI] [PubMed] [Google Scholar]
- 37.Barrett KL, Demiranda D, Katula KS. Cyclin B1 promoter activity and functional cdk1 complex formation in G1 phase of human breast cancer cells. Cell Biol Int. 2002;26:19–28. doi: 10.1006/cbir.2001.0817. [DOI] [PubMed] [Google Scholar]
- 38.Hendzel MJ, Wei Y, Mancini MA, et al. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- 39.Di MR, Fumagalli M, Cicalese A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 40.Bartkova J, Rezaei N, Liontos M, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 41.Kushner J, Bradley G, Young B, et al. Aberrant expression of cyclin A and cyclin B1 proteins in oral carcinoma. J Oral Pathol Med. 1999;28:77–81. doi: 10.1111/j.1600-0714.1999.tb02000.x. [DOI] [PubMed] [Google Scholar]
- 42.Hassan KA, el Naggar AK, Soria JC, et al. Clinical significance of cyclin B1 protein expression in squamous cell carcinoma of the tongue. Clin Cancer Res. 2001;7:2458–2462. [PubMed] [Google Scholar]
- 43.Raimondi AR, Molinolo A, Gutkind JS. Rapamycin prevents early onset of tumorigenesis in an oral-specific K-ras and p53 two-hit carcinogenesis model. Cancer Res. 2009;69:4159–4166. doi: 10.1158/0008-5472.CAN-08-4645. [DOI] [PubMed] [Google Scholar]
- 44.Nakayama T, Kaneko M, Kodama M, et al. Cigarette smoke induces DNA single-strand breaks in human cells. Nature. 1985;314:462–464. doi: 10.1038/314462a0. [DOI] [PubMed] [Google Scholar]
- 45.Brooks PJ, Theruvathu JA. DNA adducts from acetaldehyde: implications for alcohol-related carcinogenesis. Alcohol. 2005;35:187–193. doi: 10.1016/j.alcohol.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 46.Liu G, Parant JM, Lang G, et al. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nature Genet. 2004;36:63–68. doi: 10.1038/ng1282. [DOI] [PubMed] [Google Scholar]
- 47.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.