

Abstract
Adenosine provides anti-inflammatory effects in cardiovascular disease via the activation of adenosine A2A receptors; however, the physiological effect of adenosine could be limited due to its phosphorylation by adenosine kinase. We hypothesized that inhibition of adenosine kinase exacerbates extracellular adenosine levels to reduce renal inflammation and injury in streptozotocin-induced diabetes. Diabetes was induced in male C57BL/6 mice by daily injection of streptozotocin (50 mg/kg/day, i.p. for 5 days). Control and diabetic mice were then treated with the adenosine kinase inhibitor ABT702 (1.5 mg/kg, i.p two times a week for 8 weeks, n = 7–8/group) or the vehicle (5% DMSO). ABT702 treatment reduced blood glucose level in diabetic mice (~ 20%; p<0.05). ABT702 also reduced albuminuria and markers of glomerular injury, nephrinuria and podocalyxin excretion levels, in diabetic mice. Renal NADPH oxidase activity and urinary thiobarbituric acid reactive substances (TBARS) excretion, indices of oxidative stress, were also elevated in diabetic mice and ABT702 significantly reduced these changes. ABT702 increased renal endothelial nitric oxide synthase expression (eNOS) and nitrate/nitrite excretion levels in diabetic mice. In addition, the diabetic mice displayed an increase in renal macrophage infiltration, in association with increased renal NFB activation. Importantly, treatment with ABT702 significantly reduced all these inflammatory parameters (P< 0.05). Furthermore, ABT702 decreased glomerular permeability and inflammation and restored the decrease in glomerular occludin expression in vitro in high glucose treated human glomerular endothelial cells. Collectively, the results suggest that the reno-protective effects of ABT702 could be attributed to the reduction in renal inflammation and oxidative stress in diabetic mice.
Keywords: Diabetes, adenosine kinase inhibition, albuminuria, renal injury, oxidative stress, nitric oxide, macrophage infiltration
Introduction
Diabetes mellitus is now considered an epidemic disease [1,2]. Careful glycemic control is challenging and it is well known that hyperglycemia leads to several complications including nephropathy despite existing therapeutic measures [3,4]. Approximately 25 % of patients with type 1 and type 2 diabetes develop renal injury [5,6]. Diabetic renal injury is mainly characterized by albuminuria, hypertension and loss of renal function [7]. Diabetic nephropathy is the primary cause of end-stage renal disease worldwide [8]. Microvascular dysfunction in the glomerulus appears as an early pathogenic event in progression of diabetic renal complication [9,10]. Current treatment of diabetic renal injury involves blood pressure and glucose control [11]. Renin-angiotensin system blockers have been modestly successful in delaying the progression of renal injury. This necessitates the need of new therapeutic interventions that reverse early pathophysiological changes of diabetic renal complications and hence prevent the progression to end-stage renal failure.
The etiology of diabetic renal injury is multi-factorial; however, oxidative stress and inflammation play a crucial role in the progression of diabetic renal injury [12–14]. Hyperglycemia increases oxidative stress in diabetic animal models and it is well known that NAD(P)H oxidase is the a major source of superoxide production in the vasculature [15,16]. Besides its ability to elevate inflammatory cytokines production, superoxide also scavenges NO decreasing NO availability and leads to the production of peroxynitrite, which has deleterious effects on the vasculature [17–19]. On the other hand, evidence suggests that renal infiltration of monocytes/macrophages also contributes to the pathogenesis of diabetic renal injury [20,21]. The subsequent immune response to infiltrated monocytes/macrophages leads to renal fibrosis and extra cellular matrix deposition due to the release of lysosomal enzymes, superoxide and inflammatory cytokines perturbing the renal inflammatory cascade during diabetes [13,22,23]. Thus, pharmacological manipulation of inflammation is a rational approach to prevent early pathological changes of diabetic renal injury and subsequent loss of renal function.
The physiological effect of adenosine is now considered a new direction in halting the progression of organ damage in cardiovascular disease [24–26]. Adenosine has diverse functions, depending on its interaction with different receptor subtypes: A1, A2A, A2B, and A3 [24–26]. Adenosine stimulation of adenosine A2A receptor (A2A) has been shown to provide potent anti-inflammatory effects and A2A receptor are highly expressed in the glomeruli and immune cells such as monocyte/macrophages, neutrophils and T cells [24,27–29]. In the kidney, A2A receptor activation has been shown to attenuate inflammation and renal injury associated with diabetic nephropathy [24]. Agonists of A2A receptor also preserved the normal structure of podocyte foot processes, slit diaphragms, and actin cytoskeleton in puromycin-induced podocyte injury in mice [30]. Given the crucial role of inflammation in the development of diabetic renal injury and the fact that adenosine kinase could limit the extracellular adenosine levels by its conversion to adenosine mono-phosphate, we target adenosine kinase, the key enzyme in adenosine metabolism, using the selective adenosine kinase inhibitor ABT702 to increase free adenosine levels. We hypothesize that adenosine kinase inhibition attenuates diabetes-induced renal injury as the potential effects of ABT702 in diabetes-induced renal complications have not been clearly addressed. Thus, we evaluated the efficacy of ABT702 in alleviating diabetes-induced renal injury in the current study in the context of its potential effects on oxidative stress and inflammation.
Materials and methods
All procedures with animals were performed in accordance with the Public Health Service Guide for the Care and Use of Laboratory Animals (Department of Health, Education, and Welfare publication, NIH 80-23) and Georgia Regents University guidelines. Ten-week-old male C57BL/6 mice were given daily injection of streptozotocin (50 mg/kg; i.p.) for 5 days after a 4 hour fast; control animals received the vehicle (citrate buffer; 0.01 mol/L, pH: 4.5). Diabetes was confirmed by measurement of fasting blood glucose levels of >250 mg/dl three days after streptozotocin injection. Thereafter, diabetic mice were randomly subdivided to receive the adenosine kinase inhibitor 4-amino-5-(3-bromophenyl)-7-(6-morpholinopyridin-3-yl)pyrido[2, 3-d]pyrimidine {ABT702 (CAS214697-26-4), 1.5 mg/kg, i.p., Santa Cruz Biotechnology, Santa Cruz, CA} two times a week or injections of the vehicle, 5% DMSO (n = 7–8/group) for eight weeks. This dose has previously shown to lower oxidative stress, inflammation and apoptosis in diabetic retina [25]. Eight weeks after initiation of ABT702 treatment, mice were individually placed in metabolic cages for collection of 24-hour urine samples for determination of total protein, albumin, nephrin, podocalyxin, collagen, creatinine, thiobarbituric acid reactive substances (TBARS), nitrate/nitrite and monocyte chemoattractant protein-1 (MCP-1) excretions. Mice were then anesthetized with sodium pentobarbital (50 mg/kg i.p.) and blood samples obtained for glucose and creatinine determination. Kidneys were also insolated for histopathological examination, adenosine kinase levels, NADPH oxidase and P-65NFB activities and Western blotting analysis.
Renal Histopathology
Four mice were used from each group. Kidney was fixed in 4% paraformaldehyde in PBS and embedded in paraffin. Five micrometer-thick sections were obtained, deparaffinized, and stained with Masson trichrome for detection of collagen deposition. Ten microscopic images of the kidney cortex per mice were randomly taken at 200× magnification using light-microscopy and intensity of the staining was scored blindly on a scale of 1 to 10. Additional sections were used for immunohistochemical evaluation of nephrin as a marker of glomerular injury and F4/80 as a marker for activated macrophage infiltration (antibodies from Santa Cruz Biotechnology, Santa Cruz, CA). The number of F4/80 positive cells was counted in kidney sections by an evaluator who had no knowledge of the treatment groups and correlated to the area in square millimeter.
Renal NADPH oxidase and p-65NFB activities
NADPH oxidase enzymatic activity was measured by lucigenin chemiluminescence (5 µmol/L) in the presence of 100 µmol/L NADPH as previously described [31]. Briefly, the renal cortex was homogenized in ice cold buffer in the presence of protease inhibitors. 50 µL of homogenate or buffer control was added to the wells of a 96-well micro-plate (OptiPlate-96, Perkin-Elmer, Waltham, MA) and incubated at 37°C for 30 minutes in the presence of lucigenin (5 µmol/L) and NADPH. Luminescence was quantified using a Top Count Micro-Plate Scintillation and Luminescence Counter (Perkin Elmer, Waltham, MA) set to single-photon counting mode and total count per minute (cpm) was normalized to g protein.
For assessment of p65-NFB activity, a nuclear extract kit from Cayman (Ann Arbor, MI) was used to prepare nuclear extract from the kidney cortex. The nuclear extract was used for the assessment of nuclear factor kappa B (NFB) activity using the TransAM p65-NFB transcription factor assay kit (Cayman, Ann Arbor, MI) and optical density was normalized per ng of nuclear extract protein.
Homogenization of the renal cortex for protein expression using Western blotting
Renal lysates were subjected to Western blot analysis as previously described [21,31]. Briefly, kidney samples were homogenized in ice cold modified RIPA buffer supplemented with inhibitors for proteases and phosphatases. Protein concentrations were determined by standard Bradford assay (Bio-Rad, Hercules, CA) using bovine serum albumin as the standard. Kidney protein samples (30–50 µg) were separated by SDS-PAGE then transferred onto a nitrocellulose membrane and incubated with primary antibodies. Antibodies for adenosine kinase, eNOS, adenosine A1 and A2A (Santa Cruz Biotechnology, Santa Cruz, CA) and β-actin (Sigma) were detected with a horseradish peroxidase-conjugated antibody and ECL chemiluminescence (Amersham BioSciences, Buckinghamshire, UK). Intensity of immunoreactivity was measured by densitometry and β-Actin was used to verify equal protein loading (n=6/group).
Assays
Urinary total protein and albumin excretions were determined using commercially available kits from Biorad, Hercules, CA and Exocell, Philadelphia, PA, respectively. Urinary excretions of nephrin, podocalyxin and collagen were measured using ELISA kits from Exocell, Philadelphia, PA. Urinary MCP-1 excretion was assessed as a marker of inflammation using an ELISA kit from BD Bioscience, San Jose, CA. Plasma and urinary creatinine were assessed using a kit from Cayman Chemical (Ann Arbor, MI) and values were used to calculate creatinine clearance. Urinary TBARS excretion was assessed as a marker of oxidative stress (Cayman Chemical, Ann Arbor, MI). Urinary nitrate/nitrite was also assessed as an indication of available NO using a kit from Cayman Chemical, Ann Arbor, MI.
Human glomerular endothelial cells culture
Human glomerular endothelial cells (HGECs, ACBRI 128) were purchased as cryopreserved aliquot in passage 3 from Cell System (Kirkland, WA). Cells were cultured in CSC complete medium with 10% serum (4Z0-500) in a 37 °C temperature controlled incubator with 5% carbon dioxide. The cells were splitted using CSC Passage Reagent Group (4Z0-800) and were continuously grown until passage 7–8 was reached.
To determine whether ABT 702's anti-inflammatory effects could improve glomerular permeability and barrier function, HGECs were seeded on collagen/fibronectin coated membranes with 0.4 µm pores (Transwell; Corning Costar) in normal glucose media. Confluent cells were switched to 1% serum media overnight and were treated with normal (5mM glucose) or high (30 mM glucose) glucose condition with either vehicle (DMSO) or 50 µM ABT702 in the apical chambers of inserts for five days using VEGF as positive control (100 ng/ml) to increase permeability and mannitol (25 mM) to rule out the osmotic effect of high glucose on permeability (n=3/treatment). Fluorescein isothiocyanate–dextran (FITC-dextran) was then added to the apical chambers of inserts followed by obtaining aliquots from the basolateral chamber every 30 minute for 6 hours after the dextran application to measure the fluorescence intensity. The rate of diffusive flux (Po) was calculated as recently shown [32] by the following formula, Po = [(FA/t)VA/(FLA). Po is in centimeters/second; FA is basolateral fluorescence; FL is apical fluorescence; t is change in time; A is the surface area of the filter in square centimeters; and VA is the volume of the basolateral chamber in cubic centimeters. Additional cells were also treated with normal and high glucose condition with either vehicle or 50 µM ABT702 in DMSO for five days before the cells were harvested and homogenized in RIPA buffer supplemented with protease and phosphatase inhibitors for Western blotting determination of glomerular occludin and mitogen-activated protein kinase (MAPK) phosphorylation (n=3/treatment).
Data analyses
Statistical analyses were performed using Prism software (Graph Pad, San Diego, CA). Data are reported as means SEM. All data were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test for comparison of groups; p<0.05 was considered significant.
Results
Effect of ABT702 on metabolic parameters in diabetic mice
The streptozotocin-treated mice displayed a decrease (~ 20%; p<0.05) in body weight eight weeks after induction of diabetes compared to their control counterparts with a modest non-significant increase in food intake (Table 1). ABT702 did not significantly change body weight loss or food intake (Table 1). Hyperglycemia also increased water intake and urine volume compared to control mice (Table 1, P<0.05). Although ABT702 treatment attenuated the increase in water intake and urine production in diabetic mice, these changes were not significant. Blood pressure was not affected by either diabetes or ABT702 treatment (Table 1). There was a significant increase in non-fasting blood glucose level in streptozotocin-induced diabetic mice compared to control (400± 21 vs. 190± 31 mg/dl, P< 0.05). ABT702 treatment significantly reduced plasma glucose levels in diabetic mice compared to the untreated diabetic group (323±27 vs. 400±21 mg/dl, respectively); however, blood glucose levels remained higher than the control group (323±27 vs. 190±31 mg/dl, P< 0.05). Although there was no difference in creatinine clearance among mice groups, induction of diabetes increased albuminuria relative to control and ABT702 significantly reduced the increase in albuminuria in diabetic mice, however, albuminuria remained significantly elevated in ABT702 treated diabetic mice compared to control mice with or without ABT702 treatment (Table 1).
Table 1.
Effects of ABT702 treatment on body weight, urine volume, food and water intake, blood glucose, blood pressure, creatinine clearance and albuminuria in steptozotocin-induced diabetic mice.
| Group | Control | Control/ABT702 | Diabetic | Diabetic/ABT702 |
|---|---|---|---|---|
| Body weight (gm) | 30± 0.5 | 29± 0.7 | 24± 0.8* | 24± 0.6* |
| Urine volume (ml/day) | 2.8 ± 0.4 | 2.4 ± 0.4 | 17.7 ± 2.0* | 14± 2.0* |
| Water intake (ml/day) | 9.4 ± 1.0 | 9.4 ± 0.8 | 23.1 ± 2.5* | 18± 2.5* |
| Food intake (gm/day) | 9.6 ± 1.2 | 11.3 ± 0.5 | 10.6± 1.5 | 12.3± 2.2 |
| Non-fasting blood glucose (mg/dl) | 190 ± 31 | 189 ± 3 | 400 ± 21* | 323± 27*≠ |
| Creatinine clearance (ml/hour) | 4.4 ± 0.4 | 4.6 ± 0.9 | 5.2 ± 0.7 | 4.5± 0.6 |
| Systolic blood pressure (mmHg) | 125± 2 | 131± 6 | 129± 4 | 126± 2 |
| Albuminuria (µg/day) | 16 ± 2.0 | 23 ± 6.0 | 270 ± 30* | 191 ± 11*# |
Mice were placed in metabolic cages for 24 h after eight weeks of induction of diabetes and ABT702 treatment (n=7–8/group, * P < 0.05 vs. control or control/ABT702 treated mice and #P < 0.05 vs. diabetic mice).
Renal adenosine kinase expression significantly increased in control mice treated with ABT702. Induction of diabetes with streptozotocin significantly decreased renal adenosine kinase expression when compared to control untreated or treated mice with ABT702 (P<0.05). Inhibition of adenosine kinase with ABT702 partially restored the decrease in renal adenosine kinase expression in diabetic mice (P<0.05); however, renal adenosine kinase expression in diabetic/ABT702 treated group remained significantly lowered than control mice treated with ABT702 (Figure 1A). There was no difference in renal adenosine A1 receptor expression among mouse groups (Figure 1B). Diabetes mildly increased renal adenosine A2A receptor expression relative to control, although this change was not significant (Figure 1B, P< 0.06). ABT702 significantly decreased A2A receptor expression in control and diabetic mice when compared to either control or diabetic untreated mice (Figure 1C, P< 0.05).
Figure 1.
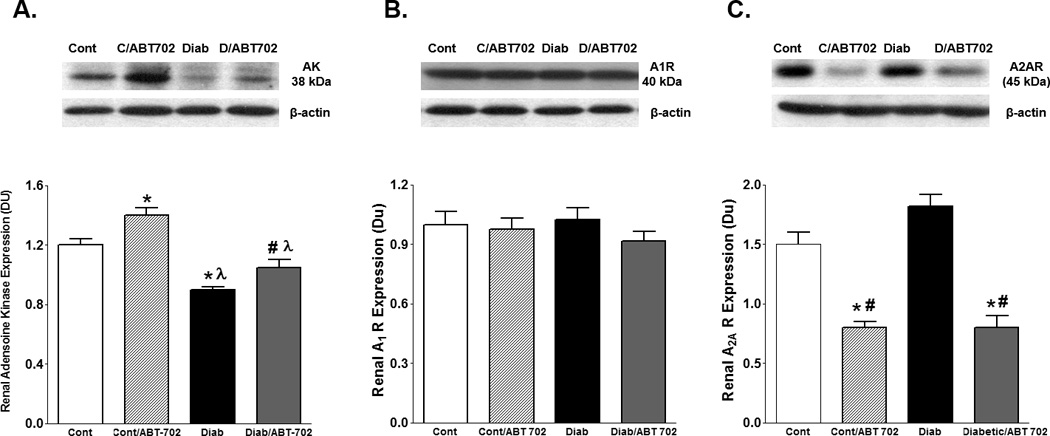
Renal adenosine kinase (A), adenosine A1 receptor (B) and adenosine A2A receptor (C) expression levels relative to β-actin in streptozotocin-induced diabetic mice. Adenosine kinase expression decreased in diabetic mice and was partially restored by ABT702 treatment. Although there was no significant change in A1 receptor expression between control and diabetic mice with or without ABT702, inhibition of adenosine kinase with ABT702 significantly reduced renal A2A receptor expression in control as well as in diabetic mice. (*P <0.05 vs. control mice, P<0.05 vs. control/ABT702 mice and #P <0.05 vs. diabetic mice, n=6/group).
The development of diabetes in mice was associated ~ 2 folds increase in total protein excretion and ~ 6 folds increase in albumin excretion when compared to control mice. ABT702 treatment resulted in a significant decrease in protein and albumin excretion in diabetic mice compared to their untreated counterparts (Figure 2A–B). We also determined whether the effect of ABT702 on albumin excretion is associated with the protection of kidney-slit diaphragm from hyperglycemia-induced podocyte loss and disruption of kidney filtration barrier. Thus, we measured urinary excretion of nephrin as a critical component of the slit diaphragm in the glomerular filtration barrier and podocalyxin excretion as a marker of podocyte protein shedding in diabetes. As shown in Figure 3, nephrin and podocalyxin excretion levels were significantly elevated in diabetic vs. control mice (P< 0.05). ABT702 reduced nephrin and podocalyxin excretion levels in diabetic mice; however, these changes were not significant (Figure 3A & B) as nephrin and podocalyxin excretion levels remained significantly higher in ABT702 treated diabetic mice than control mice (P< 0.05). These observations were also consistent with lower glomerular nephrin expression in diabetic mice vs. control and ABT702 partially restored the loss in glomerular nephrin expression (Figure 3A). Similarly, while diabetes is known to increase collagen deposition as shown in kidney sections stained with Masson trichrome (blue staining, Figure 4A) as well as increase collagen excretion (Figure 4B), ABT702 significantly reduced the increase in renal collagen deposition and collagen excretion in diabetic mice (Figure 4B).
Figure 2.
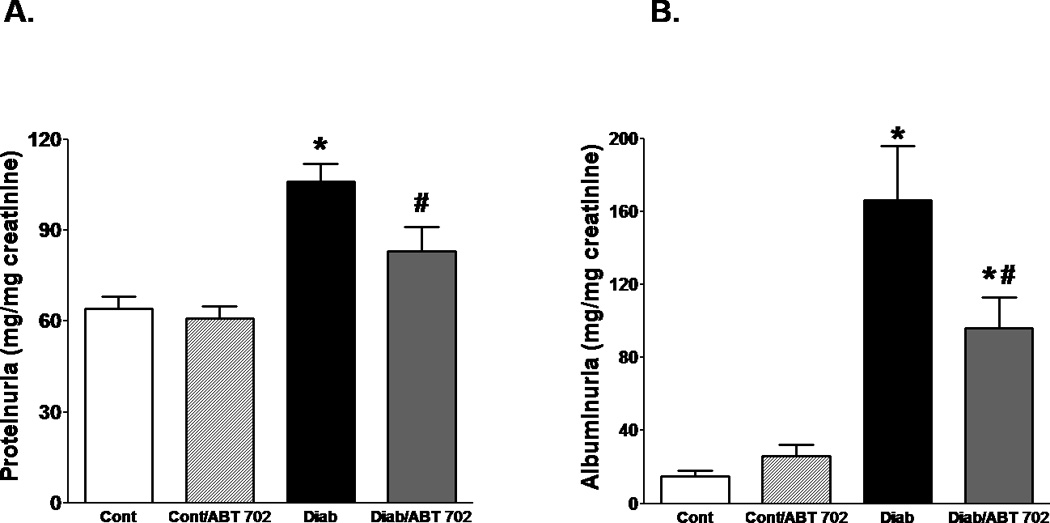
Urinary protein (A) and albumin (B) excretion levels relative to urinary creatinine as indices of renal injury in streptozotocin-induced diabetic mice. Protein and albumin excretions increased significantly in diabetic than control mice (*P <0.05 vs. control mice). Inhibition of adenosine kinase with ABT702 treatment resulted in significant reduction in these parameters in diabetic mice (#P <0.05 vs. diabetic mice; n= 7–8/group). However, albuminuria in ABT702 treated diabetic mice remained significantly higher than control non-diabetic mice.
Figure 3.
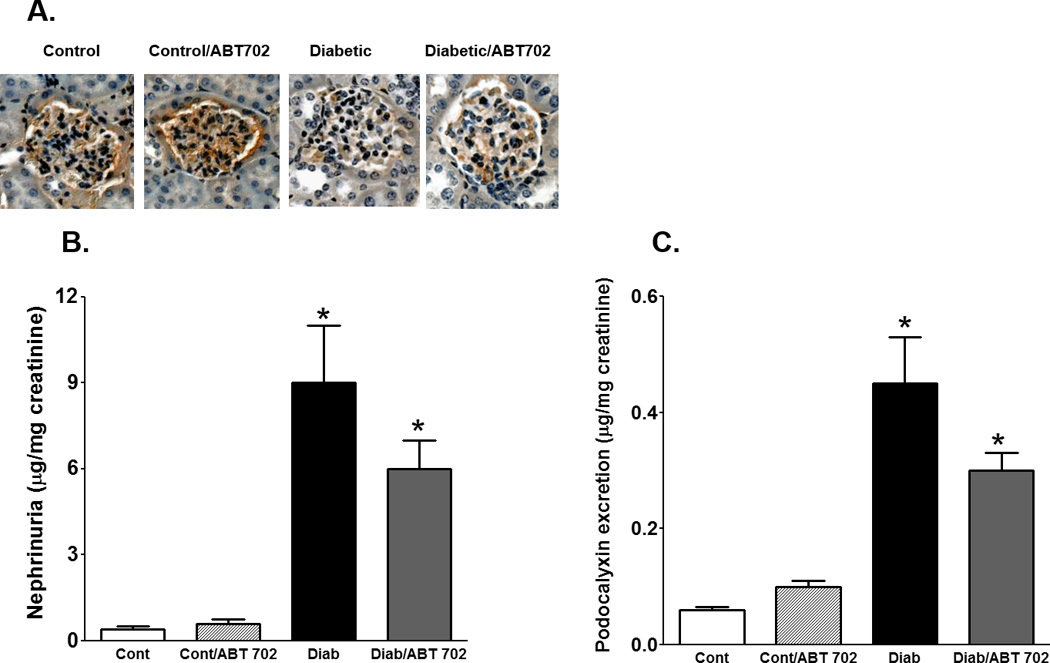
Effect of adenosine kinase inhibition on glomerular nephrin expression (A) and urinary nephrin and podocalyxin excretion levels (B–C) in streptozotocin-induced diabetic mice. Glomerular nephrin expression decreased and urinary nephrin and podocalyxin excretion levels significantly increased in diabetic when compared to control mice (*P <0.05 vs. control mice). ABT702 treatment mildly rescued the decrease in glomerular nephrin expression as well as decreased nephrin and podocalyxin excretion in diabetic mice although these changes were not significant (n=4 for nephrin expression and n=7–8/group for excretion data).
Figure 4.
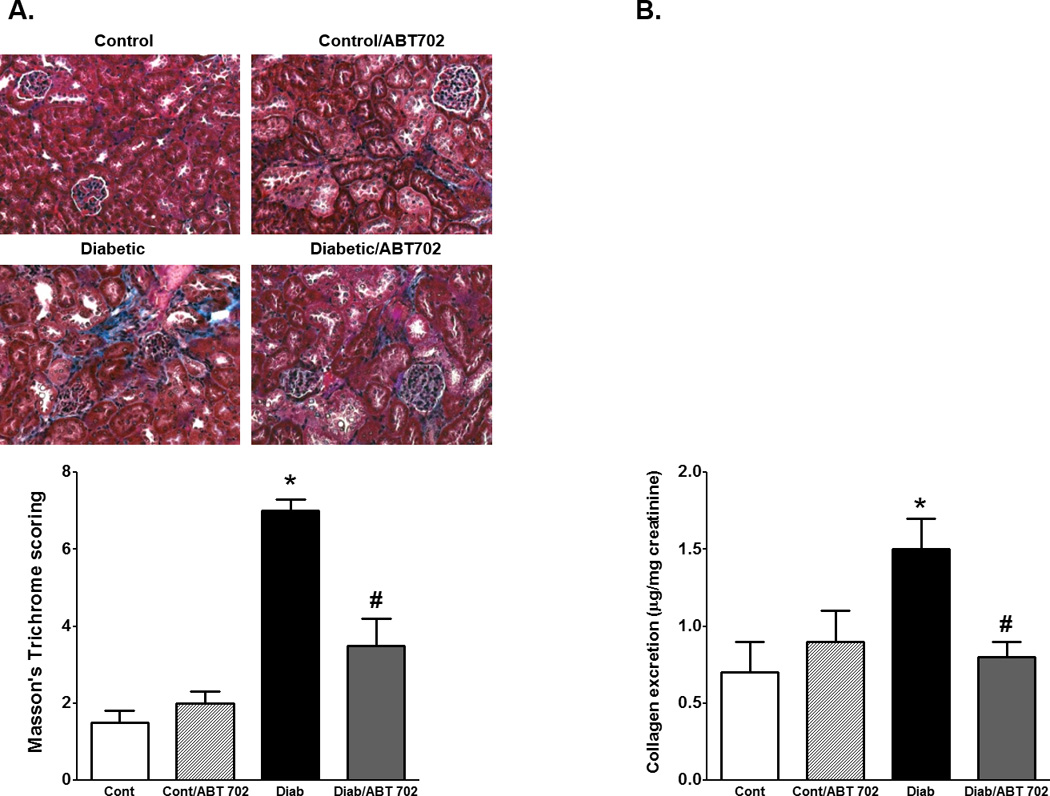
Representative images (200×) and average score for Masson’s trichrome staining (blue staining, A) and urinary collagen excretion relative to creatinine levels (B) in control and diabetic mice with or without ABT702 treatment. (*P <0.05 vs. control mice and #P <0.05 vs. diabetic mice; n= 7–8/group).
Because oxidative stress and inflammation play a crucial role in the development and progression of renal injury [17], we expanded our study to investigate whether ABT702 could protect the kidney from diabetic complication via lowering the oxidative stress. We initially determined NADPH oxidase activity where NADPH oxidase is considered the main source for superoxide production in the vasculature [33]. As shown in Figure 5A, renal NADPH oxidase activity was significantly elevated in diabetic vs. control mice and ABT702 reduced this elevation (P<0.05). Urinary TBARS levels were also assessed as an index of oxidative stress in diabetic mice. The vehicle-treated diabetic mice displayed an increase in urinary TBARS excretion levels compared with control non diabetic which was significantly lowered by ABT702 treatment (Figure 5B, P<0.05). It is well known that elevation in superoxide production could scavenge nitric oxide (NO) and reduce NO availability resulting in exacerbation in vascular injury [34]. Renal eNOS expression, the main enzyme for endothelial NO production, was significantly elevated in diabetic mice when compared to control untreated or ABT702-treated mice. ABT702 seems to potentiate the increase in eNOS expression in diabetic mice although this change was not significant when compared to diabetic untreated mice (Figure 6A). Similarly, urinary nitrate/nitrite, an indicative of functional NO [35], was elevated in diabetic mice with or without ABT702; however, this elevation was only significant in diabetic mice treated with ABT702 compared to control (P<0.05, figure 6B).
Figure 5.
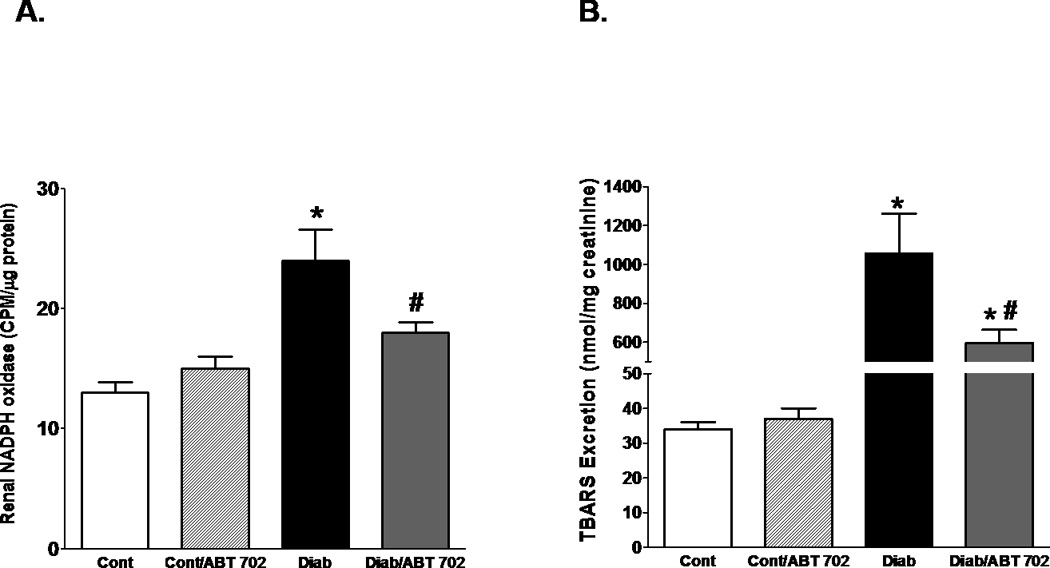
Renal NADPH oxidase activity (A) and urinary TBARS excretion (B) in streptozotocin-induced diabetic mice. Renal NADPH oxidase activity and urinary TBARS excretion were significantly elevated in diabetic than control mice (*P <0.05 vs. control mice) but were reduced with ABT702 treatment (#P <0.05 vs. diabetic mice). However, urinary TBARS excretion remained significantly higher in diabetic mice treated with ABT702 compared with control (*P <0.05 vs. control mice, 7–8/group).
Figure 6.
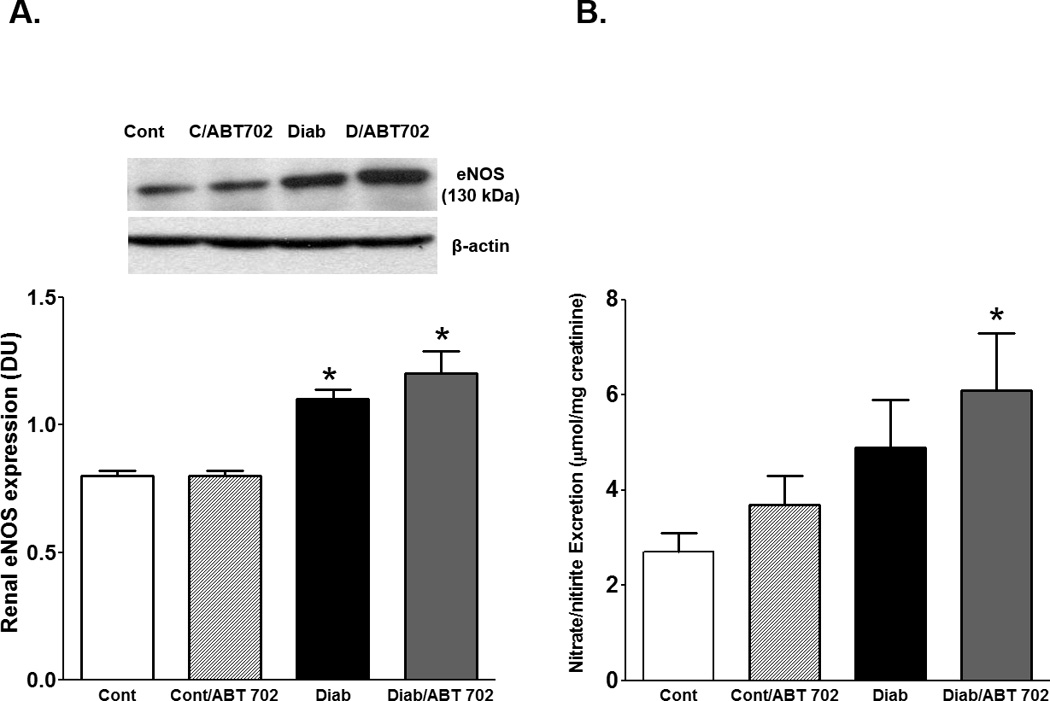
Renal eNOS expression relative to β-actin (A) and urinary nitrate/nitrite excretion (B) in streptozotocin-induced diabetic mice. Renal eNOS expression level significantly increased in diabetic with or without ABT702 treatment compared with control mice (*P <0.05 vs. control mice, n=6/group). Only ABT702 treatment of diabetic mice significantly increased urinary nitrate/nitrite compared with control mice (#P <0.05 vs. diabetic mice, n=6–7/group).
Because studies suggest a role of infiltrated macrophage in the progression of renal injury, we used immunohistochemistry to assess number of F/480+ cells as a marker of active macrophage infiltration in diabetic mice [21]. As shown in Figure 7A, macrophage infiltration was significantly elevated in diabetic mice compared to control and this change was reduced with ABT702 treatment (P<0.05). However, the number of infiltrated macrophage remained significantly higher in ABT702-treated diabetic mice than control untreated or ABT702 treated mice. We further determined the effect of ABT702 on NFKB activation as a major inflammatory signaling pathway in the pathogenesis and progression of diabetic renal injury [12]. Renal P-65 NFB was significantly elevated in diabetic vs. control mice and this effect was prevented with ABT702 treatment (Figure 7B). Because MCP-1 is a downstream cytokine for NFB activation, we also assessed urinary MCP-1 excretion as another inflammatory marker in diabetic mice after 8 weeks of ABT702 treatment. As shown in Figure 7C, urinary MCP-1 excretion was significantly higher in diabetic mice compared to non-diabetic controls and ABT702 treatment significantly reduced these changes; However, MCP-1 excretion levels in ABT702 treated diabetic mice remained significantly higher than either control untreated or ABT702 treated mice (P<0.05).
Figure 7.
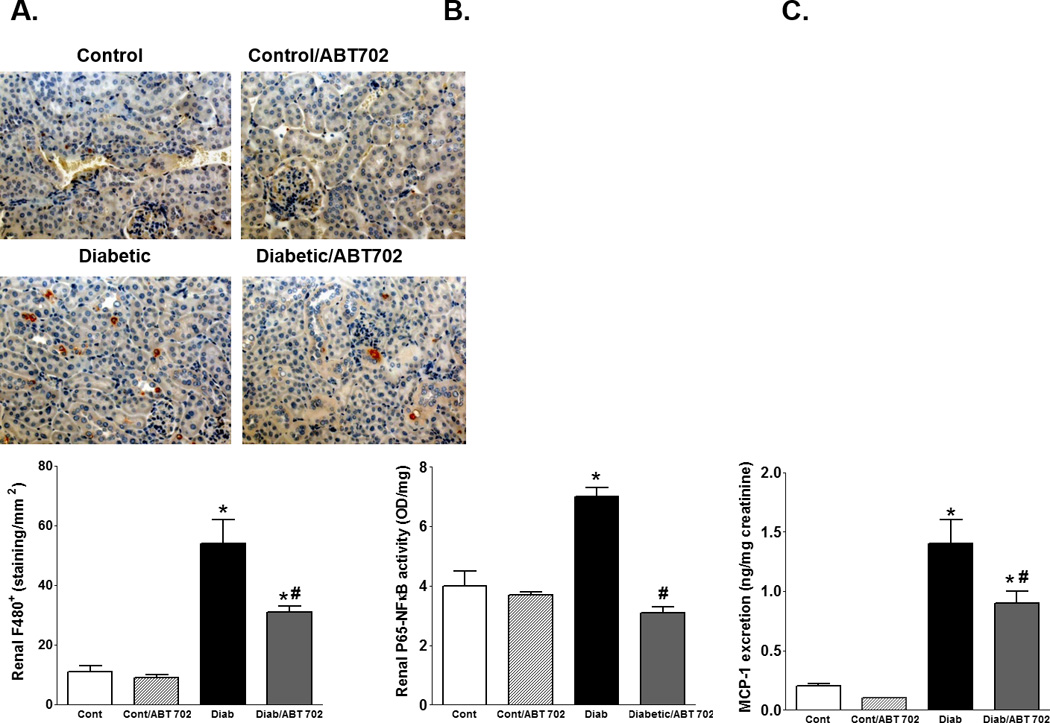
A. Representative images for F4/80 expression, an indicator for activated macrophage, at 200X and number of F4/80+ cells per mm2 (n=4) in the kidney of streptozotocin-induced diabetic mice with or without ABT702 treatment. Renal P-65NFB (B) and urinary MCP-1 excretion (C) in control, diabetic and ABT702 treated control and diabetic mice (n=7–8/group). Number of renal F4/80+ cells, renal P-65NFKB and urinary MCP-1 excretion level were significantly elevated in diabetic compared with control mice (*P <0.05 vs. control mice). ABT702 treatment significantly decreased these parameters in diabetic mice (#P <0.05 vs. diabetic mice, n=7–8/group).
Effects of high glucose on FITC-dextran permeability and MAPK phosphorylation in HGECs
We further assessed whether ABT702 could improve glomerular permeability, barrier function and inflammation in cultured HGECs under normal and high glucose conditions. Figure (8) showed that incubation of HGECs with high glucose condition (30 mM) for five days significantly increased FITC-dextran permeability and MAPK phosphorylation and decreased glomerular occludin expression compared with normal glucose condition (5 mM) (P<0.05). ABT702 prevented high glucose-induced elevation in FITC-dextran permeability and MAPK phosphorylation and restored the decrease in occludin expression in HGECs. To examine the effect of osmolality on glomerular permeability, HGECs were also cultured in 30 mM mannitol for five days with or without ABT-702 treatment. Neither mannitol alone or with ABT702 had a significant effect on glomerular permeability in HGECs (Figure 8 A).
Figure 8.
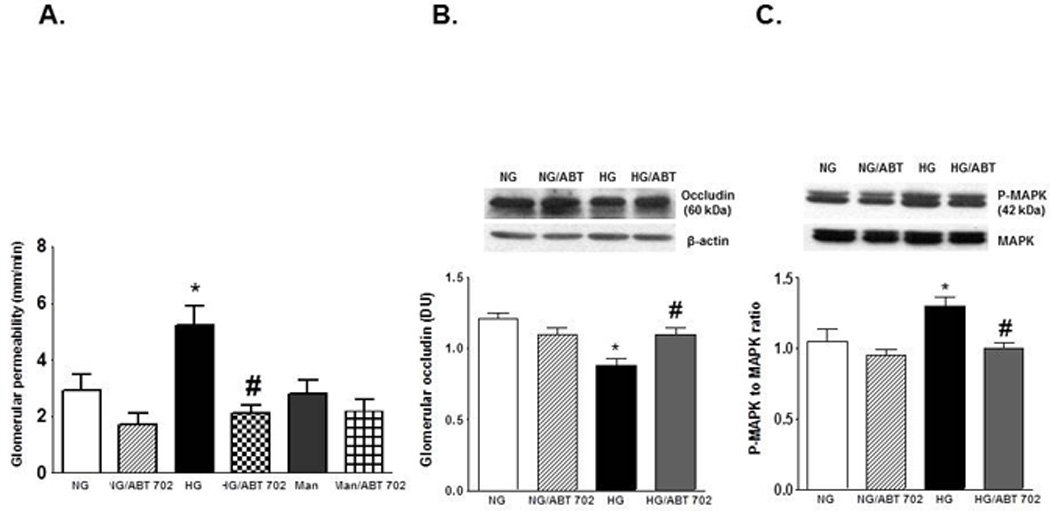
FITC-dextran permeability (A), Occludin expression relative to β-actin (B) and phospho-MAPK relative to MAPK (C) in HGECs incubated in normal (5mM) and high (30 mM) glucose condition for five days with or without ABT-702 treatment (50 µM). High glucose significantly increased FITC-dextran permeability, phospho-MAPK/MAPK ratio and decreased occludin expression compared with normal glucose condition (*P <0.05 vs. normal glucose condition) and ABT-702 treatment decreased FITC-dextran permeability and phospho-MAPK/MAPK ratio and restoring the decrease in occluding expression in high glucose treated HGECs (#P <0.05 vs. high glucose condition, n=3).
Discussion
Targeting early features of renal injury in diabetes could prevent the progression of ESRD. Adenosine signaling activation is known to provide anti-inflammatory effects [36]; however, the physiological effects of exogenous adenosine intake are limited by its hemodynamic effects (hypotension and bradycardia), rapid reuptake and subsequent intracellular metabolism by adenosine kinase [24–26,30]. Accordingly, the use of adenosine kinase inhibitors is considered a new avenue to amplify the endogenous physiological effects of adenosine while minimizing unwanted hemodynamic side effects of exogenous adenosine. The current study demonstrates that 8 week diabetic mice showed a marked albuminuria and nephrinuria together with early histological changes of diabetic renal injury such as collagen deposition and increased collagen excretion. Furthermore, diabetic renal injury was associated with activation of NADPH oxidase and oxidative stress in the kidney together with a marked degree of renal inflammation as manifested by increased macrophage infiltration, activation of NFB and increased MCP-1 excretion. Administration of the adenosine kinase inhibitor, ABT702, reduced blood glucose levels and ameliorated streptozotocin-induced albuminuria and collagen deposition together with decreased oxidative stress and renal inflammation in mice. In vitro, ABT702 also reduced the elevation in glomerular permeability and MAPK phosphorylation and restored the decrease in glomerular occludin expression in HGECs treated with high glucose condition for 5 days.
Adenosine A2A agonists have been shown to significantly reduce albuminuria in diabetic mice as well as plasma creatinine [24]. Proteinuria was greater in diabetic A2A knock-out (KO) mice than diabetic wild type (WT) mice [24]. Consistent with these findings, our laboratory initially found that proteinuria was significantly higher in diabetic A2A KO mice than diabetic WT mice (proteinuria was 248± 8 mg/day after 10 weeks of streptoztocin-induced diabetes in A2A KO mice vs. 185± 6 mg/day in diabetic WT mice, P< 0.05). The elevation in proteinuria in diabetic A2A KO mice was also associated with a 50% increase in renal ICAM-1 expression when compared to diabetic WT mice suggesting that A2A receptor stimulation contributes to kidney protection in diabetes via reduction in renal inflammation similar to what has been shown in kidney ischemia-reperfusion injury [37]. The elevation in proteinuria in diabetic A2A KO mice did not seems to be attributed to any significant increase in blood glucose and blood pressure (data are not shown). Adenosine is known to be released in response to inflammation to provide a compensatory anti-inflammatory effect via up-regulation of A2A receptor [25,36,38]. Consistent with these findings, our data showed that diabetes down-regulated adenosine kinase, suggesting increased adenosine availability, and up-regulated renal A2A receptor expression without changing A1 receptor expression compared to control. However, the current study suggests that inhibition of adenosine kinase provides renal protection against diabetic insult independent on A2A receptor up-regulation as ABT702 lowered A2A receptor expression and partially restored the decrease in adenosine kinase expression in diabetic mice kidney. Our finding is consistent with the previous finding of Elsherbiny et al. in which adenosine kinase expression decreased and A2A receptor expression increased in diabetic retina and ABT702 treatment reversed these changes [25]. Since previous studies reported increases in glomerular and urinary adenosine levels in diabetic rat [39], our data suggest that during the inflammatory condition of diabetes, adenosine kinase is down-regulated to increase extracellular adenosine levels whereas A2A receptor is up-regulated to mediate adenosine anti-inflammatory properties. Since adenosine kinase inhibition provides renal protection and reduce renal inflammation, these effects would offset the necessity to up-regulate renal A2A receptor expression to combat diabetes-induced renal inflammation.
Albuminuria is considered the earliest clinical indicator of diabetic renal injury and marked elevation of albuminuria correlates with the progression of renal disease [40,41]. In our study, adenosine kinase inhibition with ABT702 significantly reduced the increase in albuminuria in diabetic mice, which we initially thought, is due to reduction in glomerular injury and podocyte loss. Since nephrin and podocalyxin are crucial complex proteins in the assembly and reinforcement of the slit diaphragm in the kidney [42,43], assessment of nephrin and podocalyxin excretion levels could reflect glomerular injury and podocyte loss. We and other have previously demonstrated a reduction in nephrin expression in diabetic human and streptozotocin-induced diabetic rat and mice models [12,21,41]. A2A receptor agonist has been shown to restore podocin and nephrin expression in diabetes [24]; however, in our study adenosine kinase inhibition decreased A2A receptor expression in both control as well as in diabetic mice and did not significantly reduce nephrin and podocalyxin excretion in diabetic mice. Accordingly it is less likely that the reno-protective effect of ABT702 is attributed to the improvement in slit diaphragm protein in the kidney.
The extracellular adenosine levels are mainly regulated by intracellular adenosine kinase activity since adenosine degradation to inosine, by adenosine deaminase, has been previously shown to play a minor role in the regulation of adenosinergic function [44]. The mechanism (s) by which adenosine kinase inhibition reduce (s) renal injury in streptozotocin-induced diabetic renal injury is/are not known; however, we could speculate that increased adenosine levels via adenosine kinase inhibition may directly improve glucose homeostasis since previous studies suggest that adenosine agonists improve glucose homeostasis in diabetic animal models [45,46]. Although we observed a significant reduction in blood glucose levels in ABT702-treated diabetic vs. untreated diabetic mice, the reduction in blood glucose levels is less likely to be the sole reno-protective mechanism of ABT702 in diabetic mice as blood glucose remained significantly higher than control.
Previous studies suggest that TGF-β contributes to the pathogenesis of diabetic renal injury [47]. TGF-β stimulates matrix production and prevents matrix degradation, which leads to increase collagen deposition and glomerulosclerosis [48]. In the current study ABT702 significantly reduced renal collagen deposition and collagen excretion suggesting that adenosine kinase inhibition may halt the progression of renal injury by slowing the development of glomerulosclerosis in diabetic mice.
Studies support a potential role of macrophages in diabetic renal injury as increased kidney macrophages infiltration correlates with the severity of renal injury in diabetes [49,50]. Increasing evidence also indicates a role for various inflammatory molecules, including chemokines, adhesion molecules, and pro-inflammatory cytokines, in diabetic complications [50]. For example, MCP-1 is produced by mesangial and tubular epithelial cells to direct monocytes to the site of inflammation mediating renal interstitial inflammation, tubular atrophy and interstitial fibrosis [51,52]. Increased MCP-1 excretion level, which is known to reflect kidney MCP-1 production, correlates with the elevation in proteinuria in diabetes [52]. Activation of NFκB inflammatory signal is also known to exacerbate renal injury in diabetic animal models as phosphorylation of NFκB and its nuclear translocation activates down-stream inflammatory cytokines such as MCP-1 perturbing inflammatory cascade [12]. Elsherbiny et al. recently showed that ABT702 significantly reduced retinal inflammation and inhibited the elevation in ICAM-1 and TNF-α expression levels in the retina of streptozotocin-induced diabetic mice [25]. ABT702 also inhibited TNF-α release in vitro in the retinal microglia treated with Amadori-glycated albumin [25]. Consistent with these findings, the current study demonstrated that ABT702 reduced renal inflammation as evident by the reduction in renal macrophage infiltration, NFκB phosphorylation and MCP-1 excretion. Since hyperglycemia is known to increase inflammation via an MAPK/ERK-dependent signaling pathway [13,53], inhibition of adenosine kinase could improve glomerular permeability and barrier function in cultured HGECs under normal and high glucose conditions via decreased inflammation. ABT702 reduced high glucose-induced elevation in FITC-dextran permeability and MAPK phosphorylation and improved glomerular occludin expression in vitro in HGECs. Thus, the ability of adenosine kinase inhibition to reduce macrophage infiltration and cytokines levels is likely a key element in the renal protection against diabetic insult. Collectively, these data suggest that ABT702-induced inhibition of adenosine kinase provides anti-inflammatory properties which could be a potential mechanism for alleviating diabetic-induced renal injury.
Reactive oxygen species (ROS) are considered a causal link between elevated glucose levels and the development of diabetic renal injury [54,55]. ABT702 inhibited oxidative and nitrosative stress in diabetic retina as it decreased superoxide and nitrotyrosine levels [25]. Treating diabetic mice with ABT702 also significantly reduced retinal neuronal cell death [25]. Accordingly, ABT702’s anti-inflammatory effects could be attributed to its antioxidant properties. NAD(P)H oxidase is the main source of superoxide production in the vasculature and hyperglycemia is known to activate NAD(P)H oxidase generating reactive oxygen species which in turn increase lipid peroxidation and membrane damage [54,55]. Consistent with these data, ABT702 treatment reduced the elevation in urinary TBARS excretion, a marker of lipid peroxidation, and renal NAD(P)H oxidase activity in diabetic mice highlighting the potential antioxidant ability of adenosine kinase inhibition as a reno-protective mechanism in diabetes.
Increased superoxide production not only reduces NO availability but also scavenges NO, generating peroxynitrite which is known to induce deleterious effects on the vascular function and aggravate renal injury during diabetes [56,57]. In the current study, ABT702 increased eNOS expression and nitrate/nitrite excretion in diabetic mice suggesting that inhibition of adenosine kinase could also improve vascular function via the reduction in NADPH-derived superoxide production as well as the improvement in NO availability alleviating the progression of renal injury in diabetic mice.
In summary, our study provides experimental evidence that chronic administration of adenosine kinase inhibitors attenuates diabetes-induced renal injury and kidney histopathological changes by amplifying the physiological effects of extracellular adenosine. We believe that the reno-protective effect of adenosine kinase inhibition is mediated primarily by reducing blood glucose and abrogating the oxidative stress and inflammatory response associated with diabetes. These observations are of clinical relevance as adenosine kinase could be a promising target to halt the progression of diabetic renal injury given the fact that many patients with diabetes mellitus develop progressive renal injury despite current available therapeutic regimens such as ACE inhibitors and angiotensin receptor blockers.
Supplementary Material
Acknowledgement
This work was supported by grants from the American Heart Association Scientist Development Grant (SDG) to A. Elmarakby and 1R01EY023315-01 award to M. Al-Shabrawey.
References
- 1.Schneider H, Shaw J, Zimmet P. Guidelines for the detection of diabetes mellitus--diagnostic criteria and rationale for screening. Clin Biochem Rev. 2003;24:77–80. [PMC free article] [PubMed] [Google Scholar]
- 2.Lam DW, LeRoith D. The worldwide diabetes epidemic. Curr Opin Endocrinol Diabetes Obes. 2012;19:93–96. doi: 10.1097/MED.0b013e328350583a. [DOI] [PubMed] [Google Scholar]
- 3.Conserva F, Pontrelli P, Accetturo M, Gesualdo L. The pathogenesis of diabetic nephropathy: Focus on micrornas and proteomics. J Nephrol. 2013;26:811–820. doi: 10.5301/jn.5000262. [DOI] [PubMed] [Google Scholar]
- 4.Demirel F, Tepe D, Kara O, Esen I. Microvascular complications in adolescents with type 1 diabetes mellitus. J Clin Res Pediatr Endocrinol. 2013;5:145–149. doi: 10.4274/Jcrpe.994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinke JM, Mauer M International Diabetic Nephropathy Study G. Lessons learned from studies of the natural history of diabetic nephropathy in young type 1 diabetic patients. Pediatr Endocrinol Rev. 2008;5(Suppl 4):958–963. [PubMed] [Google Scholar]
- 6.Fried LF, Emanuele N, Zhang JH, Brophy M, Conner TA, Duckworth W, Leehey DJ, McCullough PA, O'Connor T, Palevsky PM, Reilly RF, Seliger SL, Warren SR, Watnick S, Peduzzi P, Guarino P Investigators VN-D. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med. 2013;369:1892–1903. doi: 10.1056/NEJMoa1303154. [DOI] [PubMed] [Google Scholar]
- 7.Fineberg D, Jandeleit-Dahm KA, Cooper ME. Diabetic nephropathy: Diagnosis and treatment. Nat Rev Endocrinol. 2013;9:713–723. doi: 10.1038/nrendo.2013.184. [DOI] [PubMed] [Google Scholar]
- 8.McKay GJ, Savage DA, Patterson CC, Lewis G, McKnight AJ, Maxwell AP, Warren UKGSG. Association analysis of dyslipidemia-related genes in diabetic nephropathy. PloS one. 2013;8:e58472. doi: 10.1371/journal.pone.0058472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salmon AH, Ferguson JK, Burford JL, Gevorgyan H, Nakano D, Harper SJ, Bates DO, Peti-Peterdi J. Loss of the endothelial glycocalyx links albuminuria and vascular dysfunction. J Am Soc Nephrol. 2012;23:1339–1350. doi: 10.1681/ASN.2012010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salmon AH, Satchell SC. Endothelial glycocalyx dysfunction in disease: Albuminuria and increased microvascular permeability. J Pathol. 2012;226:562–574. doi: 10.1002/path.3964. [DOI] [PubMed] [Google Scholar]
- 11.Masuo K, Rakugi H, Ogihara T, Esler MD, Lambert GW. Cardiovascular and renal complications of type 2 diabetes in obesity: Role of sympathetic nerve activity and insulin resistance. Curr Diabetes Rev. 2010;6:58–67. doi: 10.2174/157339910790909396. [DOI] [PubMed] [Google Scholar]
- 12.Elmarakby AA, Faulkner J, Al-Shabrawey M, Wang MH, Maddipati KR, Imig JD. Deletion of soluble epoxide hydrolase gene improves renal endothelial function and reduces renal inflammation and injury in streptozotocin-induced type 1 diabetes. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1307–R1317. doi: 10.1152/ajpregu.00759.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elmarakby AA, Ibrahim AS, Faulkner J, Mozaffari MS, Liou GI, Abdelsayed R. Tyrosine kinase inhibitor, genistein, reduces renal inflammation and injury in streptozotocin-induced diabetic mice. Vascul Pharmacol. 2011;55:149–156. doi: 10.1016/j.vph.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 14.Carmines PK. Mechanisms of altered renal microvascular function in type 1 diabetes: Potential contribution to end organ damage. Curr Vasc Pharmacol. 2013 doi: 10.2174/15701611113116660156. [DOI] [PubMed] [Google Scholar]
- 15.Shah A, Xia L, Goldberg H, Lee KW, Quaggin SE, Fantus IG. Thioredoxin-interacting protein mediates high glucose-induced reactive oxygen species generation by mitochondria and the nadph oxidase, nox4, in mesangial cells. J Biol Chem. 2013;288:6835–6848. doi: 10.1074/jbc.M112.419101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu GC, Fang F, Zhou J, Koulajian K, Yang S, Lam L, Reich HN, John R, Herzenberg AM, Giacca A, Oudit GY, Scholey JW. Deletion of p47phox attenuates the progression of diabetic nephropathy and reduces the severity of diabetes in the akita mouse. Diabetologia. 2012;55:2522–2532. doi: 10.1007/s00125-012-2586-1. [DOI] [PubMed] [Google Scholar]
- 17.Elmarakby AA, Sullivan JC. Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc Ther. 2012;30:49–59. doi: 10.1111/j.1755-5922.2010.00218.x. [DOI] [PubMed] [Google Scholar]
- 18.Onozato ML, Tojo A, Goto A, Fujita T, Wilcox CS. Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: Effects of acei and arb. Kidney Int. 2002;61:186–194. doi: 10.1046/j.1523-1755.2002.00123.x. [DOI] [PubMed] [Google Scholar]
- 19.Pacher P, Obrosova IG, Mabley JG, Szabo C. Role of nitrosative stress and peroxynitrite in the pathogenesis of diabetic complications. Emerging new therapeutical strategies. Curr Med Chem. 2005;12:267–275. doi: 10.2174/0929867053363207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kodera R, Shikata K, Kataoka HU, Takatsuka T, Miyamoto S, Sasaki M, Kajitani N, Nishishita S, Sarai K, Hirota D, Sato C, Ogawa D, Makino H. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia. 2011;54:965–978. doi: 10.1007/s00125-010-2028-x. [DOI] [PubMed] [Google Scholar]
- 21.Elmarakby AA, Faulkner J, Pye C, Rouch K, Alhashim A, Maddipati KR, Baban B. Role of haem oxygenase in the renoprotective effects of soluble epoxide hydrolase inhibition in diabetic spontaneously hypertensive rats. Clin Sci (Lond) 2013;125:349–359. doi: 10.1042/CS20130003. [DOI] [PubMed] [Google Scholar]
- 22.Mohamed R, Jayakumar C, Ranganathan PV, Ganapathy V, Ramesh G. Kidney proximal tubular epithelial-specific overexpression of netrin-1 suppresses inflammation and albuminuria through suppression of cox-2-mediated pge2 production in streptozotocin-induced diabetic mice. Am J Pathol. 2012;181:1991–2002. doi: 10.1016/j.ajpath.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan Y, Wang Y, Cai L, Cai Y, Hu J, Yu C, Li J, Feng Z, Yang S, Li X, Liang G. Inhibition of high glucose-induced inflammatory response and macrophage infiltration by a novel curcumin derivative prevents renal injury in diabetic rats. Br J Pharmacol. 2012;166:1169–1182. doi: 10.1111/j.1476-5381.2012.01854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Awad AS, Huang L, Ye H, Duong ET, Bolton WK, Linden J, Okusa MD. Adenosine a2a receptor activation attenuates inflammation and injury in diabetic nephropathy. Am J Physiol Renal Physiol. 2006;290:F828–F837. doi: 10.1152/ajprenal.00310.2005. [DOI] [PubMed] [Google Scholar]
- 25.Elsherbiny NM, Ahmad S, Naime M, Elsherbini AM, Fulzele S, Al-Gayyar MM, Eissa LA, El-Shishtawy MM, Liou GI. Abt-702, an adenosine kinase inhibitor, attenuates inflammation in diabetic retinopathy. Life Sci. 2013;93:78–88. doi: 10.1016/j.lfs.2013.05.024. [DOI] [PubMed] [Google Scholar]
- 26.Elsherbiny NM, Al-Gayyar MM. Adenosine receptors: New therapeutic targets for inflammation in diabetic nephropathy. Inflamm Allergy Drug Targets. 2013;12:153–161. doi: 10.2174/1871528111312030001. [DOI] [PubMed] [Google Scholar]
- 27.Ferenbach DA, Hughes J. Adenosine a(2a) agonists as therapy for glomerulonephritis. Kidney Int. 2011;80:329–331. doi: 10.1038/ki.2011.167. [DOI] [PubMed] [Google Scholar]
- 28.Vitzthum H, Weiss B, Bachleitner W, Kramer BK, Kurtz A. Gene expression of adenosine receptors along the nephron. Kidney Int l. 2004;65:1180–1190. doi: 10.1111/j.1523-1755.2004.00490.x. [DOI] [PubMed] [Google Scholar]
- 29.Mirabet M, Herrera C, Cordero OJ, Mallol J, Lluis C, Franco R. Expression of a2b adenosine receptors in human lymphocytes: Their role in t cell activation. J Cell Sci. 1999;112(Pt 4):491–502. doi: 10.1242/jcs.112.4.491. [DOI] [PubMed] [Google Scholar]
- 30.Awad AS, Rouse M, Liu L, Vergis AL, Rosin DL, Linden J, Sedor JR, Okusa MD. Activation of adenosine 2a receptors preserves structure and function of podocytes. J Am Soc Nephrol. 2008;19:59–68. doi: 10.1681/ASN.2007030276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhatia K, Elmarakby AA, El-Remessy AB, Sullivan JC. Oxidative stress contributes to sex differences in angiotensin ii-mediated hypertension in spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2012;302:R274–R282. doi: 10.1152/ajpregu.00546.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murakami T, Felinski EA, Antonetti DA. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J Biol Chem. 2009;284:21036–21046. doi: 10.1074/jbc.M109.016766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bengtsson SH, Gulluyan LM, Dusting GJ, Drummond GR. Novel isoforms of nadph oxidase in vascular physiology and pathophysiology. Clin Exp Pharmacol Physiol. 2003;30:849–854. doi: 10.1046/j.1440-1681.2003.03929.x. [DOI] [PubMed] [Google Scholar]
- 34.Satoh M, Fujimoto S, Haruna Y, Arakawa S, Horike H, Komai N, Sasaki T, Tsujioka K, Makino H, Kashihara N. Nad(p)h oxidase and uncoupled nitric oxide synthase are major sources of glomerular superoxide in rats with experimental diabetic nephropathy. Am J Physiol Renal Physiol. 2005;288:F1144–F1152. doi: 10.1152/ajprenal.00221.2004. [DOI] [PubMed] [Google Scholar]
- 35.Worrall NK, Boasquevisque CH, Botney MD, Misko TP, Sullivan PM, Ritter JH, Ferguson TB, Jr, Patterson GA. Inhibition of inducible nitric oxide synthase ameliorates functional and histological changes of acute lung allograft rejection. Transplantation. 1997;63:1095–1101. doi: 10.1097/00007890-199704270-00008. [DOI] [PubMed] [Google Scholar]
- 36.Milne GR, Palmer TM. Anti-inflammatory and immunosuppressive effects of the a2a adenosine receptor. ScientificWorldJournal. 2011;11:320–339. doi: 10.1100/tsw.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okusa MD, Linden J, Huang L, Rosin DL, Smith DF, Sullivan G. Enhanced protection from renal ischemia-reperfusion [correction of ischemia:Reperfusion] injury with a(2a)-adenosine receptor activation and pde 4 inhibition. Kidney Int. 2001;59:2114–2125. doi: 10.1046/j.1523-1755.2001.00726.x. [DOI] [PubMed] [Google Scholar]
- 38.Sullivan GW. Adenosine a2a receptor agonists as anti-inflammatory agents. Curr Opin Investig Drugs. 2003;4:1313–1319. [PubMed] [Google Scholar]
- 39.Cardenas A, Toledo C, Oyarzun C, Sepulveda A, Quezada C, Guillen-Gomez E, Diaz-Encarnacion MM, Pastor-Anglada M, San Martin R. Adenosine a(2b) receptor-mediated vegf induction promotes diabetic glomerulopathy. Lab Invest. 2013;93:135–144. doi: 10.1038/labinvest.2012.143. [DOI] [PubMed] [Google Scholar]
- 40.Chodavarapu H, Grobe N, Somineni HK, Salem ES, Madhu M, Elased KM. Rosiglitazone treatment of type 2 diabetic db/db mice attenuates urinary albumin and angiotensin converting enzyme 2 excretion. PloS one. 2013;8:e62833. doi: 10.1371/journal.pone.0062833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jim B, Santos J, Spath F, Cijiang He J. Biomarkers of diabetic nephropathy, the present and the future. Curr Diabetes Rev. 2012;8:317–328. doi: 10.2174/157339912802083478. [DOI] [PubMed] [Google Scholar]
- 42.Pierchala BA, Munoz MR, Tsui CC. Proteomic analysis of the slit diaphragm complex: Clic5 is a protein critical for podocyte morphology and function. Kidney Int. 2010;78:868–882. doi: 10.1038/ki.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koop K, Eikmans M, Baelde HJ, Kawachi H, De Heer E, Paul LC, Bruijn JA. Expression of podocyte-associated molecules in acquired human kidney diseases. J Am Soc Nephrol. 2003;14:2063–2071. doi: 10.1097/01.asn.0000078803.53165.c9. [DOI] [PubMed] [Google Scholar]
- 44.Pak MA, Haas HL, Decking UK, Schrader J. Inhibition of adenosine kinase increases endogenous adenosine and depresses neuronal activity in hippocampal slices. Neuropharmacology. 1994;33:1049–1053. doi: 10.1016/0028-3908(94)90142-2. [DOI] [PubMed] [Google Scholar]
- 45.Johnston-Cox H, Koupenova M, Yang D, Corkey B, Gokce N, Farb MG, LeBrasseur N, Ravid K. The a2b adenosine receptor modulates glucose homeostasis and obesity. PloS one. 2012;7:e40584. doi: 10.1371/journal.pone.0040584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rusing D, Muller CE, Verspohl EJ. The impact of adenosine and a(2b) receptors on glucose homoeostasis. J Pharm Pharmacol. 2006;58:1639–1645. doi: 10.1211/jpp.58.12.0011. [DOI] [PubMed] [Google Scholar]
- 47.Lu A, Miao M, Schoeb TR, Agarwal A, Murphy-Ullrich JE. Blockade of tsp1-dependent tgf-beta activity reduces renal injury and proteinuria in a murine model of diabetic nephropathy. Am J Pathol. 2011;178:2573–2586. doi: 10.1016/j.ajpath.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kopp JB, Factor VM, Mozes M, Nagy P, Sanderson N, Bottinger EP, Klotman PE, Thorgeirsson SS. Transgenic mice with increased plasma levels of tgf-beta 1 develop progressive renal disease. Lab Invest. 1996;74:991–1003. [PubMed] [Google Scholar]
- 49.Soetikno V, Sari FR, Veeraveedu PT, Thandavarayan RA, Harima M, Sukumaran V, Lakshmanan AP, Suzuki K, Kawachi H, Watanabe K. Curcumin ameliorates macrophage infiltration by inhibiting nf-kappab activation and proinflammatory cytokines in streptozotocin induced-diabetic nephropathy. Nutr Metab (Lond) 2011;8:35. doi: 10.1186/1743-7075-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Okada S, Shikata K, Matsuda M, Ogawa D, Usui H, Kido Y, Nagase R, Wada J, Shikata Y, Makino H. Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes. 2003;52:2586–2593. doi: 10.2337/diabetes.52.10.2586. [DOI] [PubMed] [Google Scholar]
- 51.Morii T, Fujita H, Narita T, Koshimura J, Shimotomai T, Fujishima H, Yoshioka N, Imai H, Kakei M, Ito S. Increased urinary excretion of monocyte chemoattractant protein-1 in proteinuric renal diseases. Ren Fail. 2003;25:439–444. doi: 10.1081/jdi-120021156. [DOI] [PubMed] [Google Scholar]
- 52.Morii T, Fujita H, Narita T, Shimotomai T, Fujishima H, Yoshioka N, Imai H, Kakei M, Ito S. Association of monocyte chemoattractant protein-1 with renal tubular damage in diabetic nephropathy. J Diabetes Complications. 2003;17:11–15. doi: 10.1016/s1056-8727(02)00176-9. [DOI] [PubMed] [Google Scholar]
- 53.Kim SW, Kim CE, Kim MH. Flavonoids inhibit high glucose-induced up-regulation of icam-1 via the p38 mapk pathway in human vein endothelial cells. Biochem Biophys Res Commun. 2011;415:602–607. doi: 10.1016/j.bbrc.2011.10.115. [DOI] [PubMed] [Google Scholar]
- 54.Lee DY, Wauquier F, Eid AA, Roman LJ, Ghosh-Choudhury G, Khazim K, Block K, Gorin Y. Nox4 nadph oxidase mediates peroxynitrite-dependent uncoupling of endothelial nitric-oxide synthase and fibronectin expression in response to angiotensin ii: Role of mitochondrial reactive oxygen species. J Biol Chem. 2013;288:28668–28686. doi: 10.1074/jbc.M113.470971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khazim K, Gorin Y, Cavaglieri RC, Abboud HE, Fanti P. The antioxidant silybin prevents high glucose-induced oxidative stress and podocyte injury in vitro and in vivo. Am J Physiol Renal Physiol. 2013;305:F691–F700. doi: 10.1152/ajprenal.00028.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cohen RA, Tong X. Vascular oxidative stress: The common link in hypertensive and diabetic vascular disease. J Cardiovasc Pharmacol. 2010;55:308–316. doi: 10.1097/fjc.0b013e3181d89670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thuraisingham RC, Nott CA, Dodd SM, Yaqoob MM. Increased nitrotyrosine staining in kidneys from patients with diabetic nephropathy. Kidney Int. 2000;57:1968–1972. doi: 10.1046/j.1523-1755.2000.00046.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.