

Abstract
The circadian clock allows organisms to accurately predict the earth’s rotation and modify their behavior as a result. Genetic analyses in a variety of organisms have defined a mechanism based largely on gene expression feedback loops. However, as we delve more deeply into the mechanisms of circadian timekeeping, we are discovering that post-translational mechanisms play a key role in defining the character of the clock. We are also discovering that these modifications are inextricably linked to cellular metabolism, including redox homeostasis. A robust circadian oscillation in the redox status of the peroxiredoxins (a major class of cellular antioxidants) was recently shown to be remarkably conserved from archaea and cyanobacteria all the way to plants and animals. Furthermore, recent findings indicate that cellular redox status is coupled not only to canonical circadian gene expression pathways but also to a noncanonical transcript-independent circadian clock. The redox rhythms observed in peroxiredoxins in the absence of canonical clock mechanisms may hint at the nature of this new and hitherto unknown aspect of circadian timekeeping.
Biological timekeeping allows a system to enact complex temporal programs of development and behavior. On a human scale, the most pervasive of rhythms is the circadian oscillation, which drives our sleep/wake cycle as well as a host of other metabolic and behavioral cycles. Recent evidence indicates that redox homeostasis, how an organism deals with excess oxidative potential or a deficiency thereof, displays an endogenous circadian rhythm.1,2 Here we discuss the interplay of the circadian clock with oxidation–reduction cycles within the cell and how this might hint at a central role for nontranscriptional control of the circadian clock.
A circadian clock was defined as one with a near 24 h rhythm in a constant environment that can be entrained by stimuli and is resistant to changes in temperature (i.e., it is temperature-compensated).3 This rhythm exists at multiple levels of organization, from the wheel running behavior of mice to the rhythmic addition and removal of phosphate groups in the cyanobacterial KaiABC protein complex.4
Canonical Circadian Clock: A Gene Expression Feedback Loop
The molecular underpinnings of the circadian clock have been the subject of intense scrutiny over the years, ranging from hypothesis-free genetic screens that identified the first circadian “clock” genes to targeted studies focused on deciphering the precise mechanisms of timekeeping. These studies have allowed the field to converge on the current paradigm of the gene expression feedback loop (GEFL). The principle of a GEFL is that a negative feedback loop in gene expression, i.e., a gene that suppresses its own transcription, will not reach a steady state if a delay is incorporated into the production of the repressive element. Instead, given the correct parameters of delay and instability in the repressive signal, the pathway will oscillate.5 In the literature, the term “transcription–translation feedback loop” is often favored; we propose to use the more accurate GEFL as it incorporates the substantial contribution of post-translational and epigenetic regulation of the circadian clockwork.
A genetic screen in Drosophila for mutants with short, long, or abrogated periodicity first highlighted the Period (Per) locus involved in GEFL timekeeping.6 Subsequent cloning of Per demonstrated that the protein could repress its own transcript, closing the prototypical circadian GEFL. Since these initial discoveries, a host of GEFLs have been discovered in plants, animals, bacteria, and fungi.7
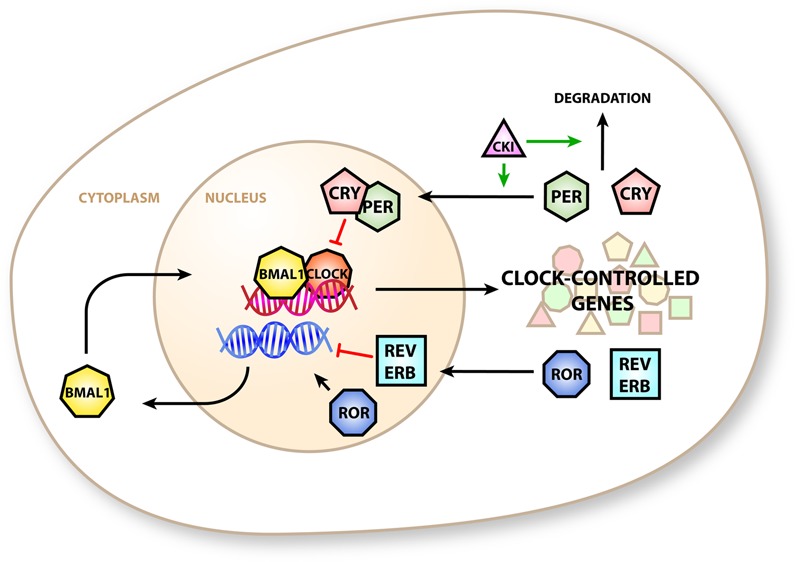
Mammals have a multiple-component GEFL, the parts of which are often termed the “canonical” clock components (Figure 1 and reviewed in ref (8)). Briefly, circadian locomotor output cycles kaput (CLOCK) and BMAL1 co-activate transcription at E-box-containing gene promoters, including the period (Per) and cryptochrome (Cry) genes. Their protein products accumulate throughout the day and translocate into the nucleus to repress their own transcription by direct interaction with the BMAL1:CLOCK complex. Throughout the night, PER and CRY are degraded, allowing the reactivation of their gene loci at dawn. A second GEFL exists in antiphase to the “peak-at-dusk” PER:CRY oscillator. Here the transcription of retinoic acid orphan receptor-α (ROR-α) and the nuclear receptor REV-ERBα is activated by the BMAL1:CLOCK complex. Their protein products activate and repress transcription of the Bmal1 locus. Gene promoters that are reactive to the core clock component rhythms translate this rhythm into the circadian gene expression program, which in turn plays a large part in generating the outputs of the circadian clock.9
Figure 1.

Canonical circadian clock in mammals. BMAL1 and CLOCK proteins co-activate E-box-containing promoters to transcribe clock-controlled genes that include the repressive PER and CRY proteins. PER and CRY form a complex that is imported into the nucleus and inhibits transcription of E-box proteins. PER activity is modulated by casein kinase 1, which promotes nuclear import and degradation. The clock-controlled genes REV-ERBα (REV-ERB) and RORα (ROR) generate additional circadian control by modulating the expression of BMAL1.
While the general principle of the GEFL is conserved, the componentry may not be.10−12 Phylogenetic analyses of clock genes suggest that circadian GEFLs evolved at least twice, once in cyanobacteria and once (or more) in eukaryotes (transcriptional components are highly dissimilar among plants, fungi, and metazoans).11 An alternative proposal is that an evolutionarily ancient oscillator exists, one which has been elaborated by the various GEFLs.1 Because this oscillator does not necessarily incorporate oscillating gene expression, it has been proposed that it could be entirely post-transcriptional in a manner akin to the KaiABC autophosphorylation–dephosphorylation paradigm already defined in cyanobacteria, and discussed below.4
Recent studies of mammalian nontranscriptional oscillators center on peroxiredoxin oxidation cycles detected across multiple genera.1,13 Whether peroxiredoxin oxidation cycles constitute a core mechanism generating oscillation or if they simply report another, cryptic, oscillation, it behooves us to first introduce this fascinating family of antioxidant proteins.
Peroxiredoxins
Peroxiredoxins make up a phylogenetically ancient family of proteins, whose primary role is associated with H2O2 detoxification.14 Peroxiredoxin enzymes work by reducing H2O2 to water. During the subsequent catalytic cycle, oxidized peroxiredoxin is normally rereduced in a fashion that ultimately consumes a reducing equivalent supplied by nicotinamide adenine dinucleotide phosphate (NADPH) (Figure 2A). Overlying the cycle of peroxiredoxin oxidation and reduction is a circadian rhythm in the redox state, which has been detected in archaea, bacteria, and eukaryotes.1,2,13,15 Strikingly, this rhythm persists (albeit perturbed) in systems in which the canonical circadian clock is either endogenously absent or pharmacologically abrogated.2,13,15
Figure 2.

Peroxiredoxin structure and function. (A) Two-Cys peroxiredoxins (PRDX) exist in dimers that can form intramolecular disulfide bridges between the peroxidatic (CysP) and resolving (CysR) cysteines. Disulfides are reduced by thioredoxin (TRX), which is in turn reduced by thioredoxin reductase (TRXR) utilizing NADPH as source of reducing power. CysR in the short-lived sulfenic form can become hyperoxidized to form sulfinic PRDX, which is recycled by sulfiredoxin (SRX). Hyperoxidized sulfonic PRDX can also form by further oxidation of PRDX, a reaction that is likely irreversible. The redox scheme of a single peroxidatic and resolving cysteine pair in each dimer is highlighted; the other pair is functionally equivalent but shown here as a disulfide and grayed out for the sake of clarity. (B) Reduced PRDX can exist as decamers or dodecamers that dynamically exchange subunits with a cellular pool of oxidized and reduced dimers. Hyperoxidation of PRDX interferes with subunit exchange and “locks” the molecules in a hyperoxidized decameric form. Hyperoxidation of decameric peroxiredoxins can induce higher-order multimerization that is associated with increased chaperone activity, although chaperone activity can also be independently modulated by post-translational modification of the protein. The cell signaling functions of peroxiredoxins are not restricted to any redox state or multimeric form.
Structurally, peroxiredoxin enzymes belong to one of two groups, the one-Cys and two-Cys peroxiredoxin enzymes, named after the number of conserved redox-active catalytic cysteine residues in their sequences.16 The mechanism of peroxiredoxin activity is conserved across the two classes, but the mechanism of recycling into the reduced state differs. In mammals, there are five two-Cys peroxiredoxins (PRDX1–5) and a single one-Cys peroxiredoxin (PRDX6).16 Peroxiredoxins are further classified by their ability to form inter- or intramolecular disulfide bonds within dimers, denoted typical (PRDX1–4) or atypical, respectively (PRDX5).16
One-Cys peroxiredoxins (such as the mammalian PRDX6) are oxidized at the catalytic cysteine residue to form a sulfenic acid cysteine (Cys-SOH). Glutathione S-transferase-π or ascorbic acid provides the reducing power to recycle the sulfenic acid residue. PRDX6 was the first peroxiredoxin to be identified as having a circadian post-translational modification.17
In two-Cys peroxiredoxins, the relatively short-lived sulfenic cysteine reacts with a second, “resolving”, cysteine to form a disulfide bridge, which is intermolecular in typical peroxiredoxins and intramolecular in atypical variants. The recycling step involves the reduction of the disulfide bridge by the thioredoxin system, utilizing NADPH as a source of reducing power. A second redox cycle can act upon the sulfenic intermediate of two-Cys peroxiredoxins. Here, the sulfenic cysteine can be further oxidized (hyperoxidized) by H2O2 to form a more stable sulfinic cysteine (Cys-SO2H), which can be recycled into the sulfenic form by the action of the ATP-dependent reductase, sulfiredoxin. The relatively slow rate of sulfinic cysteine peroxiredoxin reduction (kcat = 0.1–0.1 min–1) by sulfiredoxin allows sufficient amounts to accumulate to allow detection by Western blotting.18−20 Irreversible further oxidation of sulfinic cysteine can occur by reaction with H2O2 to form sulfonic cysteine (Cys-SO3H). This reaction is likely irreversible and effectively removes peroxiredoxin from the redox cycle.21In vivo, sulfonic peroxiredoxins (i.e., peroxiredoxins containing a sulfonic catalytic cysteine) have been detected under acute oxidative stress in HeLa cells (75–150 μM tert-butyl hydroperoxide),22 in aged rat liver23 and yeast exposed to H2O2.21 During normative cell function the sulfonic and sulfinic peroxiredoxins are present as minor species.22
The quaternary structure of peroxiredoxins is dynamic within the cell, and each conformational state may have associated functionality. Oxidized peroxiredoxin tends to favor a dimeric state, whereas reduced, hyperoxidized, and disulfide-linked peroxiredoxins can also adopt a decameric or dodecameric structure depending on context. Human red blood cell (RBC) PRDX2, for example, adopts a decameric ring structure composed of repeating dimer units, whereas bovine PRDX3 adopts an interlocking dodecameric structure; the significance of this difference is unclear.24 Reduced peroxiredoxin can also exist as a dimer, but the decameric structure is thought be strongly favored under physiological conditions16 (Figure 2B).
The (do)decamer is rapidly turned over, releasing oxidized dimers that are rapidly reduced before re-entering the oligomeric state. Hyperoxidation into the sulfinic form may freeze the dynamic cycle in the oligomeric state, promoting the formation of even larger peroxiredoxin complexes.25
Higher-order complexes, including a dodecahedral structure (12 decamers in a three-dimensional arrangement), have been detected in vitro and proposed in RBCs.26,27 Human and yeast PRDX2 have been shown to have chaperone activity when they are in the higher-order complexes.25,28 Interestingly, the human PRDX1 oligomeric state is modulated by phosphorylation of Thr90. A phospho-mimetic T90D variant of PRDX1 has increased propensity to form a higher-order complex with a concomitant rise in chaperone function and drop in peroxidase function.29 However, mutants that variously mimic sulfinic hyperoxidation, oxidation, or reduction of the catalytic cysteine and prevent decamer formation have indicated that the chaperone and peroxidase functions of two-Cys peroxiredoxin in plants do not exclusively depend upon the dimeric or decameric form or on the redox state.30 The joint antioxidant and chaperone function of peroxiredoxins may represent a concerted response to oxidative stress.
Circadian Rhythms of Peroxiredoxins
It is the hyperoxidized (sulfinic and sulfonic) forms of peroxiredoxin that have been demonstrated to oscillate by Western blotting with an antibody specific to hyperoxidized peroxiredoxin species.1,2,13 Because of the remarkable conservation of the peroxiredoxin active site, the same antibody is able to detect rhythms in mice, flies, fungi, plants, archaea, and bacteria.1,2,13
This suggests that the redox rhythm reported by peroxiredoxin hyperoxidation is very ancient, more so than sequence conservation of GEFL components would suggest the canonical clock is (Figure 3). Furthermore, the rhythm in peroxiredoxin oxidation state persists in mice, bacteria, and fungi where their GEFL components have been mutated, although the oxidation rhythm is clearly altered in comparison with wild-type controls.1,2
Figure 3.
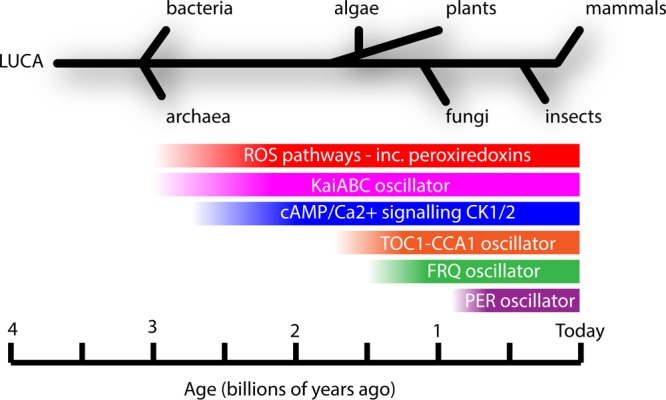
Evolution of circadian clock components. Mechanisms for mitigating oxidative stress (ROS pathways) can be dated to the great oxidation event approximately 3 billion years ago. Whereas the KaiABC oscillator and cAMP signaling pathways are of similarly ancient origin, the circadian GEFL pathways (plant TOC1-CCA1 oscillator, fungal FRQ oscillator, and metazoan PER oscillator) evolved separately and more recently. Oscillations in ROS pathway components such as peroxiredoxins may indicate that an ancient clock underlies contemporary eukaryotic canonical circadian pathways. Adapted from ref (1).
Oxidized peroxiredoxins accumulate in isolated mammalian RBCs with circadian rhythmicity, the most recent demonstration of eukaryotic circadian rhythms in the native absence of transcription,2,13 following from older observations, made with isolated platelets31 and enucleated Acetabularia,32 for example.
The fluctuation in peroxiredoxin oxidation in RBCs has been independently observed by at least two research groups utilizing different organisms (humans and mice) and different lysis conditions. A criticism of the older study by O’Neill and Reddy was the decision to lyse RBCs under nonreducing conditions without blocking reduced cysteines, using alkylating agents such as N-ethylmaleimide (NEM). This methodology produced a variety of multimeric species on a nonreducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel presumably because of additional disulfide formation occurring postlysis, because when the same samples were run under reducing conditions only a rhythmically hyperoxidized peroxiredoxin monomer was detected (Supporting Information of ref (2)). The authors chose to concentrate on this readily observable rhythm in hyperoxidized dimeric peroxiredoxin (the antibody not distinguishing between sulfinic and sulfonic forms), because it was the very existence of a rhythm in isolated human RBCs, rather than the mechanism underlying the post-translational peroxiredoxin modification, that was the focus of their investigation. The more biochemically rigorous recent study by Cho and colleagues, using NEM-treated lysates separated under nonreducing conditions, did not detect significant dimers in mouse RBCs. This means that the hyperoxidized PRX band observed previously at the molecular weight of a PRX dimer likely comprises one monomer with a hyperoxidized peroxidatic cysteine, with its reducing cysteine partaking in an intermolecular disulfide with the peroxidatic cysteine of the opposing monomer. On the basis of findings of Cho et al., it is plausible that this single intermolecular disulfide forms rapidly postlysis if NEM or sufficient reductant is not present. Despite the possibility that dimers detected in the earlier work may not have been significantly present prior to lysis, however, the rhythm in peroxiredoxin hyperoxidation is preserved, meaning that subsequent disulfide bond formation between opposing cysteines in noncovalently linked peroxiredoxin dimers in no way precludes the possibility that the other peroxidatic cysteine in the dimer was sulfinic prior to lysis.
An additional circadian rhythm in nuclear localization of PRDX2 has also been demonstrated in human keratinocytes, indicating that subcellular localization of redox factors may also be a factor in determining the character of the circadian clock.33 In addition, Kil et al. reported circadian rhythms in (mitochondrially localized) sulfinic peroxiredoxin 3 in mouse adrenal cortex, suggesting that these protein oxidation rhythms may extend to other cellular organelles.34
At present, the observation of circadian redox states in peroxiredoxins is largely phenomenological. A proteasome-mediated mechanism has been proposed in RBCs but is unlikely to be universally applicable because of the peculiarities of the system (discussed in detail below). No mechanism has been demonstrated to explain why peroxiredoxin oxidation states oscillate in such a wide range of nucleated and anucleated systems, nor has this oscillation been observed in cell-free preparations. Similarly, no causal relationship with canonical circadian clock componentry has been demonstrated. Moreover, it remains entirely plausible that circadian rhythms have evolved multiple times and have converged upon rhythmic regulation of redox and metabolism because of the advantages inherent to temporal partitioning of mutually antagonistic metabolic processes. In this case, redox rhythms result in cyclic oxidation of peroxiredoxins as a simple consequence of this protein family’s biochemistry, abundance, and ubiquity.
More intriguing perhaps is the case of nontranscriptional RBC rhythms.2,13 The RBC is in some ways an excellent model for studying the circadian clock. Mature RBCs lack organelles, including the nucleus, from which canonical circadian signals emanate. Given that RBCs are competent (1) to entrain to temperature cycles and (2) to maintain a circadian rhythm, with such a minimal set of components we can assume that all the elements of a nontranscriptional clock are contained within.2
In the RBC, some strides that enhance our understanding of the mechanism by which peroxiredoxins cycle have been taken.13 First, it was observed that ∼1% of total PRDX2 is subject to hyperoxidation into the sulfinic form over the course of this daily cycle. Second, it was found that the levels of sulfiredoxin were insufficient to allow reduction of sulfinic peroxiredoxin during the circadian cycle. Accordingly, a mutant in which sulfiredoxin was not present retained sulfinic peroxiredoxin cycles. Surprisingly, given that RBCs cannot translate new protein, sulfinic peroxiredoxin is degraded by the 20S proteasome. This leads to an age-dependent loss of PRDX2 as sulfinic protein is progressively degraded with each circadian cycle.13
It should be noted that the RBC is a highly specialized system, and anything we learn from its study must be seen with this caveat in mind. Specifically, the concentration of sulfiredoxin is lower in RBCs than in other cell types, which favors the destructive cycle of oxidation and degradation proposed by Cho and colleagues.13 It is not inconceivable that the clock apparent in RBCs simply does not function in the very different cellular environment of a more mundane nucleated cell. It was suggested that the rhythm in RBCs might be driven by autoxidation of heme. Autoxidation of hemoglobin occurs at a higher rate if the protein is in a dimeric state rather than in a cooperative tetrameric state.35 Oxidized hemoglobin is reduced primarily by NADH methemoglobin reductase, but also by NADPH methemoglobin reductase, ascorbic acid, and glutathione.36 Because hemoglobin is at high concentration in RBCs, an underlying rhythm in the cytosol that drives a subtle change in the dimer–tetramer equilibrium would be translated, by the accompanying change in the autoxidation rate, into a rhythm in the redox status of the major cellular antioxidants. An observed rhythm in NAD(P)H levels (measured by enzymatic assay) and hemoglobin oxidation (measured indirectly by front-face fluorescence) supports this hypothesis.2 Also, the level of rhythmic oxidation of PRDX2 is greatly reduced by treating the cells with carbon monoxide. This treatment effectively prevents heme oxidation and suggests that the observed rhythm in PRDX2 oxidation is dependent upon it.13 However, at the high concentrations of hemoglobin present in RBCs, the dimer will be very much a minority species and therefore would be expected to make only a very modest contribution to the overall rate of Hb autoxidation, unless the intracellular rate of heme dissociation and autoxidation is dramatically different from that measured in vitro.35
Peroxiredoxins not only may be an excellent marker for cellular rhythms but also may hint at a primordial redox oscillation that underlies contemporary circadian clocks.1 Irrespective of a basis on peroxiredoxin biochemistry, we do not know how such an oscillator might generate rhythms, why mechanistically the period should be around 24 h, or how this oscillation might be temperature-compensated. Fortunately, nature has already provided a well-characterized nontranscriptional oscillator from from which we can draw inspiration in trying to understand nontranscriptional rhythms in eukaryotic cells.
Cyanobacterial KaiABC Oscillator: A Model for Nontranscriptional Rhythms
A precedent exists for a self-sustaining nontranscriptional oscillator in the form of the cyanobacterial KaiABC oscillator. This paradigmatic example of nontranscriptional circadian oscillation was first demonstrated by Tomita et al. and was recapitulated in vitro by Nakajima et al.37,38 Here the KaiABC complex from the cyanobacterium Synechococcus elongatus was shown to undergo rhythmic autophosphorylation and dephosphorylation with circadian kinetics when the components were mixed in vitro with ATP as an energy source. Crucially, the oscillation persists without rhythmic environmental stimuli and is temperature-compensated.38
The KaiC protein forms a hexamer in the presence of ATP and is autophosphorylated at the monomer interfaces. KaiA acts as a cofactor in the phosphorylation reaction, while KaiB antagonizes this reaction. The assembly of the KaiABC complex is thought to be integral to its circadian regulation and is reviewed in depth by Johnson et al.39
Because KaiC is a broad-spectrum transcription factor that can suppress transcription from its own gene cluster, in vivo the KaiABC oscillator exists within a GEFL loop. The transcriptional activity of the KaiABC complex also works to generate the rhythmic program of gene expression essential to a circadian lifestyle. The synergistic action of two feedback loops is thought to increase robustness, although attempts to reconstitute the GEFL alone (without the post-translational feedback loop) failed to exhibit circadian rhythms.40,41
It was proposed that the catalytic cycle of peroxiredoxin hyperoxidation and recycling by sulfiredoxin may form the basis of a transcription-independent circadian clock.42 Both the oxidation of sulfenic peroxiredoxin and the converse reduction of sulfinic acid are thought to be inefficient in vivo.20,43 This mirrors slow rates of catalytic cycling observed in the KaiABC oscillator and may be instrumental in setting up the near 24 h period essential in circadian oscillators.39 This hypothesis must be updated in light of the recent observation that peroxiredoxin cycling is driven by rhythmic degradation in the mouse RBC, as is it unclear that the rate of peroxiredoxin degradation would be slow enough to allow such a long period of oscillation.13 Interestingly, both the KaiABC complex and peroxiredoxin proteins assemble into multimeric complexes in vivo, and their multimeric state is linked to their function.25,39 All currently described cellular oscillators incorporate some element of negative feedback control with delay however;5 for example, in the KaiABC oscillator, this is manifested by the inhibition of KaiC autophosphorylation by serine-only phosphorylated KaiC.39 A similar feedback system has yet to be described in peroxiredoxin redox cycles, and moreover, genetic and pharmacological manipulation of peroxiredoxin activity has only modest effects on cellular timekeeping. Post-translational modification of peroxiredoxin function has been demonstrated, however, so in vivo negative feedback is at least plausible.26 It is apparent that we currently lack a clear mechanistic hypothesis of how peroxiredoxins might generate self-sustaining rhythms from which circadian signals emanate. This does not prevent us from considering how the canonical circadian clock might be coupled to the cyclic redox state of peroxiredoxins in particular and, more generally, the cellular redox state
Coupling Cellular Rhythms to the Canonical Circadian Clock
Critical to the in vivo function of the KaiABC oscillator is its coupling of a nontranscriptional oscillation to a GEFL.40,41 This aspect is clearly missing from current data on mammalian nontranscriptional oscillators, although there is a wealth of data about how cellular redox status feeds into the canonical circadian clock and vice versa.44−47 The paradigm of post-translational control of the canonical clock is well-established48 and provides a conceptual framework within which redox signaling might couple to GEFL function.
The core clock GEFL incorporates oscillation across the entire gene expression pathway. Rhythmic marks are placed on histones,49−51 which lead to rhythmic transcription, translation of the mRNA, and functionalization of the protein.8 There is evidence that each stage of the gene expression cycle is regulated to achieve a correct circadian output. There also is evidence that GEFL need not incorporate the root of the gene expression pathway and that oscillation can occur at multiple levels. For instance, in the mouse liver, de novo transcription drives rhythms in only 22% of oscillating transcripts.51 Furthermore, approximately 20% of the oscillating proteome components have no detectable oscillation in the transcript.48 In many cases in which rhythmic transcription is described, the protein level does not oscillate appreciably.48
PER proteins provide a good example of circadian rhythmicity in protein activity, implemented by post-translational modification. The tau mutation in the gene encoding casein kinase Iε (CKIε) dramatically shortens the circadian period in Syrian hamsters.52 CKI is a kinase with multiple isoforms implicated in Wnt signaling, nucleo-cytoplasmic shuttling, DNA replication, and DNA repair pathways.53 In the context of the circadian GEFL, CKIε and CKIδ phosphorylate PER proteins and license their import into the nucleus where they repress transcription (Figure 1).54−57 Phosphorylation of PER proteins also promotes their proteasomal degradation.58 More recently, it has been shown that the related casein kinase 2 also acts upon PER protein to regulate circadian rhythms.59 The tau mutation decreases the general kinase activity of CKIε but increases its specific activity against PER protein, thereby accelerating both the import and degradation of the PER:CRY complex.60 Conversely, interfering with degradation of the PER:CRY complex function by pharmacological means causes a lengthening of period, indicating that stability of the PER/CRY complex is critical in determining cycle length.61
Post-translational regulation of the clock and enzymes such as the casein kinases, like the rhythms in peroxiredoxin oxidation, exhibits greater conservation than the canonical clock components.15,62 Accordingly, drugs that act upon post-translational control mechanisms tend to have similar effects on the circadian clock across different taxa.15
Many of these post-translational modifications are themselves circadian, creating “accessory” oscillators that cooperate in cellular timekeeping (Figure 4). In mammals, these accessory oscillators are components of cellular metabolic pathways, linking the daily rhythm to the rhythmic metabolic demands it places upon an organism.63 For instance, adenosine monophosphate-regulated kinase (AMPK) senses cellular energy levels generally64 but is also rhythmically regulated by threonine phosphorylation.9,65 Activated AMPK in turn can phosphorylate CKIε and CRY proteins and regulate the circadian clock.10,12 A similar accessory oscillation of cAMP/Ca2+ has also been described, which is of particular importance in a neuronal context (reviewed in ref (66)).
Figure 4.

Accessory circadian oscillators. Modulation of the canonical PER:CRY–BMAL:CLOCK oscillator occurs by post-translational modification of clock components. As these pathways are affected by the clock themselves, they form “accessory oscillators”. Green arrows designate control of the canonical clock, and red arrows indicate circadian control of accessory oscillators. cAMP/Ca2+ signaling oscillates in vivo and controls transcriptional output of clock-controlled genes that incorporate cAMP response elements in their promoters. AMPK senses the cellular ATP/ADP ratio, which is an output of circadian metabolism. AMPK signals to casein kinase Iε to control degradation of PER2 protein and also phosphorylates CRY proteins. The NAD+/NADH ratio is controlled by circadian transcription of the biosynthetic enzyme NAMPT. SIRT1, an NAD+-dependent histone deacetylase, controls the activity of the BMAL:CLOCK complex and can induce degradation of PER2. Peroxiredoxin (PRDX) oscillations have been detected in a variety of systems, but the extent with which and mechanism by which they interact with the circadian clock are not yet known.
Redox Control of the Circadian Clock
Cellular redox status is under the pervasive influence of the circadian clock but can also signal back to control it via a general influence upon cellular metabolism.44−47,63 If circadian peroxiredoxin oxidation oscillation, or the cryptic oscillator it reports, is coupled to the clock, this might be mediated through established redox signaling mechanisms. Additionally, the peroxiredoxins themselves might mediate coupling between the canonical circadian clock and redox oscillations.
Nicotinamide adenine dinucleotide (NAD+) is a cellular electron carrier that can also function as an enzyme substrate during the post-translational modification of proteins. NADP+ is a closely related compound functioning in biosynthetic pathways as well as redox homeostasis. Both compounds are reduced by the addition of a hydride ion (H–) to form NADH or NADPH; the phosphate group of NADPH is not thought to affect its electron transfer properties but confers substrate specificity.67 In general, the NAD+/NADH cellular ratio (>1) is higher than the NADP+/NADPH cellular ratio (<0.01), and in the cytosol, these two redox couples are not in equilibrium, allowing the cell to segregate antioxidant and biosynthetic reducing equivalents (NADPH) from those destined for mitochondrial ATP generation (NADH). Flavin adenine dinucleotide (FAD) is a redox cofactor that acts as an electron acceptor in mitochondrial oxidative phosphorylation to form FADH2, which then donates its electrons to the electron transport chain.68
Circadian oscillations in the redox state have been described in ex vivo culture, in which the ratio of oxidized FAD to reduced NADH was determined fluorometrically in oscillating suprachiasmatic nucleus slices.69 The redox rhythm was abrogated in bmal1–/– mutant mice and shown to correlate to the excitability of the neurons within the pacemaker circuitry by modulation of potassium channels.69 Regulation of the NAD+ synthetic enzyme NAMPT by the CLOCK:BMAL complex has suggested a mechanism for circadian control of redox homeostasis in mice.44,45 As previously discussed, NADH and NADPH rhythms have since been described in isolated human RBCs that cannot be explained by circadian expression of redox proteins.2
The level of NAD+ feeds back into clock component regulation and has global effects on the proteome (Figure 4).70 The class III histone deacetylase SIRT1 is NAD+-dependent and acts to generate rhythmic patterns in circadian gene histone H3 acetylation and acetylation of BMAL1.70 BMAL1 acetylation coincides with downregulation of clock-controlled genes, mediated through an increased level of recruitment of CRY1 to the BMAL1:CLOCK complex.71 Furthermore, PER2 is deacetylated by SIRT1, leading to its degradation.72 Through multiple mechanisms, SIRT1 seems to ensure high-magnitude rhythms in the circadian clock. The subtle circadian phenotype of the homozygous clock and sirt1 null mice, however, suggests that the reality must be more complex.73−75
Further interplay between the cellular redox state and the circadian clock was suggested by the observation that the affinity of BMAL1 in a complex with CLOCK (or a related protein, NPAS2) for DNA was modulated in vitro by the ratio of NAD+ to NADH or NADP+ to NADPH.46 REV-ERBβ, a heme binding protein implicated in the regulation of metabolism and the circadian clock, has also been shown to have redox sensitive cysteine residues that control the expression of target genes.47 This illustrates a mechanism by which redox balance can influence circadian clock function.47 The crystal structure of the mouse PER2:CRY1 complex may suggest a further mechanism of redox regulation of the core clock.76 The close interaction of PER2 and CRY1 is stabilized by the coordination of a zinc ion, which facilitates the reduction of a nearby disulfide bond. The likely candidates for disulfide bridge formation were identified as Cys412 and Cys363 of CRY1. Accordingly, CRY1 was found to exist in both oxidized and reduced forms when expressed in HEK293 cells. In vitro experiments indicate that nonreducing conditions do not promote the release of zinc from the complex, but this does not preclude in vivo redox sensitive zinc binding. Alternatively, as the dissociation constant of the PER2:CRY1 complex is close to native free zinc ion concentrations (in the nanomolar range), this may allow a fluctuating zinc concentration to control the function of the PER2:CRY1 complex. Zinc release has been demonstrated to support CRY1 oxidation, which would render the circadian clock zinc sensitive.76
Interestingly, in mice in which Cys414 of CRY1 has been substituted for alanine, a long period (28 h) is evident.77 It has been suggested that this mutation promotes PER1 interaction and thus has a phenotype similar to that of mutants in the BMAL:CLOCK arm of the circadian clock.76
The metabolic processes of aerobic cells produce reactive oxidative species (ROS) as an inevitable byproduct, particularly superoxide. The role of ROS in cellular signaling is under intense scrutiny at present and is thought to influence processes as diverse as stem cell proliferation and cell migration.78,79 Superoxide is rapidly converted to hydrogen peroxide (H2O2) by superoxide dismutases. Intracellular H2O2 can act as a classic second messenger within the cell by covalent modification of key molecules.79 Reduced redox sensitive cysteine residues oxidized by H2O2 cause changes to protein activity, such as in the case of phosphotyrosine phosphatases, which are inactivated by cysteine oxidation, which in turn signal to other cellular components.80 Higher concentrations of H2O2 cause glutathione oxidation, which in turn activates oxidative stress response pathways.81 Responses to oxidative stress can be mediated through gene expression, for instance via the redox sensitive cysteine-containing FOXO4 transcription factor.82 More immediately, oxidative stress can act directly to reroute metabolic flux and thereby restore redox homeostasis.83
Peroxiredoxins can facilitate signaling through a variety of mechanisms.84 There are many examples of peroxiredoxin-mediated ROS signaling pathways, many of which are mediated by protein interaction.84 While none so far described explicitly speak to modulation of the circadian GEFL, an abundance of precedents for such a mode of action does exist.
It has been proposed that an increase in the level of H2O2 of sufficient magnitude can cause sulfinic peroxiredoxin to form in vivo, deactivating a major cellular antioxidant, leading to a further increase in H2O2 levels that can act as a second messenger in signal transduction.85 The floodgate hypothesis of peroxide signaling, proposed by Wood et al.,85 has been reviewed elsewhere.79,84 Sulfonic peroxiredoxin, if it accumulates in vivo, is likely also to contribute to the floodgate effect, because it is irreversibly redox inactivated. Similarly, a study in yeast has also revealed an unexpected role of peroxiredoxin hyperoxidation (Tpx-SO2H) in cell survival. Here, thioredoxin, the major substrate for oxidized peroxiredoxin, is discharged by inactivated (sulfinic and sulfonic) peroxiredoxin, allowing it to reduce other substrates critical to cell survival.86
A further mechanism might involve the chaperone function of peroxiredoxins. As previously discussed, hyperoxidation of peroxiredoxin can induce a chaperone function in higher-order complexes. The hyperoxidation-induced chaperone function or signal transduction properties of peroxiredoxin, acting directly or via H2O2, would provide a straightforward means of propagating the circadian signal into the wider cellular system by potentiating gene expression either globally or specifically.
Conclusion
In this review, we have discussed how the canonical clock utilizes post-translational modifications in its normal function and that these modifications are closely linked to metabolism, including cellular redox homeostasis. Recent findings have demonstrated that not only is redox status linked to the canonical circadian clock, but that it may also be a conserved reporter of a noncanonical, transcription-independent circadian clock. At present, we do not know the mechanism of the nontranscriptional circadian clock in eukaryotes. It may be that a system similar to the KaiABC autophosphorylation–dephosphorylation cycles is waiting to be discovered. Alternatively, peroxiredoxin hyperoxidation cycles may be key to understanding the clockwork of this noncanonical oscillation. Our growing knowledge of ROS signaling certainly indicates that gene expression programs can be controlled by the cellular redox state at multiple levels. However, if history has taught us anything, it is likely that an entirely unexpected mechanism underlies nontranscriptional rhythms in eukaryotes.
Acknowledgments
Thanks to members of the O’Neill lab for helpful discussion.
Glossary
Abbreviations
- AMPK
adenosine monophosphate-regulated protein kinase
- BMAL1
brain and muscle Arnt-like protein-1
- CKI
casein kinase 1
- CLOCK
circadian locomotor output cycles kaput
- CRY
cryptochrome
- FAD
flavin adenine dinucleotide
- GEFL
gene expression feedback loop
- LUCA
latest universal common ancestor
- NAD+
nicotinamide adenine dinucleotide
- NADP+
nicotinamide adenine dinucleotide phosphate
- PER
period
- PRDX
peroxiredoxin
- SRX
sulfiredoxin
- TRX
thioredoxin
- TTFL
transcription–translation feedback loop.
We are supported by the Medical Research Council (MC_UP_1201/4) and the Wellcome Trust (093734/Z/10/Z).
The authors declare no competing financial interest.
References
- Edgar R.; Green E.; Zhao Y.; van Ooijen G.; Olmedo M.; Qin X.; Xu Y.; Pan M.; Valekunja U.; Feeney K. A.; Maywood E.; Hastings M. H.; Baliga N.; Merrow M.; Millar A.; Johnson C. H.; Kyriacou C.; O’Neill J. S.; Reddy A. B. (2012) Peroxiredoxins Are Conserved Markers of Circadian Rhythms. Nature 485, 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill J. S.; Reddy A. B. (2011) Circadian Clocks in Human Red Blood Cells. Nature 469, 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittendrigh C. S. (1960) Circadian Rhythms and the Circadian Organization of Living Systems. Cold Spring Harbor Symp. Quant. Biol. 25, 159–184. [DOI] [PubMed] [Google Scholar]
- Johnson C. H.; Stewart P. L.; Egli M. (2011) The Cyanobacterial Circadian System: From Biophysics to Bioevolution. Annu. Rev. Biophys. 40, 143–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák B.; Tyson J. J. (2008) Design Principles of Biochemical Oscillators. Nat. Rev. Mol. Cell Biol. 9, 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka R. J.; Benzer S. (1971) Clock Mutants of Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 68, 2112–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty C. J.; Kay S. A. (2010) Circadian Control of Global Gene Expression Patterns. Annu. Rev. Genet. 44, 419–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohawk J. A.; Green C. B.; Takahashi J. S. (2012) Central and Peripheral Circadian Clocks in Mammals. Annu. Rev. Neurosci. 35, 445–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro A.; Hayer K.; Lahens N. F.; Hogenesch J. B. (2012) CircaDB: A Database of Mammalian Circadian Gene Expression Profiles. Nucleic Acids Res. 41, D1009–D1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um J. H.; Yang S.; Yamazaki S.; Kang H.; Viollet B.; Foretz M.; Chung J. H. (2007) Activation of 5′-AMP-Activated Kinase with Diabetes Drug Metformin Induces Casein Kinase Iε (CKIε)-Dependent Degradation of Clock Protein mPer2. J. Biol. Chem. 282, 20794–20798. [DOI] [PubMed] [Google Scholar]
- Rosbash M. (2009) The Implications of Multiple Circadian Clock Origins. PLoS Biol. 7, e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamia K. A.; Sachdeva U. M.; DiTacchio L.; Williams E. C.; Alvarez J. G.; Egan D. F.; Vasquez D. S.; Juguilon H.; Panda S.; Shaw R. J.; Thompson C. B.; Evans R. M. (2009) AMPK Regulates the Circadian Clock by Cryptochrome Phosphorylation and Degradation. Science 326, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C.-S.; Yoon H. J.; Kim J. Y.; Woo H. A.; Rhee S. G. (2014) Circadian Rhythm of Hyperoxidized Peroxiredoxin II Is Determined by Hemoglobin Autoxidation and the 20S Proteasome in Red Blood Cells. Proc. Natl. Acad. Sci. U.S.A. 111, 12043–12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poynton R. A.; Hampton M. B. (2014) Peroxiredoxins as Biomarkers of Oxidative Stress. Biochim. Biophys. Acta 1840, 906–912. [DOI] [PubMed] [Google Scholar]
- O’Neill J. S.; van Ooijen G.; Dixon L.; Troein C.; Corellou F.; Bouget F.-Y.; Reddy A. B.; Millar A. (2011) Circadian Rhythms Persist Without Transcription in a Eukaryote. Nature 469, 554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A.; Karplus P. A.; Poole L. B. (2009) Typical 2-Cys Peroxiredoxins: Structures, Mechanisms and Functions. FEBS J. 276, 2469–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A. B.; Karp N. A.; Maywood E. S.; Sage E. A.; Deery M.; O’Neill J. S.; Wong G. K. Y.; Chesham J.; Odell M.; Lilley K. S.; Kyriacou C. P.; Hastings M. H. (2006) Circadian Orchestration of the Hepatic Proteome. Curr. Biol. 16, 1107–1115. [DOI] [PubMed] [Google Scholar]
- Woo H. A.; Kang S. W.; Kim H. K.; Yang K.-S.; Chae H. Z.; Rhee S. G. (2003) Reversible Oxidation of the Active Site Cysteine of Peroxiredoxins to Cysteine Sulfinic Acid. Immunoblot Detection with Antibodies Specific for the Hyperoxidized Cysteine-Containing Sequence. J. Biol. Chem. 278, 47361–47364. [DOI] [PubMed] [Google Scholar]
- Woo H. A.; Chae H. Z.; Hwang S. C.; Yang K.-S.; Kang S. W.; Kim K.; Rhee S. G. (2003) Reversing the Inactivation of Peroxiredoxins Caused by Cysteine Sulfinic Acid Formation. Science 300, 653–656. [DOI] [PubMed] [Google Scholar]
- Chang T. S.; Jeong W.; Woo H. A.; Lee S. M.; Park S.; Rhee S. G. (2004) Characterization of Mammalian Sulfiredoxin and Its Reactivation of Hyperoxidized Peroxiredoxin Through Reduction of Cysteine Sulfinic Acid in the Active Site to Cysteine. J. Biol. Chem. 279, 50994–51001. [DOI] [PubMed] [Google Scholar]
- Lim J. C.; Choi H.-I.; Park Y. S.; Nam H. W.; Woo H. A.; Kwon K.-S.; Kim Y. S.; Rhee S. G.; Kim K.; Chae H. Z. (2008) Irreversible Oxidation of the Active-Site Cysteine of Peroxiredoxin to Cysteine Sulfonic Acid for Enhanced Molecular Chaperone Activity. J. Biol. Chem. 283, 28873–28880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner E.; Luche S.; Penna L.; Chevallet M.; Van Dorsselaer A.; Leize-Wagner E.; Rabilloud T. (2002) A Method for Detection of Overoxidation of Cysteines: Peroxiredoxins Are Oxidized in Vivo at the Active-Site Cysteine During Oxidative Stress. Biochem. J. 366, 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musicco C.; Capelli V.; Pesce V.; Timperio A. M.; Calvani M.; Mosconi L.; Zolla L.; Cantatore P.; Gadaleta M. N. (2009) Accumulation of Overoxidized Peroxiredoxin III in Aged Rat Liver Mitochondria. Biochim. Biophys. Acta 1787, 890–896. [DOI] [PubMed] [Google Scholar]
- Cao Z.; Cao Z.; Roszak A. W.; Roszak A. W.; Gourlay L. J.; Gourlay L. J.; Lindsay J. G.; Lindsay J. G.; Isaacs N. W.; Isaacs N. W. (2005) Bovine Mitochondrial Peroxiredoxin III Forms a Two-Ring Catenane. Structure 13, 1661–1664. [DOI] [PubMed] [Google Scholar]
- Barranco-Medina S.; Lázaro J.-J.; Dietz K.-J. (2009) The Oligomeric Conformation of Peroxiredoxins Links Redox State to Function. FEBS Lett. 583, 1809–1816. [DOI] [PubMed] [Google Scholar]
- Woo H. A.; Yim S. H.; Shin D. H.; Kang D.; Yu D.-Y.; Rhee S. G. (2010) Inactivation of Peroxiredoxin I by Phosphorylation Allows Localized H2O2 Accumulation for Cell Signaling. Cell 140, 517–528. [DOI] [PubMed] [Google Scholar]
- Meissner U.; Schröder E.; Scheffler D.; Martin A. G.; Harris J. R. (2007) Formation, TEM Study and 3D Reconstruction of the Human Erythrocyte Peroxiredoxin-2 Dodecahedral Higher-Order Assembly. Micron 38, 29–39. [DOI] [PubMed] [Google Scholar]
- Park J.; Lee S.; Lee S.; Kang S. W. (2014) 2-Cys Peroxiredoxins: Emerging Hubs Determining Redox Dependency of Mammalian Signaling Networks. Int. J. Cell Biol. 2014, 715867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H. H.; Kim S. Y.; Park S. K.; Jeon H. S.; Lee Y. M.; Jung J. H.; Lee S. Y.; Chae H. B.; Jung Y. J.; Lee K. O.; Lim C. O.; Chung W. S.; Bahk J. D.; Yun D.-J.; Cho M. J.; Lee S. Y. (2006) Phosphorylation and Concomitant Structural Changes in Human 2-Cys Peroxiredoxin Isotype I Differentially Regulate Its Peroxidase and Molecular Chaperone Functions. FEBS Lett. 580, 351–355. [DOI] [PubMed] [Google Scholar]
- König J.; König J.; Galliardt H.; Galliardt H.; Jütte P.; Jütte P.; Schäper S.; Schäper S.; Dittmann L.; Dittmann L.; Dietz K.-J.; Dietz K.-J. (2013) The Conformational Bases for the Two Functionalities of 2-Cysteine Peroxiredoxins as Peroxidase and Chaperone. J. Exp. Bot. 64, 3483–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radha E.; Hill T. D.; Rao G. H.; White J. G. (1985) Glutathione Levels in Human Platelets Display a Circadian Rhythm in Vitro. Thromb. Res. 40, 823–831. [DOI] [PubMed] [Google Scholar]
- Woolum J. C. (1991) A Re-Examination of the Role of the Nucleus in Generating the Circadian Rhythm in Acetabularia. J. Biol. Rhythms 6, 129–136. [DOI] [PubMed] [Google Scholar]
- Avitabile D.; Ranieri D.; Nicolussi A.; D’Inzeo S.; Capriotti A. L.; Genovese L.; Proietti S.; Cucina A.; Coppa A.; Samperi R.; Bizzarri M.; Laganà A.; Torrisi M. R. (2014) Peroxiredoxin 2 Nuclear Levels Are Regulated by Circadian Clock Synchronization in Human Keratinocytes. Int. J. Biochem. Cell Biol. 53C, 24–34. [DOI] [PubMed] [Google Scholar]
- Kil I. S.; Lee S. K.; Ryu K. W.; Woo H. A.; Hu M.-C.; Bae S. H.; Rhee S. G. (2012) Feedback Control of Adrenal Steroidogenesis via H2O2-Dependent, Reversible Inactivation of Peroxiredoxin III in Mitochondria. Mol. Cell 46, 584–594. [DOI] [PubMed] [Google Scholar]
- Griffon N.; Baudin V.; Dieryck W.; Dumoulin A.; Pagnier J.; Poyart C.; Marden M. C. (1998) Tetramer-Dimer Equilibrium of Oxyhemoglobin Mutants Determined From Auto-Oxidation Rates. Protein Sci. 7, 673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita A.; Nakayama Y.; Kitayama T.; Tomita M. (2007) Simulation Study of Methemoglobin Reduction in Erythrocytes. Differential Contributions of Two Pathways to Tolerance to Oxidative Stress. FEBS J. 274, 1449–1458. [DOI] [PubMed] [Google Scholar]
- Tomita J.; Nakajima M.; Kondo T.; Iwasaki H. (2005) No Transcription-Translation Feedback in Circadian Rhythm of KaiC Phosphorylation. Science 307, 251–254. [DOI] [PubMed] [Google Scholar]
- Nakajima M.; Imai K.; Ito H.; Nishiwaki T.; Murayama Y.; Iwasaki H.; Oyama T.; Kondo T. (2005) Reconstitution of Circadian Oscillation of Cyanobacterial KaiC Phosphorylation in Vitro. Science 308, 414–415. [DOI] [PubMed] [Google Scholar]
- Johnson C. H.; Egli M. (2014) Metabolic Compensation and Circadian Resilience in Prokaryotic Cyanobacteria. Annu. Rev. Biochem. 83, 221–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwicker D.; Lubensky D. K.; ten Wolde P. R. (2010) Robust Circadian Clocks From Coupled Protein-Modification and Transcription-Translation Cycles. Proc. Natl. Acad. Sci. U.S.A. 107, 22540–22545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng S.-W.; Mukherji S.; Moffitt J. R.; de Buyl S.; O’Shea E. K. (2013) Robust Circadian Oscillations in Growing Cyanobacteria Require Transcriptional Feedback. Science 340, 737–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A. B.; Rey G. (2013) Metabolic and Nontranscriptional Circadian Clocks: Eukaryotes. Annu. Rev. Biochem. 83, 165–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K. S. (2002) Inactivation of Human Peroxiredoxin I During Catalysis as the Result of the Oxidation of the Catalytic Site Cysteine to Cysteine-Sulfinic Acid. J. Biol. Chem. 277, 38029–38036. [DOI] [PubMed] [Google Scholar]
- Ramsey K. M.; Yoshino J.; Brace C. S.; Abrassart D.; Kobayashi Y.; Marcheva B.; Hong H.-K.; Chong J. L.; Buhr E. D.; Lee C.; Takahashi J. S.; Imai S.-I.; Bass J. (2009) Circadian Clock Feedback Cycle Through NAMPT-Mediated NAD+ Biosynthesis. Science 324, 651–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y.; Sahar S.; Astarita G.; Kaluzova M.; Sassone-Corsi P. (2009) Circadian Control of the NAD+ Salvage Pathway by CLOCK-SIRT1. Science 324, 654–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter J.; Reick M.; Wu L. C.; McKnight S. L. (2001) Regulation of Clock and NPAS2 DNA Binding by the Redox State of NAD Cofactors. Science 293, 510–514. [DOI] [PubMed] [Google Scholar]
- Gupta N.; Ragsdale S. W. (2011) Thiol-Disulfide Redox Dependence of Heme Binding and Heme Ligand Switching in Nuclear Hormone Receptor Rev-Erbβ. J. Biol. Chem. 286, 4392–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles M. S.; Cox J.; Mann M. (2014) In-Vivo Quantitative Proteomics Reveals a Key Contribution of Post-Transcriptional Mechanisms to the Circadian Regulation of Liver Metabolism. PLoS Genet. 10, e1004047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmers C.; Schmitz R. J.; Nathanson J.; Yeo G.; Ecker J. R.; Panda S. (2012) Circadian Oscillations of Protein-Coding and Regulatory RNAs in a Highly Dynamic Mammalian Liver Epigenome. Cell Metab. 16, 833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valekunja U. K.; Edgar R. S.; Oklejewicz M.; van der Horst G. T. J.; O’Neill J. S.; Tamanini F.; Turner D. J.; Reddy A. B. (2013) Histone Methyltransferase MLL3 Contributes to Genome-Scale Circadian Transcription. Proc. Natl. Acad. Sci. U.S.A. 110, 1554–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike N.; Yoo S.-H.; Huang H.-C.; Kumar V.; Lee C.; Kim T.-K.; Takahashi J. S. (2012) Transcriptional Architecture and Chromatin Landscape of the Core Circadian Clock in Mammals. Science 338, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph M. R.; Menaker M. (1988) A Mutation of the Circadian System in Golden Hamsters. Science 241, 1225–1227. [DOI] [PubMed] [Google Scholar]
- Knippschild U.; Gocht A.; Wolff S.; Huber N.; Löhler J.; Stöter M. (2005) The Casein Kinase 1 Family: Participation in Multiple Cellular Processes in Eukaryotes. Cell. Signalling 17, 675–689. [DOI] [PubMed] [Google Scholar]
- Akashi M.; Tsuchiya Y.; Yoshino T.; Nishida E. (2002) Control of Intracellular Dynamics of Mammalian Period Proteins by Casein Kinase Iε (CKIε) and CKIδ in Cultured Cells. Mol. Cell. Biol. 22, 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vielhaber E.; Eide E.; Rivers A.; Gao Z. H.; Virshup D. M. (2000) Nuclear Entry of the Circadian Regulator mPER1 Is Controlled by Mammalian Casein Kinase Iε. Mol. Cell. Biol. 20, 4888–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q.-J.; Maywood E. S.; Bechtold D. A.; Lu W.-Q.; Li J.; Gibbs J. E.; Dupré S. M.; Chesham J. E.; Rajamohan F.; Knafels J.; Sneed B.; Zawadzke L. E.; Ohren J. F.; Walton K. M.; Wager T. T.; Hastings M. H.; Loudon A. S. I. (2010) Entrainment of Disrupted Circadian Behavior Through Inhibition of Casein Kinase 1 (CK1) Enzymes. Proc. Natl. Acad. Sci. U.S.A. 107, 15240–15245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchegaray J.-P.; Yu E. A.; Indic P.; Dallmann R.; Weaver D. R. (2010) Casein Kinase 1δ (CK1δ) Regulates Period Length of the Mouse Suprachiasmatic Circadian Clock in Vitro. PLoS One 5, e10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide E. J.; Woolf M. F.; Kang H.; Woolf P.; Hurst W.; Camacho F.; Vielhaber E. L.; Giovanni A.; Virshup D. M. (2005) Control of Mammalian Circadian Rhythm by CKIε-Regulated Proteasome-Mediated PER2 Degradation. Mol. Cell. Biol. 25, 2795–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya Y.; Akashi M.; Matsuda M.; Goto K.; Miyata Y.; Node K.; Nishida E. (2009) Involvement of the Protein Kinase CK2 in the Regulation of Mammalian Circadian Rhythms. Sci. Signaling 2, ra26. [DOI] [PubMed] [Google Scholar]
- Gallego M.; Eide E. J.; Woolf M. F.; Virshup D. M.; Forger D. B. (2006) An Opposite Role for Tau in Circadian Rhythms Revealed by Mathematical Modeling. Proc. Natl. Acad. Sci. U.S.A. 103, 10618–10623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badura L.; Swanson T.; Adamowicz W.; Adams J.; Cianfrogna J.; Fisher K.; Holland J.; Kleiman R.; Nelson F.; Reynolds L.; St Germain K.; Schaeffer E.; Tate B.; Sprouse J. (2007) An Inhibitor of Casein Kinase Iε Induces Phase Delays in Circadian Rhythms Under Free-Running and Entrained Conditions. J. Pharmacol. Exp. Ther. 322, 730–738. [DOI] [PubMed] [Google Scholar]
- Hastings M. H.; Maywood E. S.; O’Neill J. S. (2008) Cellular Circadian Pacemaking and the Role of Cytosolic Rhythms. Curr. Biol. 18, R805–R815. [DOI] [PubMed] [Google Scholar]
- Bass J. (2012) Circadian Topology of Metabolism. Nature 491, 348–356. [DOI] [PubMed] [Google Scholar]
- Jordan S. D.; Lamia K. A. (2013) AMPK at the Crossroads of Circadian Clocks and Metabolism. Mol. Cell. Endocrinol. 366, 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um J. H.; Pendergast J. S.; Springer D. A.; Foretz M.; Viollet B.; Brown A.; Kim M. K.; Yamazaki S.; Chung J. H. (2011) AMPK Regulates Circadian Rhythms in a Tissue- and Isoform-Specific Manner. PLoS One 6, e18450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill J. S.; Reddy A. B. (2012) The Essential Role of cAMP/Ca2+ Signalling in Mammalian Circadian Timekeeping. Biochem. Soc. Trans. 40, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belenky P.; Bogan K. L.; Brenner C. (2007) NAD+ Metabolism in Health and Disease. Trends Biochem. Sci. 32, 12–19. [DOI] [PubMed] [Google Scholar]
- How Cells Obtain Energy From Food (2002) Mol. Biol. Cell.
- Wang T. A.; Yu Y. V.; Govindaiah G.; Ye X.; Artinian L.; Coleman T. P.; Sweedler J. V.; Cox C. L.; Gillette M. U. (2012) Circadian Rhythm of Redox State Regulates Excitability in Suprachiasmatic Nucleus Neurons. Science 337, 839–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y.; Kaluzova M.; Grimaldi B.; Sahar S.; Hirayama J.; Chen D.; Guarente L. P.; Sassone-Corsi P. (2008) The NAD+-Dependent Deacetylase SIRT1 Modulates CLOCK-Mediated Chromatin Remodeling and Circadian Control. Cell 134, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama J.; Sahar S.; Grimaldi B.; Tamaru T.; Takamatsu K.; Nakahata Y.; Sassone-Corsi P. (2007) CLOCK-Mediated Acetylation of BMAL1 Controls Circadian Function. Nature 450, 1086–1090. [DOI] [PubMed] [Google Scholar]
- Asher G.; Gatfield D.; Stratmann M.; Reinke H.; Dibner C.; Kreppel F.; Mostoslavsky R.; Alt F. W.; Schibler U. (2008) SIRT1 Regulates Circadian Clock Gene Expression Through PER2 Deacetylation. Cell 134, 317–328. [DOI] [PubMed] [Google Scholar]
- Fogg P. C. M.; O’Neill J. S.; Dobrzycki T.; Calvert S.; Lord E. C.; McIntosh R. L. L.; Elliott C. J. H.; Sweeney S. T.; Hastings M. H.; Chawla S. (2014) Class IIa Histone Deacetylases Are Conserved Regulators of Circadian Function. J. Biol. Chem. 289, 34341–34348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debruyne J. P.; Noton E.; Lambert C. M.; Maywood E. S.; Weaver D. R.; Reppert S. M. (2006) A Clock Shock: Mouse CLOCK Is Not Required for Circadian Oscillator Function. Neuron 50, 465–477. [DOI] [PubMed] [Google Scholar]
- Chang H.-C.; Guarente L. (2013) SIRT1 Mediates Central Circadian Control in the SCN by a Mechanism That Decays with Aging. Cell 153, 1448–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalen I.; Reischl S.; Wallach T.; Klemz R.; Grudziecki A.; Prabu J. R.; Benda C.; Kramer A.; Wolf E. (2014) Interaction of Circadian Clock Proteins CRY1 and PER2 Is Modulated by Zinc Binding and Disulfide Bond Formation. Cell 157, 1203–1215. [DOI] [PubMed] [Google Scholar]
- Okano S.; Akashi M.; Hayasaka K.; Nakajima O. (2009) Unusual Circadian Locomotor Activity and Pathophysiology in Mutant CRY1 Transgenic Mice. Neurosci. Lett. 451, 246–251. [DOI] [PubMed] [Google Scholar]
- Dickinson B. C.; Dickinson B. C.; Chang C. J.; Chang C. J. (2011) Chemistry and Biology of Reactive Oxygen Species in Signaling or Stress Responses. Nat. Chem. Biol. 7, 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sies H. (2014) Role of Metabolic H2O2 Generation: Redox Signaling and Oxidative Stress. J. Biol. Chem. 289, 8735–8741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karisch R.; Fernandez M.; Taylor P.; Virtanen C.; St-Germain J. R.; Jin L. L.; Harris I. S.; Mori J.; Mak T. W.; Senis Y. A.; Östman A.; Moran M. F.; Neel B. G. (2011) Global Proteomic Assessment of the Classical Protein-Tyrosine Phosphatome and “Redoxome”. Cell 146, 826–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle-Donne I.; Rossi R.; Colombo G.; Giustarini D.; Milzani A. (2009) Protein S-Glutathionylation: A Regulatory Device From Bacteria to Humans. Trends Biochem. Sci. 34, 85–96. [DOI] [PubMed] [Google Scholar]
- Dansen T. B.; Smits L. M. M.; van Triest M. H.; de Keizer P. L. J.; van Leenen D.; Koerkamp M. G.; Szypowska A.; Meppelink A.; Brenkman A. B.; Yodoi J.; Holstege F. C. P.; Burgering B. M. T. (2009) Redox-Sensitive Cysteines Bridge P300/CBP-Mediated Acetylation and FoxO4 Activity. Nat. Chem. Biol. 5, 664–672. [DOI] [PubMed] [Google Scholar]
- Ralser M.; Wamelink M. M.; Kowald A.; Gerisch B.; Heeren G.; Struys E. A.; Klipp E.; Jakobs C.; Breitenbach M.; Lehrach H.; Krobitsch S. (2007) Dynamic Rerouting of the Carbohydrate Flux Is Key to Counteracting Oxidative Stress. J. Biol. 6, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall L. M., Ferrer-Sueta G., and Denicola A. (2013) Peroxiredoxins as Preferential Targets in H2O2-Induced Signaling. In Methods in Enzymology, Vol. 527, pp 41–63, Elsevier, Amsterdam. [DOI] [PubMed] [Google Scholar]
- Wood Z. A.; Poole L. B.; Karplus P. A. (2003) Peroxiredoxin Evolution and the Regulation of Hydrogen Peroxide Signaling. Science 300, 650–653. [DOI] [PubMed] [Google Scholar]
- Day A. M.; Brown J. D.; Taylor S. R.; Rand J. D.; Morgan B. A.; Veal E. A. (2012) Inactivation of a Peroxiredoxin by Hydrogen Peroxide Is Critical for Thioredoxin-Mediated Repair of Oxidized Proteins and Cell Survival. Mol. Cell 45, 398–408. [DOI] [PubMed] [Google Scholar]