

Abstract
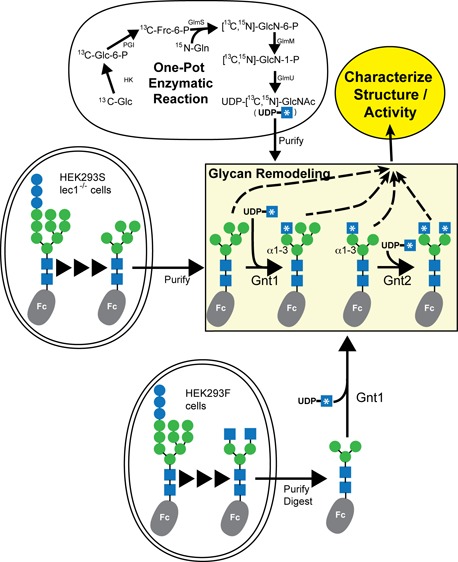
Asparagine-linked (N) glycosylation is a common eukaryotic protein modification that affects protein folding, function, and stability through intramolecular interactions between N-glycan and polypeptide residues. Attempts to characterize the structure–activity relationship of each N-glycan are hindered by inherent properties of the glycoprotein, including glycan conformational and compositional heterogeneity. These limitations can be addressed by using a combination of nuclear magnetic resonance techniques following enzymatic glycan remodeling to simultaneously generate homogeneous glycoforms. However, widely applicable methods do not yet exist. To address this technological gap, immature glycoforms of the immunoglobulin G1 fragment crystallizable (Fc) were isolated in a homogeneous state and enzymatically remodeled with [13C,15N]-N-acetylglucosamine (GlcNAc). UDP-[13C,15N]GlcNAc was synthesized enzymatically in a one-pot reaction from [13C]glucose and [15N-amido]glutamine. Modifying Fc with recombinantly expressed glycosyltransferases (Gnt1 and Gnt2) and UDP-[13C,15N]GlcNAc resulted in complete glycoform conversion as judged by mass spectrometry. Two-dimensional heteronuclear single-quantum coherence spectra of the Gnt1 product, containing a single [13C,15N]GlcNAc residue on each N-glycan, showed that the N-glycan is stabilized through interactions with polypeptide residues. Similar spectra of homogeneous glycoforms, halted at different points along the N-glycan remodeling pathway, revealed the presence of an increased level of interaction between the N-glycan and polypeptide at each step, including mannose trimming, as the N-glycan was converted to a complex-type, biantennary form. Thus, conformational restriction increases as Fc N-glycan maturation proceeds. Gnt1 and Gnt2 catalyze fundamental reactions in the synthesis of every glycoprotein with a complex-type N-glycan; thus, the strategies presented herein can be applied to a broad range of glycoprotein studies.
Protein asparagine-linked (N) glycosylation is a cotranslational event that confers a wide range of properties to the underlying polypeptide, including enhanced folding and stability, favorable pharmacokinetic properties, decoration with specific epitopes central to function (reviewed in ref (1)), and allosteric modulation of protein function.2 Many of these properties can be attributed to intramolecular interactions between N-glycan and polypeptide epitopes.3 Investigations of the structure–activity relationships of glycoprotein glycans must surmount two challenges: glycan compositional and conformational heterogeneity.
Unlike the template-dependent biosynthesis of nucleic acids or proteins, N-glycans are synthesized, ligated, and remodeled by glycosylhydrolases and glycosyltransferases that operate without a template. This complex biosynthesis generates significant compositional heterogeneity (reviewed in refs (1) and (4)). Immature N-glycans with a high mannose (Man) content are transferred to the nascent polypeptide chain during import into the lumen of the endoplasmic reticulum from lipid-linked donor molecules. For glycans destined to be complex-type, a single Man residue is removed followed by export to the Golgi, where three more Man residues are removed to form a Man5 structure (Figure 1). In the next step, an N-acetylglucosamine (GlcNAc) residue forms the base of the first N-glycan branch upon addition to the C2 hydroxyl of the Manα1–3 residue by the glycosyltransferase enzyme Gnt1. The final hydrolysis step removes two of the five remaining Man residues. Gnt2 then adds a GlcNAc residue to the C2 hydroxyl of the remaining Manα1–6 residue to form the base of a second branch. The steps that follow do not proceed to completion for every glycan, resulting in significant heterogeneity. The most common modifications are fucosylation, synthesis of additional GlcNAc branches, and transfer of galactose (Gal) and sialic acid residues. Lastly, the glycoprotein is exported to the cell surface where glycosyltransferase- or glycosylhydrolase-mediated modifications may occur.
Figure 1.

Native IgG1 Fc N-glycan processing in the Golgi. Conversions catalyzed by the enzymes indicated above the solid arrows and labeled with black type largely proceed to completion. Reactions catalyzed by enzymes denoted with gray type a dashed arrow modify some but not all of the secreted IgG1. Glycoforms studied here by nuclear magnetic resonance are underlined. Carbohydrate residues are numbered according to ref (30) and represented using the CFG convention and shown in the inset49 (GlcNAc, N-acetylglucosamine). Glycosidic linkages of the human IgG1 Fc N-glycan are indicated.
Glycoengineered proteins, including antibodies and antibody fragments, are of great interest because of the potential to enrich them with glycoforms with elevated therapeutic properties5 or to obtain homogeneous preparations for detailed studies. Many routes that show promising results have been reported, including, but not limited to, purely synthetic methods,6−8 enzymatic remodeling of the glycan termini in vitro with sugar nucleotides,9−12 approaches to enzymatically transfer a synthetic carbohydrate,13−16 and genetic manipulation of expression host organisms.17−24 Each method faces its unique challenges, and a single method for each situation has not been found.
N-Glycans are mobile moieties that often inhibit crystallization. Thus, N-glycans are enzymatically removed for studies of protein structure as a matter of routine. Solution nuclear magnetic resonance (NMR) spectroscopy offers a significant advantage in that proteins, highly concentrated in a buffered solution, are not required to crystallize and can be studied with complete carbohydrates (reviewed in ref (3)). Solution NMR spectroscopy offers another advantage over solid-state methods: the entire ensemble of interconverting conformations contributes to the observable signals. While X-ray crystallography is adept at capturing high-resolution images of low-energy conformations at low temperatures, NMR can capture subtle shifts in conformational equilibria. NMR methods for studying N-glycans often incorporate 13C-labeled sugars into N-glycans with glycosyltransferase enzymes after purification and have utilized terminal galactose and sialic acid transfer reactions,25−27 though an expression-based method to label the entire glycan is also available.28,29
This work describes a novel in vitro route to UDP-[13C,15N]GlcNAc and stepwise, site-specific labeling of the immunoglobulin G1 (IgG1) fragment crystallizable (Fc) N-glycan. One significant advantage of this approach is facile assignment of NMR resonances due to the stepwise labeling approach. This permits an assessment of N-glycan structures isolated at multiple crucial points along the N-glycan remodeling pathway as it would occur in the Golgi. The nomenclature used to describe carbohydrate residues of the Fc N-glycan is highlighted in Figure 1.30 For example, “(5′)GlcNAc” refers to the N-acetylglucosamine residue at the nonreducing end of the GlcNAcβ1–2Manα1–6Manβ1–4GlcNAcβ1–4GlcNAcβ-N moiety.
IgG1 is a critical defense protein that recognizes specific pathogen epitopes through tight binding, antigen recognition domains and triggers a pro-inflammatory, pathogen-destroying immune response through the Fc region.31 Interactions between IgG1 Fc and certain cell surface receptors, the low-affinity Fc γ receptors, require Fc N-glycosylation at Asn297 of the Fc Cγ2 domain.32,33 Transient, intramolecular interactions between the Fc N-glycan and polypeptide are central to this requirement,2,34,35 though the structure–activity relationship of this phenomenon remains undefined. This report covers new methods for addressing this relationship that are broadly applicable to structure–function studies of N-glycans.
Experimental Procedures
Materials
All materials were purchased from Sigma-Aldrich unless otherwise noted. Structure figures were prepared using PyMOL (Schrödinger LLC).
Protein Expression
An expression plasmid encoding the GlmS enzyme (glutamine:fructose-6-phosphate transaminase, EC 2.6.1.16) from Escherichia coli was prepared by amplifying and cloning the GlmS open reading frame from the pMA1 phagemid36 into the NcoI and XhoI restriction sites of the pET21d plasmid (Merck Millipore). The final cloned open reading frame encoded GlmS with N- and C-terminal tags: M+A+C2-E609+LEHHHHHH. Plasmid preparation was verified by DNA sequencing (Iowa State University DNA Facility). GlmS was expressed in transformed E. coli BL21star(DE3) cells carrying the GlmS:pET21d vector in the presence of ampicillin (50 mg/L). Expression was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) once the culture density reached an OD600 of 0.7, and cells were incubated for 20 h at 18 °C in an orbital shaking incubator. Cells were harvested in 50 mL aliquots by centrifugation; the spent medium was decanted and the pellet frozen and stored at −80 °C. Cells from a single frozen aliquot were lysed in 10 mL of 25 mM 4-morpholinepropanesulfonic acid (MOPS), 100 mM sodium chloride, 5 mM β-mercaptoethanol, and 1 mM ethylenediaminetetraacetic acid (EDTA) (pH 7.2), with four or five passages through an EmulsiFlex-C5 homogenizer (Avestin) operating at 15000 psi. Insoluble debris was removed by centrifugation at 25000g for 1 h. The clarified lysate was washed extensively in a 10 kDa cutoff Amicon centrifugal filter unit in the same buffer (without EDTA) to remove salts and concentrated to ∼1 mL. GlmS-containing washed cell lysate was prepared fresh for each reaction. GlmS proved to be unstable during purification, and removing the C-terminal six-His tag failed to improve stability.
A vector containing the GlmM (phosphoglucosamine mutase, EC 5.4.2.10) open reading frame from Bacillus anthracis cloned into the pDEST17 plasmid37 was transformed into E. coli BL21star(DE3) cells. Protein expression was induced after a culture grown at 37 °C in Luria-Bertani medium reached an OD600 of 0.6 with 0.5 mM IPTG. Cells were incubated for 20 h at 18 °C in an orbital shaking incubator and then separated from the growth medium by centrifugation. Cells were lysed using the homogenizer described above in a buffer containing 50 mM 2-amino-2-hydroxymethylpropane-1,3-diol (Tris), 200 mM sodium chloride, and 10 mM imidazole (pH 8.2) and then centrifuged at 25000g for 1 h to remove insoluble debris. Clarified lysate containing GlmM was loaded directly on a Ni2+-NTA column (Qiagen) using a Biologic LP chromatography system (Bio-Rad) and eluted with a linear gradient from 10 to 250 mM imidazole in the same buffer. Fractions containing GlmM were pooled, concentrated, and loaded on a Superdex 200 column (GE Healthcare) equilibrated with 25 mM Tris and 100 mM sodium chloride (pH 8.2). GlmM eluted as a sharp peak. Positive fractions were pooled and concentrated to 250 μM GlmM as judged by A280, diluted to 25% (v/v) glycerol, and frozen at −80 °C.
A vector containing the open reading frame encoding the bifunctional GlmU [glucosamime-1-phosphate-N-acetyltransferase (EC 2.3.1.157) and UDP-N-acetylglucosamine diphosphorylase (EC 2.7.7.23)] from E. coli cloned into the pET21b plasmid38 was transformed into E. coli BL21star(DE3) cells. Protein expression was performed largely as described from GlmM, except cells were lysed with a buffer containing 50 mM Tris, 500 mM sodium chloride, and 10 mM imidazole (pH 8.2); gel-filtration chromatography was performed in a buffer containing 10 mM Tris, 100 mM sodium chloride, and 5 mM β-mercaptoethanol (pH 8.2). GlmU eluted as a sharp peak. Positive fractions were pooled and concentrated to 190 μM GlmU as judged by A280, diluted to 25% (v/v) glycerol, and frozen at −80 °C.
IgG1 Fc was expressed in HEK293F and HEK293S(lec1–/–) cells and purified as previously described.2 The luminal domains of Gnt1 and Gnt2 were expressed as GFP fusions at the N-terminus using the pGen2 vector12 in HEK293F or HEK293S(lec1–/–) cells in the same manner. Crude expression medium was passed over a Ni2+-NTA column using gravity. Next, the column was washed with 12 column volumes of 50 mM Tris, 500 mM sodium chloride, and 30 mM imidazole (pH 8.0) and then eluted with the same buffer containing 250 mM imidazole. Fractions containing the desired protein were washed extensively with 50 mM 2-(N-morpholino)ethanesulfonic acid10 and 100 mM potassium chloride (pH 6.25), concentrated with a 10 kDa cutoff centrifugal filter to 25–40 μM protein as judged by A280, diluted to 50% (v/v) glycerol, and frozen at −80 °C.
One-Pot UDP-[13C,15N]GlcNAc Synthesis and Partial Purification
Buffer, small molecule, and protein components were combined in a single tube to final concentrations of 100 mM MOPS (pH 7.2), 100 mM sodium chloride, 5 mM magnesium chloride, 1 mM dithiothreitol, 1 mM α-d-glucose 1,6-bisphosphate, 5 mM d-[13CU]glucose, 10 mM adenosine triphosphate, 25 mM [15N-amido]glutamine, 10 mM acetyl-coenzyme A, 10 mM uridine triphosphate, 17 units of hexokinase (from Saccharomyces cerevisiae), 18 units of phosphoglucose isomerase (from S. cerevisiae), a 10% (v/v) final concentration of washed cell lysate containing recombinant EcGlmS, 4 μM BaGlmM, and 2.4 μM EcGlmU in water. The reaction mixture was incubated at room temperature for 24 h in a closed tube in the dark. Reaction purification was performed on the basis of a published method.39 Briefly, reaction products were applied to a diethylaminoethylene-cellulose column equilibrated in 10 mM triethylammonium bicarbonate (TEAB) (pH 7.0). The column was washed with 2 column volumes of 10 mM TEAB and 4 column volumes of 50 mM TEAB and eluted with 4 column volumes of 100 mM TEAB. Fractions from the 100 mM elution were pooled, lyophilized, resuspended in H2O, and lyophilized again to remove the remaining TEAB. The concentration of the purified material was determined by comparing 1H signals of the compound to that of an internal 0.5 mM 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) standard.
Preparing N-Man5 N-Glycans
IgG1 Fc (13 mg/mL) expressed in HEK293S(lec1–/–) cells was incubated with 50 mM MES (pH 6.25), 100 mM potassium chloride, 20 mM manganese chloride, 1 mM UDP-[13C,15N]GlcNAc, and 5 μM Gnt1 at room temperature for 24 h in the dark. The reaction mixture was exchanged into an NMR buffer [10 mM MOPS (pH 7.2), 100 mM potassium chloride, and 0.5 mM DSS in >98% deuterium oxide] using a 10 kDa cutoff centrifugal concentrator.
Preparing N1F and N2F N-Glycans
IgG1 Fc (14 mg/mL) expressed in HEK293F cells was incubated with 50 mM sodium citrate and 40 units of an N-acetylglucosaminidase (New England Biolabs) for 48 h at 37 °C. IgG1 Fc was then purified using a Protein A column as described previously.30 Next, a Gnt1-catalyzed reaction was performed as described above to incorporate a single [13C,15N]GlcNAc residue. The second residue was added using 6 mg/mL IgG1 Fc, 50 mM MES (pH 6.25), 100 mM potassium chloride, 20 mM manganese chloride, 1 mM UDP-[13C,15N]GlcNAc, and 4 μM Gnt2 at room temperature for 48 h in the dark. The reaction mixture was exchanged into the same NMR buffer described above using a 10 kDa cutoff centrifugal concentrator. Reactions were monitored by permethylation and matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) analysis at each step as previously described40 using a Voyager-DE PRO instrument (Applied Biosystems).
Endoglycosidase Treatments To Cleave N-Glycans
A pET:GFP-EndoF1 plasmid for expression in E. coli was provided by K. Moremen (University of Georgia, Athens, GA) and expressed and purified using standard protocols (Qiagen). Purified EndoF1 (10 μM) was added to 60 μM IgG1 Fc in a 50 mM phosphate buffer (pH 6.0) and incubated for 12 h at 37 °C.
NMR Spectroscopy
NMR spectra were recorded using 5 mm Shigemi NMR tubes in a spectrometer equipped with a cryogenically cooled probe and an Avance II console (Bruker) and operating at 50 °C and 16.4 T. Fc dimer concentrations were between 60 and 100 μM in a final volume of 300 μL. The pulse sequence for the 1H–13C heteronuclear single-quantum coherence (HSQC) spectra of Fc did not include a sensitivity enhancement element or coherence selection gradients to minimize the loss of broad peaks. Data were analyzed using Topspin (version 3.2), NMRviewJ (One Moon Scientific), and NMRPipe.41 Chemical shifts were referenced directly (1H) and indirectly (13C and 15N) to the internal DSS methyl peak at 0.07 ppm (1H).
Results and Discussion
One-Pot Synthesis of UDP-[13C,15N]-N-Acetylglucosamine
Synthesis of UDP-GlcNAc from glucose by eukaryotic and prokaryotic organisms proceeds along similar metabolic pathways; however, the prokaryotic system includes a bifunctional enzyme (GlmU) that catalyzes the last two steps (Figure 2A).42−44 Because of its relative simplicity, the prokaryotic pathway was recapitulated in vitro using a combination of commercially available and laboratory-expressed enzymes with off-the-shelf metabolites, including [13CU]glucose and [15N-amido]glutamine. Recovery of the starting [13CU]glucose in the form of UDP-[13C,15N]GlcNAc, following purification, was ∼18% (moles of UDP-[13C,15N]GlcNAc per mole of [13CU]glucose). A higher conversion would likely be achieved by increasing the concentrations of certain donor substrates, including [15N-amido]glutamine and acetyl-CoA; however, the cost of greater metabolite concentrations is unlikely to be offset by the increased yield. A 1 mL reaction mixture, starting with 1 mg of [13CU]glucose, produced 0.55 mg of UDP-[13C,15N]GlcNAc. This is enough UDP-[13C,15N]GlcNAc for the Gnt1-catalyzed labeling of at least 13 mg of IgG1 Fc [a typical sample for two-dimensional (2D) NMR analysis contains 1 mg of IgG1 Fc].
Figure 2.
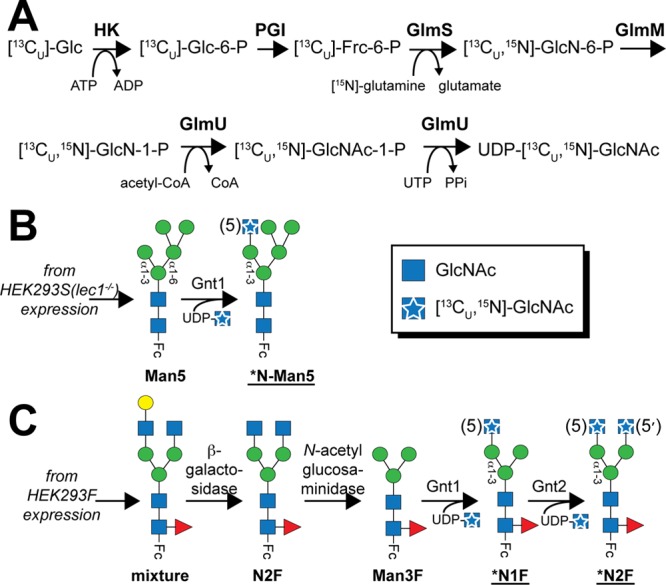
Schemes for in vitro enzymatic conversions described in this study. (A) A one-pot synthesis of UDP-[13C,15N]GlcNAc utilizes enzymes from bacterial pathways and [13C]glucose. Carbohydrate remodeling started with Fc bearing either a mannose-type (B) or a complex-type (C) N-glycan. [13C,15N]GlcNAc is shown as a blue square with a white star in the cartoon figures and by “*N” in the glycan name; residue numbers corresponding to the convention introduced in Figure 1 are given in parentheses.
A 1H–13C heteronuclear single-quantum coherence (HSQC) spectrum of the purified material revealed chemical shifts that were identical to database values for UDP-GlcNAc45(Figure S1 of the Supporting Information). A 1H–15N HSQC spectrum also showed a single, intense peak for the amide moiety (Figure S1B of the Supporting Information). This preparation proved to be sufficiently pure for glycosyltransferase-catalyzed sugar additions even though minor impurities were observed in the NMR spectra. This one-pot synthetic route permits an additional labeling strategy to incorporate a 13C-labeled acetate moiety from acetyl-CoA. Methyl groups have beneficial spin relaxation properties in magnetic resonance applications that permit measurements on very large (∼1 MDa) or dynamic systems.46,47 Furthermore, the synthesis of the methyl donor in this reaction, (13C-acetyl)-CoA, is easily achieved.48
Beyond the applications to N-glycans, UDP-[13C,15N]GlcNAc is a “gateway” nucleotide sugar that can be converted to many other products, too diverse to completely describe here. Potential immediate applications include using UDP-[13C,15N]GlcNAc as a substrate in glycosaminoglycan or sialic acid biosynthesis.49 Alternatively, a simple single enzyme-catalyzed epimerization leads to UDP-[13C,15N]-N-acetylgalactosamine50 that serves as the basis of eukaryotic O-GalNAc glycans (including mucins and many others) and is critical for glycosaminoglycan and glycosphingolipid biosynthesis.49
The enzymatic method for preparing UDP-GlcNAc described here offers marked benefits over previously described methods with respect to isotope labeling for NMR and MS-based studies. Other enzymatic methods for synthesizing UDP-GlcNAc that start from GlcNAc or GlcN have been described.51−55 However, isotope-labeled GlcNAc and GlcN can be cost prohibitive and are available with only limited labeling patterns, unlike the scheme presented here that can be used to produce a wide array of custom labeling patterns starting with inexpensive starting materials, including glucose and glutamine. Numerous chemical methods are also available and can be adapted for the synthesis of GlcNAc analogues; however, these methods are less efficient than the one-pot enzymatic method presented here (not limited to refs (56−58)).
Gnt1-Catalyzed Conversion of the Man5 N-Glycan
Addition of a GlcNAc residue in a β1–2 linkage to the (3)Man residue, catalyzed by Gnt1 (also known as lec1 or MGAT1), is a crucial step in hybrid and complex-type N-glycan maturation (Figure 1). A Gnt1-deficient HEK293 cell line (HEK293S or lec1–/–) halts this process and produces glycoproteins with nearly homogeneous Man5 N-glycans.59,60 Glycans on IgG1 Fc expressed using this cell line are thus auspicious substrates for investigating Gnt1 activity in vitro. As expected on the basis of published reports,61,62 MS analysis of IgG1 Fc incubated with UDP-GlcNAc and Gnt1 revealed complete conversion of Fc-Man5 to the Fc-N-Man5 glycoform (Figure 3; experimental mass of 1825.3 Da, observed mass of 1824.9 Da).
Figure 3.
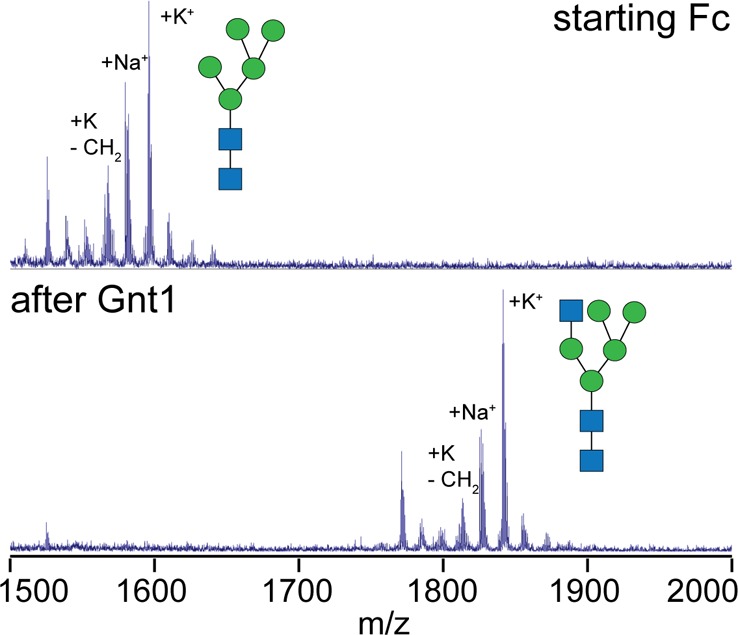
Gnt1-catalyzed remodeling of lec1–/–-expressed IgG1 Fc with a Man5 N-glycan as monitored by MALDI-MS. Enzymatic remodeling occurs when the N-glycans are attached to the Fc polypeptide; however, the analysis shown here includes N-glycan removal followed by permethylation.
Enzymatic Conversion to a Complex-Type Fc N-Glycan
HEK293F cells, unlike the HEK293S (lec1–/–) cells, have the capacity to generate complex-type polysaccharides and express Fc with a fucosylated biantennary N-glycan that varies with respect to the amount of terminal galactose incorporated (Figure 4). Exoglycosidase treatment removed terminal sugars and resulted in Fc with a primarily Man3F glycan (Figure 4; experimental mass of 1345.7 Da, observed mass of 1345.5). This material was then used as a substrate for modification by Gnt1, the result being Fc containing the (5)-[13C,15N]GlcNAc residue (*N1F; Figures 2C and 4; experimental mass of 1597.8 Da, observed mass of 1597.3 Da). The fact that Gnt1 could recognize a Man3F substrate was unknown; Gnt1 was shown to modify a polysaccharide with an identical display of three Man residues tethered through the β-Man to a GlcNAc,61,62 but because the Fut8 core fucosyltransferase modifies N-glycans only after Gnt1 in vivo(63)(Figure 1), it was unknown if the presence of the (0)fucose(Fuc) residue would prevent this modification in vitro.
Figure 4.

Generation of *N1F and *N2F Fc glycoforms from HEK293F-expressed Fc was confirmed using MALDI-MS analysis. These steps are shown in Figure 2C. Enzymatic remodeling occurs when the N-glycans are attached to the Fc polypeptide; however, the analysis shown here includes N-glycan removal followed by permethylation.
The *N2F glycoform was prepared from the *N1F material following a reaction catalyzed by Gnt2, which likewise forms a GlcNAcβ1–2Man linkage, except Gnt2 modifies the (3′)Man residue rather than the (3)Man modified by Gnt1 (Figures 2C and 4; experimental mass of 1850.0 Da, observed mass of 1850.1 Da). On the basis of MALDI-MS analysis, the Gnt1- and Gnt2-catalyzed reactions proceeded nearly to completion [>95% conversion (Figure 4)].
This approach represents a significant advance to in vitro enzymatic N-glycan remodeling. In contrast to the methods described in the introductory section, this method rebuilds N-glycans from a paucimannose (Man3) core N-glycan that is present in all eukaryotic N-glycans and permits incorporation of 13C or 15N labels at each step. Thus, it is expected that all eukaryotic N-glycans could be remodeled in this manner with suitable exoglycosidases, many of which are commercially available. The robust nature of this approach is reflected in the high conversion of the IgG1 Fc glycoprotein, a notably difficult protein to remodel enzymatically.11,12,64 Furthermore, this method utilizes commercially available sugar nucleotides (if stable isotope-enriched sugars are not required) in place of synthetic oligosaccharide precursors for the case of transglycosylation (see ref (64)).
NMR Analysis of [13C,15N]GlcNAc-Labeled Fc Glycoforms
NMR spectra of IgG1 Fc following Gnt1-catalyzed remodeling of the Man5 glycan using UDP-[13C,15N]GlcNAc revealed peaks for each 1H–13C and 1H–15N moiety (Figure 5). The peaks were relatively intense and narrow, considering the glycans are part of the ∼55 kDa Fc. This property indicates the presence of significant GlcNAc motion relative to the polypeptide domains and is consistent with similar measurements of galactose and sialic acid residues on the 3–4–5 branch of the N-glycan [as opposed to the 3′–4′–5′ branch (Figure 1)].12,25,341JC–C couplings from a one-dimensional (1D) 13C-observe experiment agreed with the resonance assignments based on an assignment of β-GlcNAc (Figure 5B). Similar spectra were observed at 25, 37, and 50 °C, and peak positions in duplicate samples were reproduced.
Figure 5.

1H–13C HSQC spectra of IgG1 Fc with a Man5 N-glycan following addition of [13C,15N]GlcNAc. (A) A 2D 1H–13C HSQC spectrum of the *N-Man5 N-glycan following EndoF1-catalyzed hydrolysis is shown as gray contours. Blue contours show the positions of peaks from IgG1 Fc bearing a *N-Man5 N-glycan. Arrows indicate the direction of peak movement because of interactions with the Fc polypeptide. Peak labels that correspond to a figure of β-linked GlcNAc are shown (inset) and refer to the carbon position of the 1H–13C peak. 1JC–C couplings are not resolved because of the limited resolution in the 13C dimension. (B) 1D 13C-observe NMR spectrum of *N-Man5 Fc. 1JC–C values are indicated. (C) 2D 1H–15N HSQC spectra before and after N-glycan hydrolysis with the same colors used in panel A.
The Fc polypeptide influences the position and line shapes of GlcNAc peaks. Endoglycosidase F1 treatment hydrolyzed the glycosidic linkage between the (1)GlcNAc and (2)GlcNAc residues, releasing the glycan from its covalent attachment to Fc. As a result, resonances of the released N-glycan were different from those of the Fc-conjugated material [average Δ1H = 0.098 ppm (Figure 5)], even though the Fc polypeptide was still present in the experiment. It was previously shown that the N-glycan termini (residues 6 and 6′) exchange between a restricted and free state on a microsecond time scale, which results in a single observable peak in NMR spectra that represents the population-weighted average of the two states.34 Mutating the polypeptide surface to prevent interaction resulted in the predominance of the free state.2 On the basis of these reports, it is likely that peaks corresponding to the (5)GlcNAc residue also represent the population-weighted average of two distinct states. Thus, the farther the peak is shifted from that of the free state (seen with the hydrolyzed N-glycan), the greater the restriction mediated by intramolecular interactions between N-glycan and polypeptide residues. It is not clear how the Fc polypeptide influences the frequencies of (5)GlcNAc resonances in the restricted state, though electric field effects likely contribute.65 Multiple charged surface residues lie in the proximity of the (5)GlcNAc residue, including lysine 334, which is within 8 Å (Figure S2 of the Supporting Information).
A 2D 1H–13C spectrum of the *N1F glycoform reveals a pattern of peaks similar to that observed with *N-Man5 (Figure 6, red and blue contours, respectively). A spectrum of the *N2F glycoform was likewise similar, but not identical, with respect to the position of peaks corresponding to the (5)GlcNAc residue (Figures 6 and 7, black contour). As the glycoform advanced from *N-Man5 to *N1F to *N2F, the deviation of the peak positions as compared to the released glycan (shown with an “X” in Figure 6) likewise increased. A small shift in peak positions was observed between spectra of *N-Man5 and *N1F (average Δ1H = 0.016 ppm), though a larger shift was seen between *N1F and *N2F (average Δ1H = 0.034 ppm). This indicates the (5′)GlcNAc residue (*N1F → *N2F) had a greater effect on resonance frequency than removing the two Man residues [*N-Man5 → *N1F (see Figure 6)]. Changes in resonance frequencies of the (5)GlcNAc residue likely reflect changes in structure of the N-glycan. Peaks observed with the *N-Man5, *N1F, and *N2F Fc glycoforms shifted away, in a stepwise manner, from peaks observed in spectra of a hydrolyzed N-glycan. This shift indicated that N-glycan conformation becomes more restricted as maturation, mediated by glycan-modifying enzymes in the Golgi, proceeds. This conclusion is supported by previous work showing similar directions of chemical shift changes for galactose and sialic acid resonances upon temperature changes.12,34
Figure 6.
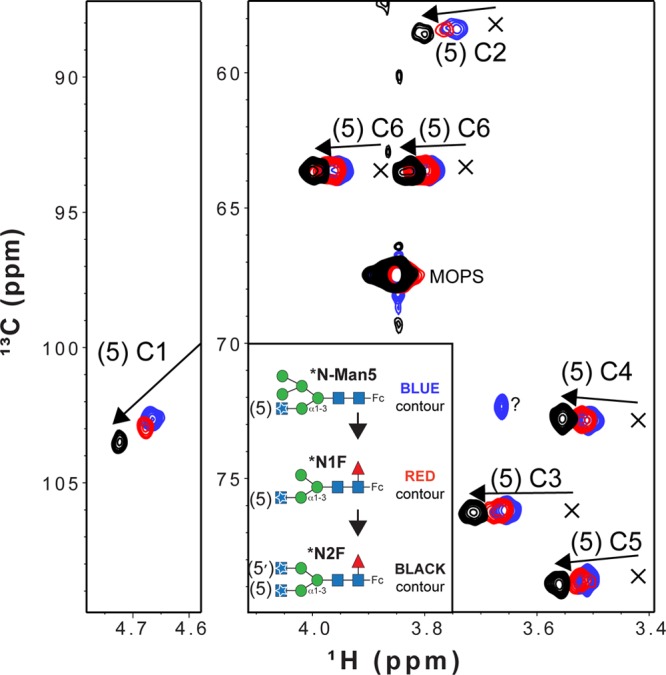
Overlay of 1H–13C HSQC spectra collected with purified IgG1 Fc glycoforms that reveals shifts of peaks away from that of a hydrolyzed N-glycan. The positions of peaks from an Fc N-glycan following EndoF-catalyzed hydrolysis are indicated with X’s. Arrows show the direction of peak movement as the N-glycan matures. The C1 peak for the hydrolyzed glycan was obscured by residual water in the sample and was not observed.
Figure 7.
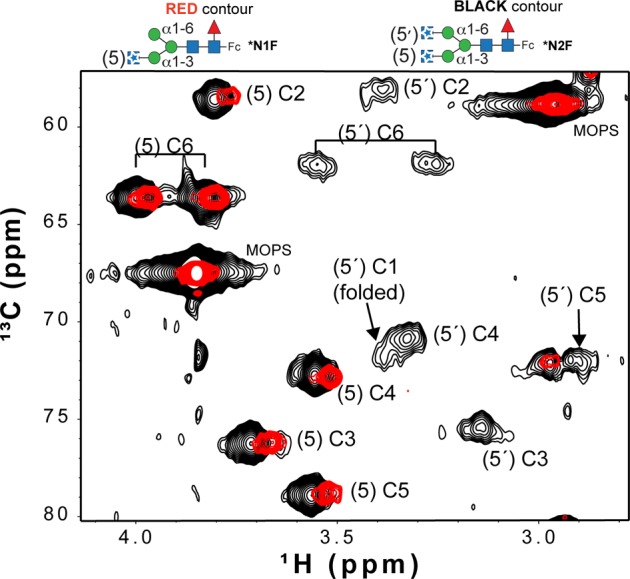
Broad, low-intensity peaks appear in a 1H–13C HSQC spectrum following Gnt2-catalyzed addition of a second [13C,15N]GlcNAc. The resonance assignments for the (5′)GlcNAc C3, C4, and C5 peaks were obtained by comparison to a report by Yamaguchi et al.28
Peak line widths also reflected changes in the N-glycan. NMR line widths are a direct reflection of transverse relaxation rates of nuclear orientations in an NMR experiment. These values decrease with the tumbling rate of the nucleus and can also be increased by conformational exchange (Rex) on the microsecond to millisecond time scale (contributions from magnetic field inhomogeneity and chemical shift anisotropy likewise increase line widths but are not considered here).66 Line widths of (5)GlcNAc resonances from the 1H–13C HSQC spectra, measured without applying a line-broadening function during processing, increased following attachment of the N-glycan to Fc and indicated structural differences between the different glycoforms. The line width of (5)GlcNAc H6, after removing the effect of 2&3JHH coupling, showed a representative response and increased from 7 Hz (hydrolyzed *N-Man5) to 14 Hz (*N-Man5-Fc) to 19 Hz (*N1F-Fc) to 14 Hz (*N2F-Fc) as shown in Figure S3 of the Supporting Information. These values are much smaller than the expected value of 35 Hz for the same nucleus at the same magnetic field strength tumbling with an apparent molecular mass of 55 kDa. The value of 35 Hz is based on contributions of nearby nuclei to the line width and excludes effects of chemical shift anisotropy (CSA) and Rex.66 It is unclear why the values measured from the *N1F glycoform are larger than those of the *N-Man5 and *N2F forms, but this may reflect changes in exchange rates.
Interactions between the N-glycan and polypeptide surface can explain both the peak position and line width differences in the spectra of Fc glycoforms. Phe241 is located adjacent to the β-Man residue in Fc models determined by X-ray crystallography67,68(Figure 8) and was shown to restrict N-glycan motion.2 Covalent attachment of the N-glycan through Asn297 places the glycan in a prime position to make this crucial contact. A smaller shift is observed between the *N-Man5 and *N1F glycoforms as two Man residues are removed and a (0)Fuc is added (Figure 1). The Fuc is not expected to influence movement of the N-glycan branch termini greatly because it does not appear to interact with the Fc polypeptide.69 Man residues interfere with contacts along the polypeptide surface and likely result in a relatively small stabilization upon removal. Bowden et al.70 observed the C2 hydroxyl of the Man residue at the nonreducing terminus of the Manα1–6Manα1–6Manβ moiety in the Man5 N-glycan (see Figure 1) was positioned to prevent an interaction with F243 and shifted the entire N-glycan away from the polypeptide surface.
Figure 8.
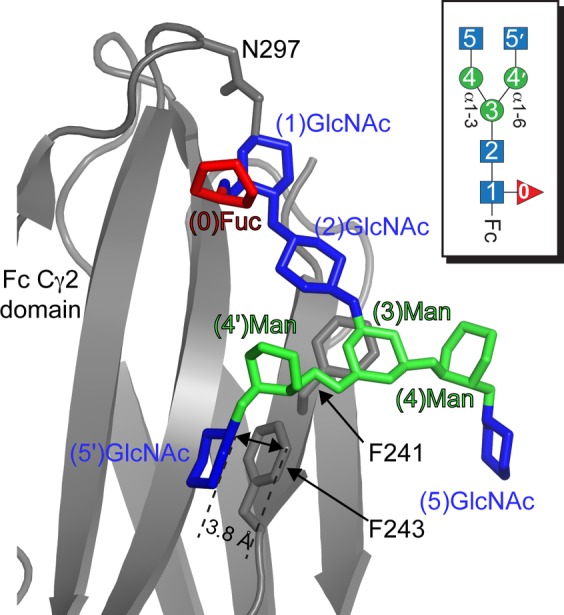
Structural model of the IgG1 Fc–N-glycan interface (based on Protein Data Bank entry 4ku1(71)). F243 directly contacts the (5′)GlcNAc residue. The (5)GlcNAc does not appear to make a direct contact with the polypeptide surface. Carbohydrate residues are shown in a stick model with the color of each residue corresponding to the key for each residue shown in Figure 1. Only ring atoms of the carbohydrates are shown for the sake of simplicity, and galactose residues present in the original Protein Data Bank model are not shown for the sake of clarity.
The (5′)GlcNAc, however, can adopt a conformation suitable for contact with F2432,67,68 (Figure 8). This is supported by the relatively greater shift of (5)GlcNAc peaks following Gnt2-catalyzed glycan modification, and in the appearance of the (5′)GlcNAc resonance that appeared to be broad and weak (1H6 line width of ∼67 Hz) >0.5 ppm (1H) upfield of the (5)GlcNAc peaks (Figure 7). A 1H–15N HSQC spectrum of the *N2F glycoform likewise shows two distinct peaks corresponding to acetamide moieties of the 5 and 5′ [13C,15N]GlcNAc residues (Figure S4 of the Supporting Information). This dramatic shift of all (5′)GlcNAc resonances is consistent with close contact of the (5′)GlcNAc residue with the polypeptide for at least some period of time. NMR spectra of the (6′)-13C-Gal residue of Fc with a G2F glycoform (Figure 1) showed similar peak displacements and reduced intensities, which was due to a transient interaction with the polypeptide surface.34 Furthermore, line widths and positions of these peaks from Fc with double F241S and F243S mutations overlapped with an N-glycan on a trypsinized Fc glycopeptide.2
Summary
A one-pot, purely enzymatic method was used to produce UDP-[13C,15N]GlcNAc efficiently from [13C]glucose and [15N]glutamine. IgG1 Fc N-glycans were enzymatically remodeled from a natural immature form and an unnatural core N-glycan, common to all eukaryotic N-glycans, to high conversion using UDP-[13C,15N]GlcNAc. NMR spectra recorded at different points along the N-glycan remodeling pathway reveal enhanced interactions of the carbohydrate with the polypeptide surface. These small changes in equilibria cannot be observed by X-ray crystallography. These methods are broadly applicable beyond IgG1 Fc and provide spectroscopic probes for characterizing N-glycan structure and motion.
Acknowledgments
I am indebted to Dr. Kelley Moremen (University of Georgia) for providing the Gnt1:pGen2, Gnt2:pGen2, and EndoF1 expression plasmids, Dr. Marie-Ange Badet-Denisot (ICSN-CNRS) for providing the pMA1 phagemid, Dr. Lesa J. Beamer (University of Missouri, Columbia, MO) for providing the BaGlmM:pDEST17 plasmid, and Dr. Jian Liu (University of North Carolina at Chapel Hill, Chapel Hill, NC) for providing the EcGlmU:pET21b plasmid. I also thank Dr. Bruce Fulton (Iowa State University) for help with the NMR instrumentation and Mr. Quinlin M. Hanson for preparing the EndoF1 enzyme.
Supporting Information Available
2D 1H–13C and 1H–15N HSQC spectra of UDP-α-d-[13C,15N]GlcNAc, Fc model showing the location of charged residues, representative line shapes and peak information extracted from 2D NMR measurements, and a 1H–15N HSQC-TROSY spectrum of (15N-Y;2×[13C,15N]-GlcNAc)-IgG1 Fc (*N2F glycoform). This material is available free of charge via the Internet at http://pubs.acs.org.
This work was financially supported by National Institutes of Health Grant K22AI099165 and by funds from the Roy J. Carver Department of Biochemistry, Biophysics and Molecular Biology at Iowa State University.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Moremen K. W.; Tiemeyer M.; Nairn A. V. (2012) Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subedi G. P.; Hanson Q. M.; Barb A. W. (2014) Restricted motion of the conserved immunoglobulin G1 N-glycan is essential for efficient FcγRIIIa binding. Structure 22, 1478–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barb A. W.; Borgert A. J.; Liu M.; Barany G.; Live D. (2010) Intramolecular Glycan-Protein Interactions in Glycoproteins. Methods Enzymol. 477, 365–388. [DOI] [PubMed] [Google Scholar]
- Aebi M. (2013) N-linked protein glycosylation in the ER. Biochim. Biophys. Acta 1833, 2430–2437. [DOI] [PubMed] [Google Scholar]
- Anthony R. M.; Nimmerjahn F.; Ashline D. J.; Reinhold V. N.; Paulson J. C.; Ravetch J. V. (2008) Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science 320, 373–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.; Dong S.; Shieh J. H.; Peguero E.; Hendrickson R.; Moore M. A.; Danishefsky S. J. (2013) Erythropoietin derived by chemical synthesis. Science 342, 1357–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne R. J.; Wong C. H. (2010) Advances in chemical ligation strategies for the synthesis of glycopeptides and glycoproteins. Chem. Commun. 46, 21–43. [DOI] [PubMed] [Google Scholar]
- Yamamoto N.; Tanabe Y.; Okamoto R.; Dawson P. E.; Kajihara Y. (2008) Chemical synthesis of a glycoprotein having an intact human complex-type sialyloligosaccharide under the Boc and Fmoc synthetic strategies. J. Am. Chem. Soc. 130, 501–510. [DOI] [PubMed] [Google Scholar]
- Raju T. S.; Briggs J. B.; Chamow S. M.; Winkler M. E.; Jones A. J. (2001) Glycoengineering of therapeutic glycoproteins: In vitro galactosylation and sialylation of glycoproteins with terminal N-acetylglucosamine and galactose residues. Biochemistry 40, 8868–8876. [DOI] [PubMed] [Google Scholar]
- Hodoniczky J.; Zheng Y. Z.; James D. C. (2005) Control of recombinant monoclonal antibody effector functions by Fc N-glycan remodeling in vitro. Biotechnol. Prog. 21, 1644–1652. [DOI] [PubMed] [Google Scholar]
- Kobata A. (2008) The N-linked sugar chains of human immunoglobulin G: Their unique pattern, and their functional roles. Biochim. Biophys. Acta 1780, 472–478. [DOI] [PubMed] [Google Scholar]
- Barb A. W.; Meng L.; Gao Z.; Johnson R. W.; Moremen K. W.; Prestegard J. H. (2012) NMR characterization of immunoglobulin G Fc glycan motion on enzymatic sialylation. Biochemistry 51, 4618–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou G.; Ochiai H.; Huang W.; Yang Q.; Li C.; Wang L. X. (2011) Chemoenzymatic synthesis and Fcγ receptor binding of homogeneous glycoforms of antibody Fc domain. Presence of a bisecting sugar moiety enhances the affinity of Fc to FcγIIIa receptor. J. Am. Chem. Soc. 133, 18975–18991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz F.; Huang W.; Li C.; Schulz B. L.; Lizak C.; Palumbo A.; Numao S.; Neri D.; Aebi M.; Wang L. X. (2010) A combined method for producing homogeneous glycoproteins with eukaryotic N-glycosylation. Nat. Chem. Biol. 6, 264–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Giddens J.; Fan S. Q.; Toonstra C.; Wang L. X. (2012) Chemoenzymatic glycoengineering of intact IgG antibodies for gain of functions. J. Am. Chem. Soc. 134, 12308–12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E. L.; Giddens J. P.; Iavarone A. T.; Godula K.; Wang L. X.; Bertozzi C. R. (2014) Chemoenzymatic Fc glycosylation via engineered aldehyde tags. Bioconjugate Chem. 25, 788–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umana P.; Jean-Mairet J.; Moudry R.; Amstutz H.; Bailey J. E. (1999) Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat. Biotechnol. 17, 176–180. [DOI] [PubMed] [Google Scholar]
- Yamane-Ohnuki N.; Kinoshita S.; Inoue-Urakubo M.; Kusunoki M.; Iida S.; Nakano R.; Wakitani M.; Niwa R.; Sakurada M.; Uchida K.; Shitara K.; Satoh M. (2004) Establishment of FUT8 knockout Chinese hamster ovary cells: An ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol. Bioeng. 87, 614–622. [DOI] [PubMed] [Google Scholar]
- Cox K. M.; Sterling J. D.; Regan J. T.; Gasdaska J. R.; Frantz K. K.; Peele C. G.; Black A.; Passmore D.; Moldovan-Loomis C.; Srinivasan M.; Cuison S.; Cardarelli P. M.; Dickey L. F. (2006) Glycan optimization of a human monoclonal antibody in the aquatic plant Lemna minor. Nat. Biotechnol. 24, 1591–1597. [DOI] [PubMed] [Google Scholar]
- Li H.; Sethuraman N.; Stadheim T. A.; Zha D.; Prinz B.; Ballew N.; Bobrowicz P.; Choi B. K.; Cook W. J.; Cukan M.; Houston-Cummings N. R.; Davidson R.; Gong B.; Hamilton S. R.; Hoopes J. P.; Jiang Y.; Kim N.; Mansfield R.; Nett J. H.; Rios S.; Strawbridge R.; Wildt S.; Gerngross T. U. (2006) Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nat. Biotechnol. 24, 210–215. [DOI] [PubMed] [Google Scholar]
- Palmberger D.; Wilson I. B.; Berger I.; Grabherr R.; Rendic D. (2012) SweetBac: A new approach for the production of mammalianised glycoproteins in insect cells. PLoS One 7, e34226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis D. L. (2009) Baculovirus-insect cell expression systems. Methods Enzymol. 463, 191–222. [DOI] [PubMed] [Google Scholar]
- Loos A.; Gruber C.; Altmann F.; Mehofer U.; Hensel F.; Grandits M.; Oostenbrink C.; Stadlmayr G.; Furtmuller P. G.; Steinkellner H. (2014) Expression and glycoengineering of functionally active heteromultimeric IgM in plants. Proc. Natl. Acad. Sci. U.S.A. 111, 6263–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth A. M.; Kuo C. W.; Khoo K. H.; Jarvis D. L. (2014) A new insect cell glycoengineering approach provides baculovirus-inducible glycogene expression and increases human-type glycosylation efficiency. J. Biotechnol. 182–183, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y.; Kato K.; Shindo M.; Aoki S.; Furusho K.; Koga K.; Takahashi N.; Arata Y.; Shimada I. (1998) Dynamics of the carbohydrate chains attached to the Fc portion of immunoglobulin G as studied by NMR spectroscopy assisted by selective 13C labeling of the glycans. J. Biomol. NMR 12, 385–394. [DOI] [PubMed] [Google Scholar]
- Macnaughtan M. A.; Tian F.; Liu S.; Meng L.; Park S.; Azadi P.; Moremen K. W.; Prestegard J. H. (2008) 13C-sialic acid labeling of glycans on glycoproteins using ST6Gal-I. J. Am. Chem. Soc. 130, 11864–11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya Y.; Satoh T.; Kato K. (2014) Recent advances in glycoprotein production for structural biology: Toward tailored design of glycoforms. Curr. Opin. Struct. Biol. 26C, 44–53. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y.; Walchli M.; Nagano M.; Kato K. (2009) A 13C-detection NMR approach for large glycoproteins. Carbohydr. Res. 344, 535–538. [DOI] [PubMed] [Google Scholar]
- Kamiya Y.; Yanagi K.; Kitajima T.; Yamaguchi T.; Chiba Y.; Kato K. (2013) Application of Metabolic 13C Labeling in Conjunction with High-Field Nuclear Magnetic Resonance Spectroscopy for Comparative Conformational Analysis of High Mannose-Type Oligosaccharides. Biomolecules 3, 108–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barb A. W.; Brady E. K.; Prestegard J. H. (2009) Branch-specific sialylation of IgG-Fc glycans by ST6Gal-I. Biochemistry 48, 9705–9707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K., Travers P., Walport M., and Janeway C. (2012) Janeway’s Immunobiology, 8th ed., Garland Science, New York. [Google Scholar]
- Lux A.; Yu X.; Scanlan C. N.; Nimmerjahn F. (2013) Impact of immune complex size and glycosylation on IgG binding to human FcγRs. J. Immunol. 190, 4315–4323. [DOI] [PubMed] [Google Scholar]
- Jefferis R. (2009) Recombinant antibody therapeutics: The impact of glycosylation on mechanisms of action. Trends Pharmacol. Sci. 30, 356–362. [DOI] [PubMed] [Google Scholar]
- Barb A. W.; Prestegard J. H. (2011) NMR analysis demonstrates immunoglobulin G N-glycans are accessible and dynamic. Nat. Chem. Biol. 7, 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Baruah K.; Harvey D. J.; Vasiljevic S.; Alonzi D. S.; Song B. D.; Higgins M. K.; Bowden T. A.; Scanlan C. N.; Crispin M. (2013) Engineering hydrophobic protein-carbohydrate interactions to fine-tune monoclonal antibodies. J. Am. Chem. Soc. 135, 9723–9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obmolova G.; Badet-Denisot M. A.; Badet B.; Teplyakov A. (1994) Crystallization and preliminary X-ray analysis of the two domains of glucosamine-6-phosphate synthase from Escherichia coli. J. Mol. Biol. 242, 703–705. [DOI] [PubMed] [Google Scholar]
- Mehra-Chaudhary R.; Neace C. E.; Beamer L. J. (2009) Crystallization and initial crystallographic analysis of phosphoglucosamine mutase from Bacillus anthracis. Acta Crystallogr. F65, 733–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.; Bridges A.; Liu J. (2006) Determination of the substrate specificities of N-acetyl-d-glucosaminyltransferase. Biochemistry 45, 12358–12365. [DOI] [PubMed] [Google Scholar]
- Kelly T. M.; Stachula S. A.; Raetz C. R.; Anderson M. S. (1993) The firA gene of Escherichia coli encodes UDP-3-O-(R-3-hydroxymyristoyl)-glucosamine N-acyltransferase. The third step of endotoxin biosynthesis. J. Biol. Chem. 268, 19866–19874. [PubMed] [Google Scholar]
- Anumula K. R.; Taylor P. B. (1992) A Comprehensive Procedure for Preparation of Partially Methylated Alditol Acetates from Glycoprotein Carbohydrates. Anal. Biochem. 203, 101–108. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Mengin-Lecreulx D.; van Heijenoort J. (1993) Identification of the glmU gene encoding N-acetylglucosamine-1-phosphate uridyltransferase in Escherichia coli. J. Bacteriol. 175, 6150–6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengin-Lecreulx D.; van Heijenoort J. (1994) Copurification of glucosamine-1-phosphate acetyltransferase and N-acetylglucosamine-1-phosphate uridyltransferase activities of Escherichia coli: Characterization of the glmU gene product as a bifunctional enzyme catalyzing two subsequent steps in the pathway for UDP-N-acetylglucosamine synthesis. J. Bacteriol. 176, 5788–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooday B. W. (1977) Biosynthesis of the fungal wall: Mechanisms and implications. The first Fleming Lecture. J. Gen. Microbiol. 99, 1–11. [DOI] [PubMed] [Google Scholar]
- Ulrich E. L.; Akutsu H.; Doreleijers J. F.; Harano Y.; Ioannidis Y. E.; Lin J.; Livny M.; Mading S.; Maziuk D.; Miller Z.; Nakatani E.; Schulte C. F.; Tolmie D. E.; Kent Wenger R.; Yao H.; Markley J. L. (2008) BioMagResBank. Nucleic Acids Res. 36, D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay L. E.; Prestegard J. H. (1987) Methyl-Group Dynamics from Relaxation of Double Quantum Filtered NMR Signals: Application to Deoxycholate. J. Am. Chem. Soc. 109, 3829–3835. [Google Scholar]
- Kay L. E. (2011) Solution NMR spectroscopy of supra-molecular systems, why bother? A methyl-TROSY view. J. Magn. Reson. 210, 159–170. [DOI] [PubMed] [Google Scholar]
- Wilson I. B. (1952) Preparation of Acetyl Coenzyme-A. J. Am. Chem. Soc. 74, 3205–3206. [Google Scholar]
- Varki A. (2009) Essentials of glycobiology, 2nd ed., Cold Spring Harbor Laboratory Press, Plainview, NY. [PubMed] [Google Scholar]
- Creuzenet C.; Belanger M.; Wakarchuk W. W.; Lam J. S. (2000) Expression, purification, and biochemical characterization of WbpP, a new UDP-GlcNAc C4 epimerase from Pseudomonas aeruginosa serotype O6. J. Biol. Chem. 275, 19060–19067. [DOI] [PubMed] [Google Scholar]
- Zhao G.; Guan W.; Cai L.; Wang P. G. (2010) Enzymatic route to preparative-scale synthesis of UDP-GlcNAc/GalNAc, their analogues and GDP-fucose. Nat. Protoc. 5, 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Fan L.; Wei P.; Huang L.; Cai J.; Xu Z. (2010) Efficient production of uridine 5′-diphospho-N-acetylglucosamine by the combination of three recombinant enzymes and yeast cells. Prep. Biochem. Biotechnol. 40, 294–304. [DOI] [PubMed] [Google Scholar]
- Okuyama K.; Hamamoto T.; Ishige K.; Takenouchi K.; Noguchi T. (2000) An efficient method for production of uridine 5′-diphospho-N-acetylglucosamine. Biosci., Biotechnol., Biochem. 64, 386–392. [DOI] [PubMed] [Google Scholar]
- Inoue K.; Nishimoto M.; Kitaoka M. (2011) One-pot enzymatic production of 2-acetamido-2-deoxy-d-galactose (GalNAc) from 2-acetamido-2-deoxy-d-glucose (GlcNAc). Carbohydr. Res. 346, 2432–2436. [DOI] [PubMed] [Google Scholar]
- Zhai Y.; Liang M.; Fang J.; Wang X.; Guan W.; Liu X. W.; Wang P.; Wang F. (2012) NahK/GlmU fusion enzyme: Characterization and one-step enzymatic synthesis of UDP-N-acetylglucosamine. Biotechnol. Lett. 34, 1321–1326. [DOI] [PubMed] [Google Scholar]
- Rao A. K.; Mendicino J. (1978) Synthesis of UDP-N-[1-14C]acetyl d-glucosamine and UDP-N-[1-14C]acetyl-d-galactosamine from [1-14C]acetate. Anal. Biochem. 91, 490–495. [DOI] [PubMed] [Google Scholar]
- Masuko S.; Bera S.; Green D. E.; Weiwer M.; Liu J.; DeAngelis P. L.; Linhardt R. J. (2012) Chemoenzymatic synthesis of uridine diphosphate-GlcNAc and uridine diphosphate-GalNAc analogs for the preparation of unnatural glycosaminoglycans. J. Org. Chem. 77, 1449–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner G. K.; Pesnot T.; Field R. A. (2009) A survey of chemical methods for sugar-nucleotide synthesis. Nat. Prod. Rep. 26, 1172–1194. [DOI] [PubMed] [Google Scholar]
- Reeves P. J.; Callewaert N.; Contreras R.; Khorana H. G. (2002) Structure and function in rhodopsin: High-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc. Natl. Acad. Sci. U.S.A. 99, 13419–13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves P. J.; Kim J. M.; Khorana H. G. (2002) Structure and function in rhodopsin: A tetracycline-inducible system in stable mammalian cell lines for high-level expression of opsin mutants. Proc. Natl. Acad. Sci. U.S.A. 99, 13413–13418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheimer C. L.; Hill R. L. (1981) Purification and characterization of a rabbit liver α1–3 mannoside β1–2 N-acetylglucosaminyltransferase. J. Biol. Chem. 256, 799–804. [PubMed] [Google Scholar]
- Fujiyama K.; Ido Y.; Misaki R.; Moran D. G.; Yanagihara I.; Honda T.; Nishimura S.; Yoshida T.; Seki T. (2001) Human N-acetylglucosaminyltransferase I. Expression in Escherichia coli as a soluble enzyme, and application as an immobilized enzyme for the chemoenzymatic synthesis of N-linked oligosaccharides. J. Biosci. Bioeng. 92, 569–574. [DOI] [PubMed] [Google Scholar]
- Longmore G. D.; Schachter H. (1982) Product-identification and substrate-specificity studies of the GDP-l-fucose:2-acetamido-2-deoxy-β-d-glucoside (FUC goes to Asn-linked GlcNAc) 6-α-l-fucosyltransferase in a Golgi-rich fraction from porcine liver. Carbohydr. Res. 100, 365–392. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Li C.; Huang W.; Li B.; Strome S.; Wang L. X. (2008) Glycoengineering of human IgG1-Fc through combined yeast expression and in vitro chemoenzymatic glycosylation. Biochemistry 47, 10294–10304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hass M. A.; Jensen M. R.; Led J. J. (2008) Probing electric fields in proteins in solution by NMR spectroscopy. Proteins 72, 333–343. [DOI] [PubMed] [Google Scholar]
- Cavanagh J. (2007) Protein NMR Spectroscopy: Principles and Practice, 2nd ed., Academic Press, Amsterdam. [Google Scholar]
- Huber R.; Deisenhofer J.; Colman P. M.; Matsushima M.; Palm W. (1976) Crystallographic structure studies of an IgG molecule and an Fc fragment. Nature 264, 415–420. [DOI] [PubMed] [Google Scholar]
- Deisenhofer J. (1981) Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-Å resolution. Biochemistry 20, 2361–2370. [PubMed] [Google Scholar]
- Matsumiya S.; Yamaguchi Y.; Saito J.; Nagano M.; Sasakawa H.; Otaki S.; Satoh M.; Shitara K.; Kato K. (2007) Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J. Mol. Biol. 368, 767–779. [DOI] [PubMed] [Google Scholar]
- Bowden T. A.; Baruah K.; Coles C. H.; Harvey D. J.; Yu X.; Song B. D.; Stuart D. I.; Aricescu A. R.; Scanlan C. N.; Jones E. Y.; Crispin M. (2012) Chemical and structural analysis of an antibody folding intermediate trapped during glycan biosynthesis. J. Am. Chem. Soc. 134, 17554–17563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M.; Walker R. C.; Lanzilotta W. N.; Prestegard J. H.; Barb A. W. (2014) Immunoglobulin G1 Fc domain motions: Implications for Fc engineering. J. Mol. Biol. 426, 1799–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.