

Abstract
A total of 15,590 unique zebrafish EST clusters from two cDNA libraries have been identified. Most significantly, only 22% (3437) of the 15,590 unique clusters matched 2805 (of 15,200) clusters in the Danio rerio UniGene database, indicating that our EST set is complementary to the existing ESTs in the public database and will be invaluable in assisting the annotation of genes based on the upcoming zebrafish genome sequence. Blast search showed that 7824 of our unique clusters matched 6710 known or predicted proteins in the nonredundant database. A cDNA microarray representing ∼3100 unique zebrafish cDNA clusters has been generated and used to profile the gene expression patterns across six different embryonic stages (cleavage, blastula, gastrula, segmentation, pharyngula, and hatching). Analysis of expression data using K-means clustering revealed that genes coding for muscle-specific proteins displayed similar expression patterns, confirming that the coordinate gene expression is important for myogenesis. Our results demonstrate that the combination of microarray technology with the zebrafish model system can provide useful information on how genes are coordinated in a genetic network to control zebrafish embryogenesis and can help to identify novel genes that are important for organogenesis.
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to EMBL under accession nos. AL901610–AL928536.]
In recent years, zebrafish has been adopted as a model system for studies of vertebrate development because of its unique features favorable for genetic studies compared with other vertebrate systems. These features include a reasonably short lifetime, a large number of progenies, external fertilization and embryonic development, and translucent embryos (Talbot and Hopkins 2000). The relative ease of obtaining haploid and homozygous diploid individuals by gynogenesis offers another advantage for genetic analysis (Streisinger et al. 1981, 1986). Thus far, genetic linkage maps with a high density of markers covering the whole genome are readily available in the database (Knapik et al. 1998; Postlethwait et al. 1998; Gates et al. 1999; Shimoda et al. 1999). Zebrafish has also been regarded as an excellent model system for the studies of human disease because many zebrafish mutants with phenotypes such as disorders in hematopoiesis, cardiovascular generation, and kidney development are reminiscent of human disease states (for review, see Dooley and Zon 2000). The upcoming completion of sequencing of the zebrafish genome will no doubt facilitate our genetics and genomics studies in zebrafish in the future.
Two large-scale screens using chemical mutagens have been performed and >1500 phenotypic mutants corresponding to over 500 genes have been generated to identify genes required for the early developmental processes in zebrafish (Driever et al. 1996; Haffter et al. 1996). So far, >50 mutant genes have been cloned via candidate gene approaches (Schulte-Merker et al. 1994), positional cloning (Zhang et al. 1998), or synteny conservation strategies (Karlstrom et al. 1999). Recently, a large-scale insertional mutagenesis screen using a retroviral vector system has also been initiated (Amsterdam et al. 1999; Golling et al. 2002). In addition to the forward genetics approach, several other approaches have also been developed for the studies of gene function. These approaches include morpholino-mediated gene “knockdown” system (Nasevicius and Ekker 2000), ribozyme-mediated gene “knockdown” system (Xie et al. 1997), RNAi-mediated gene silencing system (Li et al. 2000), and photo-mediated gene activation of caged RNA/DNA system (Ando et al. 2001).
Although many genes important for cell signaling or organogenesis have been studied in zebrafish, global analysis of gene expression will likely provide more information regarding how gene regulation is coordinated in this genetic network. In addition, systematical analysis of the differences in the patterns of gene expression among different stages or between a mutant and the wild-type control will also provide an opportunity to identify those functional but unknown genes that might be important for controlling embryogenesis and organogenesis. The ease of harvesting a large amount of near-synchronized embryos provides an excellent opportunity to obtain mRNA from embryos from different embryonic stages for the study of gene expression patterns. Here we report that a total of 26,927 ESTs (length >200 bp, average length 473 ± 95 bp) have been obtained from two zebrafish cDNA libraries, one a primary library (Z1) and the other a normalized library (Z2). We also report the result from clustering analysis of these ESTs and summarize the result from blast search against a public database. To have a global view of gene expression profiles during zebrafish embryogenesis, we generated a cDNA microarray carrying ∼3100 unique cDNA sequences and used it to hybridize with samples prepared from the stages of cleavage, blastula, gastrula, segmentation, pharyngula, and hatching, respectively, against the sample prepared from unfertilized egg. A group of genes displaying similar patterns of up-regulated gene expression from the pharyngula stage onward was grouped by K-means clustering. Sequence blasts have revealed that a portion of these genes encode well-known muscle-specific proteins, indicating that the expression of these genes is coordinated during myogenesis. RNA gel blot hybridization experiments have confirmed their temporal expression patterns, and in situ hybridization experiments have revealed that most of these genes are indeed expressed in muscle tissue. Using a set of known genes as trainers, Support Vector Machine (SVM) has predicted some putative unknown genes that might be components in muscle tissue. Indeed, one of them, cDNA 160-D11 (a putative uncharacterized gene), was proved to express specifically in muscle tissue. Our work presents a good example in which the combination of microarray technology with the zebrafish model system will not only consolidate our existing knowledge, but will also help us to identify novel factors that might be important for organogenesis. It also provides us with a global view on how genes are coordinated to form a genetic network to control zebrafish embryogenesis.
RESULTS
ESTs From the Z1 Library
Total RNA was extracted from zebrafish at different growth stages (for details, see Methods; Kimmel et al. 1995). The unamplified primary double-stranded cDNA (ds-cDNA) derived from the mRNA sample was used for the construction of our Z1 cDNA library. Plasmid DNA from an individual clone randomly picked from the Z1 library was subjected to EST sequencing. As shown in Table 1, 2556 unique cDNA clusters were identified from a total of 11,908 individual clones with length >200 bp. Overall redundancy is close to 78% (unique clusters against total sequence runs, the same following), which is normally seen for nonnormalized cDNA libraries (statistics from ftp://ftp.ncbi.nlm.nih.gov/repository/Unigene/Dr.seq.uniq). Statistical analysis showed that only 1660/2556 clusters are each represented by a single clone and the rest are each represented by >2 clones (Table 2). Notably, 16 genes, accounting only for 6.3% (16/2556) of unique cDNA clusters, are each represented by >100 clones (Table 2) and contribute significantly to the high redundancy (3638/11908 = 31%). Eleven of these 16 genes share homology with genes of known/putative function in the nonredundant (nr) database, and their protein products are involved in different cellular activities (Supplementary Table 1). The cluster for the Y-box binding protein gene (also known as nuclease-sensitive element binding protein; Didier et al. 1988; Gai et al. 1992; Grant and Deeley 1993) contains 648 individual clones, indicating a high abundance of the transcript of this gene in our mRNA sample (Supplementary Table 1). Five of the 16 high-abundance genes do not match any known gene or predicted protein (Supplementary Table 1). Because the majority of our ESTs in Z1 contain a poly(A) tail and because sequencing was done from the 3′ end, we cannot exclude the possibility that the sequences obtained for these five genes may not include the ORF if they have a long 3′-untranslated region (3′-UTR) sequence.
Table 1.
Summary of Statistics of ESTs Obtained From Two Libraries
| Total unique clusters identified | 15,590a |
| From Z1 library | 2556 (of 11,908) |
| From Z2 library | 13,308 (of 15,019) |
| Clusters matching sequences in the zebrafish UniGene database | 3437b |
| Clusters having hits in nr database | 7824 (6710 distinct hitsc) |
| Clusters having no hits in nr database | 7766 |
Supplementary Table 2. Mass alignment program, Tigr-Assembler.
Supplementary Table 3.
Supplementary Table 4.
Table 2.
Redundancy Comparison Between Z1 and Z2 Libraries
| Number of EST clones in a cluster | ||||||||||
| >100 | 50–99 | 2–49 | 7 | 6 | 5 | 4 | 3 | 2 | 1 | |
| Number of clusters in Z1 | 16 | 17 | 863 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 1660 |
| Number of clusters in Z2 | 0 | 0 | 1 | 0 | 7 | 19 | 190 | 1240 | 11,851 | |
(n.d.) Not determined.
ESTs From the Normalized cDNA Library (Z2)
To reduce such high redundancy during EST sequencing and to speed up the identification of unique cDNAs, we constructed a normalized cDNA library (Z2), as illustrated in Figure 1 (for details, see Methods). The strategy used to construct our normalized cDNA library was based on the principle that, during cDNA annealing, rare cDNA transcripts anneal less rapidly than do abundant cDNA species; thus, the single-stranded fraction of cDNA (ss-cDNA) becomes progressively more normalized during the course of annealing (Ko 1990; Patanjali et al. 1991). To determine the normalization efficiency, we hybridized ∼10,000 clones of each library constructed from the prenormalized and the normalized cDNA, respectively, with probes derived from the β-tubulin gene (Fig. 2C). A dramatic reduction in the number of positive clones was observed in the normalized cDNA library (∼8; NIII) when compared with the prenormalized library (∼67; SI). On the other hand, the number of clones for the relative low-expressing gene RAG1 (recombination activating gene 1) was increased from zero (in SI) to four (in NIII). Clones from this high-quality library (Z2) were used in our new EST sequencing project. As shown in Table 1, 13,308 unique cDNA clusters were obtained from a total of 15,019 ESTs of length >200 bp. Overall redundancy in the Z2 library is only ∼12% and this rate is significantly lower than that in the Z1 library (Table 1). Statistical analysis showed that 11851/13308 clusters are each represented by a single clone and the rest are each represented by more than two clones (Table 2). The cluster for the beta-lactamase gene contains seven individual clones and is most abundant in the Z2 library, which significantly contrasts with the large number of redundant clones in the Z1 library (Table 2), further demonstrating the effectiveness of normalization.
Figure 1.

Procedure for cDNA normalization (for details, see Methods).
Figure 2.

Construction of the normalized cDNA library (Z2). (A) Size selection of cDNA for normalization. (Left) Total cDNA before size selection, (right) size-selected cDNA. (B) Separation of ss-cDNA and ds-cDNA on hydroxyapatite column (HA column). ss-cDNA is eluted at 100 min and ds-cDNA is eluted at 150 min. (C) Colony hybridization to examine the normalization efficiency using the β-tubulin gene as a probe. (SI) Library constructed from prenormalized cDNA; (NIII) library constructed from cDNA after three rounds of normalization.
Statistical Analysis of ESTs From Z1 and Z2 Libraries
Combining Z1 and Z2 libraries, a total of 15,590 of unique cDNA clusters were identified (Table 1; Supplementary Table 2). Large-scale EST sequencing for zebrafish has also being carried out at Washington University (WashU) headed by Dr. S. Johnson, and the total EST collection is approaching 200,000 (http://zfish.wustl.edu/). A portion of these EST sequences was subjected to clustering analysis and the result was deposited in the UniGene database in NCBI (ftp://ftp.ncbi.nlm.nih.gov/repository/Unigene/Dr.seq.uniq). As released on June 14, 2002, a total of 15,200 clusters were identified from a total of 188,259 ESTs and other sequences (∼90% redundancy). Surprisingly, only 22% of our unique clusters (3437/15590) matched 2805/15,200 D. rerio UniGene clusters (Table 1, Supplementary Table 3) in this UniGene database. This ratio is much lower than that obtained from comparison between the EST sequences from the zebrafish embryonic inner ear (44%) and the UniGene database (Coimbra et al. 2002). Thus, it is reasonable to estimate that, combining our and WashU's EST set, ∼27,000 unique clusters have been identified and these ESTs will be invaluable in assisting with the annotation of the zebrafish genome in the near future. Because many cDNAs contain more than one EcoRI site and because EcoRI fragments were used to construct the normalized cDNA library, the final number of unique clusters is likely slightly overestimated. This is reflected in the fact that 3437 of our EST clusters matched only 2805 Unigene clusters in the public database.
The translations in the six phases of all 15,590 unique clusters in our database were compared with nr (released on 13 May 2002; ftp://ftp.ncbi.nlm.nih.gov/blast /db/nr). As shown in Table 1, 7824/15590 unique clusters in total have hits (P<10−8) in nr and these hits represent 6710 distinctive genes that include 5180 known genes either corresponding to D. rerio known genes or to known genes with a wide range of functions in other organisms (Supplementary Table 4). As expected, genes encoding for metabolic enzymes (including hydroxylase, oxidase, reductase, dehydrogenase, synthase, metabolic kinase and phosphatase, transferase, and protein degradation enzymes) are most abundant in our EST set, and in total 1058/6710 (∼16%) were identified (Table 3). A significant number of genes (∼257/6710) were found to encode products for protein, DNA, and RNA biosynthesis (ribosomal proteins, polymerase, and initiation and elongation factors; Table 3). Genes encoding for protein kinase (194/6710) and phosphatase (66/6710) are also well represented (Table 3). Significantly, many transcription factor genes (no less than 248/6710, including ∼77 zinc finger proteins and ∼57 homeobox proteins) and receptor genes (no less than 218/6710) are well represented in our EST set (Table 3). The remaining 1530/6710 genes correspond to putative, unknown, or unnamed genes predicted from cDNA or genomic sequences. There were 7766/15590 unique cDNA clusters that did not have a hit in the database. Because the size of cDNA was selected between 0.5 and 2.0 kb after cDNA was synthesized using oligo(dT), for those genes with long 3′-UTRs, the sequences obtained might not include their ORF sequence. Therefore, it is likely that the number of sequences with no hit in the nr database is overestimated.
Table 3.
Classification of Blast Resulta
| Protein category | Number of distinctive hits |
| Enzyme for metabolism | |
| Carboxylase | 15 |
| Dehydrogenase | 87 |
| Hydroxylase | 19 |
| Isomerase | 16 |
| Metabolic kinase | 73 |
| Metabolic phosphatase | 21 |
| Nuclease | 14 |
| Oxidase | 38 |
| Protein degradation enzyme | 77 |
| Reductase | 40 |
| Synthase | 52 |
| Transferase | 128 |
| Others | 478 |
| Protein, DNA, and RNA biosynthesis | |
| Ribosomal protein | 153 |
| DNA polymerase | 5 |
| RNA polymerase | 19 |
| Initiation, elongation, splicing, and | |
| other factors | 80 |
| Cellular signaling | |
| Receptor and related | 218 |
| Protein kinase | 194 |
| Protein phosphatase | 66 |
| G-protein and exchange factors | 37 |
| Transcription factor | |
| Zinc finger protein | 77 |
| Homeobox protein | 57 |
| Other | 114 |
| Protein degradation | |
| Proteasome related | 23 |
| Ubiquitination related | 32 |
| Membrane protein | |
| Channel protein | 45 |
| Other membrane protein | 74 |
| Cell structure and cell division protein | |
| Actin | 15 |
| Tubulin | 22 |
| Dynein | 10 |
| Cyclin | 7 |
| Carrier and transporter | 106 |
| Muscle specific protein | 85 |
| Unclassified | 2797 |
| Unknown and unnamed protein | 1530 |
Details in Supplementary Table 4.
Generation of Zebrafish cDNA Microarray
PCR products generated from 11,480 individual clones from the Z1 library were arrayed on glass slides, and, at the same time, these clones were subjected to DNA sequencing. In total, sequences of 10,518 EST clones were obtained (Supplementary Sequence File: in FASTA format) and the alignment result revealed that this set of ESTs represents 3100 unique cDNA clusters (Supplementary Table 5). Blast search using six frames of all ESTs revealed that 4519 ESTs had hits (P < 10−5) in the nr protein database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/nr) and these 4519 hits represent 728 distinct proteins (Supplementary Table 6). Both muscle-specific proteins (Table 4) and ribosomal proteins (Supplementary Table 7) are well represented in this EST set. The remaining 5999 did not have a hit in the database. Nevertheless, for reasons mentioned earlier, the number of sequences on the array with no hits in our blast search is likely to be overestimated.
Table 4.
Representatives of Muscle Proteins
| Myosin-binding protein C [Gallus gallus] |
| Cytoskeletal beta actin [Kenyan clawed frog] |
| Muscle actin [Lethenteron japonicum] |
| Cardiac tropomyosin [Xenopus laevis] |
| Muscle-type pyruvate kinase [Gallus gallus] |
| Parvalbumin alpha (A1) |
| Similar to actin-related protein 2/3 [Homo sapiens] |
| Alpha-tropomyosin [Danio rerio] |
| Fast skeletal muscle troponin I [chicken] |
| Muscle glycogen phosphorylase [Homo sapiens] |
| Slow-type myosin-binding protein C [Homo sapiens] |
| Muscle-specific creatine kinase [Danio rerio] |
| Cardiac troponin T [Danio rerio] |
| Alpha-smooth muscle actin [Oryctolagus cuniculus] |
| Calcium-binding parvalbumin B |
| Cytoskeletal actin [Lytechinus pictus] |
| Muscle-specific cytosolic thyroid hormone-binding protein (pyruvate kinase) |
| CsMA-1 |
| Myosin light chain [Danio rerio] |
| Vimentin [Danio rerio] |
| Fast myotomal muscle actin [Salmo salar] |
| Myosin-binding protein-H [Gallus gallus] |
| Skeletal alpha1 actin [Danio rerio] |
| Parvalbumin [Danio rerio] |
| Fast skeletal muscle troponin T [Danio rerio] |
| Fast skeletal muscle troponin C [Danio rerio] |
| Skeletal muscle tropomyosin NM [Homo sapiens] |
Gene Expression Profiling During Zebrafish Embryogenesis
Zebrafish embryogenesis occurs during the first 3 d after fertilization and this period can be broadly categorized into seven stages: zygote, cleavage, blastula, gastrula, segmentation, pharyngula, and hatching (Kimmel et al. 1995). By the end of the third day, most of the major organ systems in the organism, including brain, nervous system, muscle, heart, blood, gut, liver, eyes, and ears have already been initiated and some are even properly formed from the totipotent fertilized egg (Kimmel et al. 1995). Under optimal growth conditions, the time span between each sequential stage is almost invariable, and the accomplishment of embryogenesis is the result of precise coordination of the unidirectional transitions between each stage under the control of a precise genetic network.
Total RNA was extracted, respectively, from unfertilized eggs (E0) and near-synchronized embryos at the stage of cleavage (E2), blastula (E3), gastrula (E4), segmentation (E5), pharyngula (E6), and hatching (E7) for mRNA preparations. Because it represents the maternal transcripts of the basal level from where de novo embryonic gene expression at different stages can easily be compared, the unfertilized egg (E0) was used as a reference sample against which samples from other stages were compared. In each case, the experiment was repeated, reversing the fluorescent dye. For each slide, the fluorescence intensities of the two probes hybridized to each spot were normalized and the ratios were then calculated (Supplementary Table 5). To identify those genes whose expression is regulated during embryogenesis, we determined the number of clones showing a ratio (against E0) greater or less than twofold (log value >1 or <−1) for each stage (Table 5). For example, at the E3 stage, 356 of 3100 total clusters examined showed a significant increase (log ratio ≥1) and 160 showed a significant decrease (log ratio ≤−1) in their expression levels, indicating that activities at this stage might not need the de novo expression of many genes (Table 5). On the other hand, at the E5 stage, 811 clusters were up-regulated (log ratio ≥1) and, meanwhile, 872 clusters were down-regulated (log ratio ≤−1; Table 5). The marked changes in expression patterns of this great number of genes were likely related to the active morphogenesis occurring during segmentation. At this stage, the somites are developing, and the rudiments of the primary organs such as brain and optic primordium become visible (Kimmel et al. 1995).
Table 5.
Genes Showing Significant Changes in Expression During Embryogenesis
| Log ratio | E2/E0 | E3/E0 | E4/E0 | E5/E0 | E6/E0 | E7/E0 |
| >+1a | 206 | 356 | 545 | 811 | 871 | 848 |
| <−1a | 197 | 160 | 579 | 872 | 1015 | 807 |
Expression data for all 11,480 individuals are deposited in Supplementary Table 5.
Number of unique cDNA clusters.
To study the expression pattern of each individual gene during embryogenesis in detail, we analyzed normalized data (Supplementary Table 5) from 24 microarray experiments (4 experiments for each stage) using hierarchical clustering and K-means clustering methods (EPCLUST: http://ep.ebi.ac.uk/EP/EPCLUST/). Hierarchical clustering and K-means clustering grouped genes with similar expression patterns that displayed dynamic changes of expression. In general, expression of most genes on the array examined displayed one of the following expression patterns: up-regulated, down-regulated, and fluctuating (data not shown). We focused on groups displaying patterns of either up- or down-regulated. All expression data were clustered into 20 groups using the K-means clustering method (http://ep.ebi.ac.uk/EP/EPCLUST/). Seven groups were selected to form five different expression patterns (Fig. 3), including early up-regulated (49 cDNA clusters from E3 and E4 stages; Fig. 3A; Supplementary Fig. 1), middle up-regulated (350 cDNA clusters from E4 and E5 stages; Fig. 3B; Supplementary Fig. 2), late up-regulated (64 cDNA clusters from E6 and E7 stages; Fig. 3C; Supplementary Fig. 3), early down-regulated (151 cDNA clusters from E3 and E4 stages; Fig. 3D; Supplementary Fig. 4), and late down-regulated (251 cDNA clusters from E4 and E5 stages; Fig. 3E; Supplementary Fig. 5). The early down-regulated group contains genes encoding for proteins such as claudin-like protein ZF-A89 and ZF-A9 (Fig. 3D; Supplementary Fig. 4). The late down-regulating subgroup includes genes encoding for proteins such as the p32 subunit of splicing factor SF2, single-stranded D-box binding factor 2, and GAP-associated phosphoprotein p62 (Fig. 3E; Supplementary Fig. 5). As ribosomal genes (Supplementary Table 7) and muscle genes are well represented in this EST set (Table 4), we examined their expression patterns from the E2 to E7 stages. The expression of most ribosomal genes belong to the middle up-regulated genes, indicating the initiation of active protein biosynthesis at this stage (Fig. 3B; Supplementary Fig. 2), whereas a significant number of muscle genes fall in the late up-regulated group (Fig. 3C; Supplementary Fig. 3), indicating the maturation of myogenesis at this stage. The coordinate actions displayed by muscle genes and ribosomal genes indicate that coordinate regulation of gene expression is not only important for organogenesis but also for subcellular organogenesis.
Figure 3.

Examples of gene expression profiling during zebrafish embryogenesis using the K-means clustering method. For hierarchical and K-means clustering, Euclidean distance was applied to divide all expression data into 20 clusters. Of that, seven were selected to form five clusters (groups). The five groups were subjected to screening to get rid of the redundant cluster ID and annotation ID before the final presentation. (A) Patterns for genes significantly up-regulated at the E3 stage. (B) Patterns for genes significantly up-regulated at the E4 or E5 stages. The expression of most genes for ribosomal proteins displayed such patterns (Supplementary Fig. 2). (C) Patterns for genes significantly up-regulated at the E6 stage. The expression of many genes for muscle-specific proteins displayed such patterns (Supplementary Fig. 3). (D) Patterns for genes down-regulated at the E3 or E4 stages. (E) Patterns for genes down-regulated at the E5 or E6 stages. Samples of the various stages for microarray probe labeling and Northern blots were collected on the basis of the staging series as described in Kimmel et al. (1995). One to two stages were collected to represent one period of embryos. Altogether, seven periods were collected. E1, the zygotic period, was omitted in the experiment. (E0) Unfertilized; (E2) cleavage (four- to eight-cell); (E3) blastula (4–4⅓ h); (E4) gastrula (5¼ to 5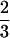 h); (E5) segmentation (6 to 10 somites); (E6) pharyngula (24 h); (E7) hatching (72 h). M. thermautotrophicus = Methanothermobacter thermautotrophicus.
h); (E5) segmentation (6 to 10 somites); (E6) pharyngula (24 h); (E7) hatching (72 h). M. thermautotrophicus = Methanothermobacter thermautotrophicus.
Coordinate Expression of Muscle Genes
The abundance of muscle genes on our microarray provided us with a chance to study their coordinate actions and myogenesis. In zebrafish, myogenesis starts at the segmentation stage and strong evidence has shown that myogenic transcriptional factor MyoD (Weinberg et al. 1996), Engrailed (Halpern et al. 1993), and Hedgehog (Roy et al. 2001) are involved in controlling muscle initiation and differentiation. Zebrafish embryonic muscle contains three distinct fiber types: muscle pioneer slow muscle, nonpioneer slow muscle fibers, and fast muscle fibers (Barresi et al. 2000). Ten zebrafish muscle-specific genes have been identified and analyzed previously (Xu et al. 2000) and the reported result strongly indicated that the expression of these genes is coordinately regulated. Blast search revealed that 27 unique clusters on our array are likely genes encoding different muscle proteins, including skeletal α-actin, myosin light chain, troponin, tropomyosin, and other regulatory proteins (vimentin, desmin, etc.). Consistent with a previous report (Xu et al. 2000), hierarchical and K-means clustering showed that many known muscle genes in our data set displayed a similar expression pattern (Fig. 3C; Supplementary Fig. 3). The expression patterns of six well-known muscle genes from microarray hybridization were shown as examples in Figure 4. Their expression patterns from E2 to E7 stages were further confirmed via RNA gel blot hybridization and their identities as muscle-specific genes were verified via in situ hybridization (Fig. 4).
Figure 4.

Coordinated expression of genes for muscle-specific proteins. Expression patterns of six muscle-specific genes (clone ID: parvalbumin, 109-C11; skeletal alpha1 actin, 092-G12; fast skeletal muscle troponin C, 068-F03; creatine kinase, 144-H03; myosin light chain 2, 077-D05; tropomyosin alpha chain, 098-G11; putative novel muscle gene, 160-D11; all from the Z1 library) are shown. (Left panel) Patterns obtained from microarray hybridization; (middle panel) patterns obtained from RNA gel blot hybridization; (right panel) in situ hybridization to confirm the tissue-specific expression of these six genes in WT embryos.
Prediction of Putative Unknown Muscle Genes Using Support Vector Machine (SVM)
The SVM method is mainly used to predict putative functional unknown genes that might function in the same pathway or be coregulated in the same organ or tissue or subcellular organelle as are those known genes based on their expression patterns. SVM uses a training set to specify in advance which data should cluster together (Brown et al. 2000). This method is mainly based on the assumption that genes of similar function display similar expression patterns (Eisen et al. 1998). A set of known ESTs (2500) was used as a training set to allow the SVM to learn to discriminate between muscle and nonmuscle genes (Table 6). To examine the quality of training, we tested the trained SVM on another set of known ESTs (1387). As shown in Table 6, the majority of predicted positives are true positives (44/54), whereas the majority of predicted negatives are true negatives (1286/1333), proving that the training was fairly successful. When the expression data of the remaining unknown ESTs (7562) were subjected to SVM analysis, 110 candidates, corresponding to 56 unique cDNA clusters and 30 unsequenced cDNAs, were predicted as true positives (Table 6; Supplementary Fig. 6). One such cDNA (clone ID 160-D11) was chosen for further studies because a preliminary blast search using the available sequence of 160-D11 did not identify any hit in GenBank.
Table 6.
SVM Analysis of Expression Data
| Training set | Number of ESTs | 2500 |
| Number of positive | 157 | |
| Number of negative | 2341 | |
| Number of positive support vectors | 131 | |
| Number of negative support vectors | 139 | |
| Testing set | Number of ESTs | 1387 |
| True positive | 44 | |
| False positive | 10 | |
| True negative | 1286 | |
| False negative | 47 | |
| Prediction set | Number of ESTs | 7562 |
| Number of positive predicted | 110a | |
| Number of negative predicted | 7452 |
Parameter of the program, C = 10; Gaussian radial basis function kernel with δ = 0.01. (SVM) Support vector machine.
80 of the 110 predicted positive ESTs were sequenced and they belong to 56 unique cDNA clusters. The remaining 30 cDNAs were not sequenced. Details in Supplementary Figure 6. Known muscle ESTs were used as positives and selected known nonmuscle ESTs were used as negatives.
A probe derived from 160-D11 was used for RNA gel blot hybridization and a transcript of size 6.7 kb was identified. Examination of transcript levels of 160-D11 at different stages (E2–E7) showed that its expression pattern changed in a similar way as did other muscle-specific genes (Fig. 4), which is consistent with the pattern obtained from microarray hybridization. Furthermore, in situ hybridization showed that 160-D11 was specifically expressed in muscle (Fig. 4). Because of the large size of transcript (6.7 kb) for 160-D11 as revealed by Northern analysis, repeated attempts to get the full-length sequence for 160-D11 through 5′ racing were unsuccessful. Nevertheless, we managed to get 2.5 kb of 160-D11 cDNA sequence and blast search did not identify any homology with any known gene in the database. Based on the fact that the size of the 160-D11 transcript is ∼6.7 kb, we could not exclude the possibility that the sequence obtained does not contain the ORF, and thus we could not fully exclude the possibility that 160-D11 is a known gene.
DISCUSSION
In this report, we described the EST sequencing results from Z1 and Z2 libraries. Clustering analysis showed that clone redundancy in Z1 is much higher than in Z2. Apparently, the low redundancy in Z2 resulted from successful normalization of cDNA derived from our mRNA sample. As indicated from screening 10,000 colonies, the number of beta-tubulin and RAG1 clones was down approximately eightfold and up approximately fourfold, respectively. Presumably, after normalization, most genes would be more or less evenly represented in the cDNA pool used for library construction. If we could propose that there were 30,000–40,000 expressed genes represented in our RNA sample collection, mathematically, randomly picking 20,000 clones from this normalized library (we picked 200 × 96 clones) would avoid the problem caused by random cloning and likely provide us with a high chance of obtaining the clones representing unique ESTs, although, inevitably, some clones were redundant. We believe this is the case; as proof, when compared with the public Unigene database, 3437 of our unique EST clusters matched 2805 of the Unigene cluster. The Unigene cluster normally covers a significant length of the corresponding cDNA. Had the problem of random cloning been a contributing factor, a higher number (at least 2 × 2805) of our ESTs would have been expected. In addition, as further evidence, when all unique ESTs were used to blast the nr database, we got 7824 hits representing 6710 distinctive genes. As a significant number of genes are deposited as full length in nr, had the random cloning caused a problem, many fewer distinctive genes would be identified. Considering cost and efficiency, the high quality of the normalized cDNA library (Z2) makes it ideal for EST sequencing. The lower rate of overlapping with the Unigene set in the public database, the wide-range coverage of genes involved in diverse cellular activities, and the large number of novel sequences in our EST database provide us with a rich resource to generate the zebrafish cDNA microarray. Because all individual clones have been stored carefully, EST clones could be accurately retrieved and provided on the basis of microarray results. We are currently generating a zebrafish cDNA microarray using ESTs representing our unique cDNA clusters to provide a public service to the fish community worldwide.
The accomplishment of embryogenesis is marked by the formation of almost all necessary organs and tissues for a living embryo. This process is under the control of a genetic network that relies on the precise coordinate actions of many genes that are essential for normal development. For decades, great efforts and also great achievements have been made to identify genes important for organogenesis and to study how these genes interact with one another to control embryogenesis. Because of some of its unique features favorable for genetics studies, zebrafish has recently been chosen as the model system to study vertebrate development (Talbot and Hopkins 2000). Although forward genetics is still the most powerful tool to identify genes important for embryogenesis, global analysis of gene expression patterns will no doubt enable us to gain insight into how gene regulation is coordinated in the genetic network. In addition, systematic analysis of expression data (e.g., comparison among different developmental stages or mutant vs. wild-type control) using well-developed bioinformatic tools will provide us with an opportunity to identify novel genes of previous unknown function that are potentially important for organogenesis. Microarray has successfully been used to study the coordinate actions of gene expression during cell division in yeast (Cho et al. 1998), for circadian and for mesoderm development in Drosophila (Furlong et al. 2001; McDonald and Rosbash 2001), and for many other developmental processes. As a test, 11,480 EST sequences from the Z1 library, representing 3100 unique cDNA clusters, have been fabricated on the glass slide. This microarray has been used to profile the gene expression patterns from E2 to E7 stages during embryogenesis. On analysis of the expression pattern displayed by redundant clones, a reproducible result was obtained (data not shown). Because ribosomal proteins and muscle-specific proteins are well represented in this EST set, we focused on their expression patterns. Clustering analysis clearly showed that the expression of most ribosomal genes increased significantly at the segmentation stage (E5), indicating that coordinate actions of genes are important for the genesis of subcellular organelles (ribosomes). Clustering analysis also showed that the expression of muscle genes was tightly coordinated, indicating that coordinate gene expression is important for myogenesis. Although it is expected that SVM will help us to observe the coordinate actions for ribosomal and muscle genes, its use to identify putative novel muscle genes is an example of the exploration of our expression data. In fact, a putative unknown gene has been identified as a muscle-specific component that awaits functional revelation.
METHODS
Construction of a Primary cDNA Library (Z1) and a Normalized cDNA Library (Z2)
Total RNA was extracted from zebrafish (local wild type) at different stages (for Z1, stages used were 12-h-, 2-d-, 4-d-, 1-wk-, 2-wk-, 1-mo-, 2-mo-, and 3-mo-postfertilization, respectively; for Z2, stages used were 4-h-, 2-d-, 4-d-, 1-wk-, 2-wk-, 1-mo-, 2-mo-, and 3-mo- postfertilization, respectively; Kimmel et al. 1995), using the Tri Reagent according to the manufacturer's protocol (Molecular Research Centre). An equal amount of total RNA from each of the stages was mixed for mRNA purification using an mRNA purification kit (Promega). For the Z1 library, both the first- and second-stranded cDNA was synthesized using a cDNA synthesis kit (GibcoBRL; primer for first-strand cDNA: 5′-GAGAGAGAGAGAGA GAGAGAACTAGATCTCGAGTTTTTTTTTTTTTTTTTT; adapters are composed of 10- and 14-mer oligonucleotides, which are complementary to each other with EcoRI cohesive end: 5′-OH-AATTCGGCACGAGG-3′ and 3′-GCCGTGCTCC-P-5′). A size selection was performed for cDNAs of 0.5–2.0 kb before cloning them unidirectionally into predigested vector (5′-EcoRI and 3′-XhoI sites; ZAP Express cDNA Synthesis kit, Stratagene). Transformed cells were plated at ∼3000 cfu per 24 × 24 cm2 Qtray (Genetix), and grown overnight. An individual colony was selected and placed (Qpix from Genetix) in 96-well deep well plates with 1 mL LB/ampicillin per well. The overnight cultures were divided into three portions (Biomek FX, Beckman): (1) 100 μL for PCR, (2) 50 μL for glycerol stock, and (3) the remainder for minipreps.
For the Z2 library, oligo(dT)20-V (V = G, C, A) was used as primer for the first-strand cDNA synthesis and the second strand was synthesized as described (GibcoBRL). Two primers, LLR1A (5′-gagatattagaattctactc-3′) and LLR1B (complementary strand 5′-gagtagaattctaatat-3′; Ko 1990), were annealed at equal molar ratio and used as adaptors to ligate to the blunt-ended ds-cDNA, and the product was subjected to size selection. Total cDNA-adaptor ligated mix was loaded on an agarose gel (1%) for size fractioning, and gel containing a cDNA size between 0.5 kb and 2.0 kb was purified. The gel-purified cDNA was amplified via PCR (denaturation at 94°C, 30 sec; annealing using a temperature gradient from 47°C to 50°C, 2 min; extension at 72°C, 3 min; 20 cycles) and the PCR product was pooled and concentrated for the first-round denaturation/reassociation step (1 μg PCR product in 50 μL reassociation buffer containing 0.3 M sodium phosphate, 0.4 M EDTA, 0.04% SDS at pH 6.8). After denaturation at 100°C for 5 min, DNA was immediately transferred to 65°C and allowed to reassociate for 24 h and then quenched on ice. The yielded mixture of ss- and ds-cDNA was separated on a 1-cm hydroxyapatite (Bio-Gel HTP gel # 130–0520, DNA grade) jacketed column maintained at 65°C using the AKTA FPLC (Amersham Pharmacia Biotech) system described following. The reassociated DNA was diluted in 1 mL column equilibration buffer A (10 mM sodium phosphate, 0.1% SDS at pH 6.8, 65°C) and loaded onto the preequilibrated HA column. The column was washed with 3 column volume (CV) of buffer A, then eluted with a continuous gradient buffer from 0%–100% Buffer B (0.4 M sodium phosphate, 0.1% SDS at pH 6.8, 65°C) over 10 CV, followed by 4 CV of buffer B to wash the column. ssDNA eluted at ∼120 mM sodium phosphate and dsDNA at ∼300 mM sodium phosphate under these conditions. Fractions containing ssDNA were pooled and concentrated using a Centricon-YM30 filter cartridge and the obtained ssDNA was used for the second-round PCR. Two more rounds of normalization were repeated and the final PCR products were digested with EcoRI and ligated to predigested pBluescript SK+ vector for library construction. Colony picking and bacteria culturing were done in the same way as described earlier.
High-Throughput Sequencing and Sequence Assembling
Minipreps were carried out in 96-well format using the conventional alkaline/SDS lysis method using robotics Biomek FX (Beckman) followed by ethanol precipitation. Vector T3-primer was used to determine the EST sequence from each clone using either the Big Dye terminator cycle sequencing kit (Perkin Elmer) or DYEnamic ET terminator cycle sequencing kit (Amersham Pharmacia Biotech). All sequences obtained were subjected to mass editing (http://www.mrc-lmb.cam.ac.uk/pubseq/manual/pregap4_unix_toc.html) for vector and adaptor sequence clipping and elimination of low-quality or short sequences. For clustering 26,927 ESTs, the Tigr-Assembler program was used (http://www.tigr.org/software/assembler). For clustering 10,518 ESTs on microarray, the Staden Package GAP4 program was used (http://www.mrc-lmb.cam.ac.uk/pubseq/manual/gap4_unix_toc.html).
Sequence Comparison Against Public Databanks
The 15,590 unique clusters were used as queries for BLASTN searches against the section D. rerio UniGene (ftp://ftp.ncbi.nlm.nih.gov/repository/Unigene/Dr.seq.uniq.) containing 15,200 unique clusters (released on June 14, 2002). Sequences are considered identical if the blast E value is <10−50 (Coimbra et al. 2002). The consensus sequence of each of the 15,590 unique clusters was translated into six frames and then compared with nr (released on May 13, 2002; ftp://ftp.ncbi.nlm.nih.gov/blast/db/nr). Only blast E values of <10−8 were considered significant (Makabe et al. 2001).
Microarray Preparation
DNA Samples
The bacterial pellet spun down from the 100-μL overnight culture was resuspended in 50 μL of sterile water and incubated at 95°C for 10 min. The denatured cell suspension was spun again; 3 μL of the supernatant was used in a 100-μL PCR reaction containing the following components: 0.32 μM of each primer (T3 and T7), 0.2 mM of each deoxynucleotide, 1 × PCR buffer (500 mM KCl, 100 mM Tris-HCl at pH 9.0, 1% Triton-X-100, 15 mM MgCl2), and 1 μL Taq polymerase. The 96-well reaction was run using an amplification program of 3 min denaturation at 94°C, 35 cycles of 1 min at 94°C, 1 min at 50°C, and 1.5 min at 72°C, and terminated by a 10-min extension at 72°C. PCR products were first evaporated (70°C, 2 h) to smaller volumes (∼50 μL), followed by the standard 3M NaOAc/ethanol precipitation. The final DNA pellet was resuspended in 15 μL 1.5 M betaine/3 × SSC. For array DNA sample selection, 0.5 μL sample was run on a gel to identify products displaying a single, strong band (>0.15 μg/μL) and products with more than one fragment or a low yield (<0.15 μg/μL) or no amplified inserts were eliminated. Qualified samples (PCR products) were transferred from 96-well plates to 384-well plates. Positive and negative controls were added directly to 384-well plates on the basis of their designated positions on the array (see Controls).
Array Printing
Microscope slides were coated with Poly-L-Lysine following the instructions by Sigma, with some modifications. Slides were cleaned for 1 h in washing solution (2.5 N NaOH, 60% ethanol), washed in distilled water (five changes of water, 1–2 min each), and then immersed for 1 h in coating solution (0.01% Poly-L-Lysine, 0.1M PBS). After coating, the slides were washed with water and dried by centrifugation (5 min, 500 rpm), followed by a further incubation at 45°C for 10 min. The coated slides were cured for at least 2 wk in a drying cabinet before printing. DNA samples (PCR products) were arrayed from 384-well plates with an arrayer (Pixsys 5500XL Arrayer, Cartesian) loaded with 32 pins (TeleChem International). Each pin generated a subgrid of 19 × 19 spots (with a centro to centro (CTC) of 200 μm) and as a result the array carried 11,552 elements (i.e., 19 dots × 19 dots × 32 subgrids). On completion of arraying (∼10–12 h), DNA was fixed by rehydration over a water bath (65°C) for 3 sec and snap-dried on a heating block (75°C) for 3 sec, followed by UV cross-linked at 65 mJ of energy. After that, the remaining functional groups of Poly-L-Lysine on the slides were blocked by treating the slides with blocking solution (150 mM succinic anhydride in 1-methyl-2-pyrrolidinone, buffered with 85 mM sodium borate at pH 8.0) for 30 min. After washing with distilled water, the bound DNA samples were denatured for 2 min in distilled water (95°C), rinsed with 95% ethanol, and then finally dried by centrifugation (15 min, 500 rpm).
Controls
Two positive controls (D. rerio β-actin and histone H3; constructs were kindly provided by Dr. V. Korzh, IMCB, Singapore) and 10 negative controls (Arabidopsis GA1, GA4, GAI, SM2, SM1, and COI1, plexA, pBluescript KS, pGEMT, λDNA) were placed on our array. They were each spotted on six subgrids of an array, at the four corners of each (i.e., six spots per control across the whole array). These control inserts were amplified using the universal primer pairs on their respective vectors, followed by similar precipitation and resuspension in spotting buffer as described earlier. Of the 10 negative controls, the first 4 were also used as spiking controls for data normalization.
Fluorescent Probe Preparation
Embryo
Fish embryos from each of the six different developmental stages, namely, cleavage (E2), gastrula (E3), blastula (E4), segmentation (E5), pharyngula (E6), and hatching (E7), were collected, respectively, on the basis of their developmental morphology when incubated in 28.5°C (Kimmel et al. 1995). The reference sample (E0, unfertilized eggs) was collected by squeezing the stomach of anaesthetized 3–4-month-old female fish. To minimize the variation introduced into each developmental stage because of asynchronization of growth during development, we made efforts to ensure that embryos within a stage differed not more than 45 min from one another by selecting embryos under a dissecting microscope.
mRNA and Fluorescent Probe Preparation
Total RNA was extracted using Trizol (Molecular Research Centre) followed by mRNA purification (PolyA Tract, Promega). The quality of each mRNA sample was confirmed in a reverse transcription (Superscript II, Life Technologies) test reaction in the presence of DIG-dUTP, following the manufacturer's instruction (Roche Molecular Biochemicals). The labeled ss-DNA products were purified using microcon-YM30 (Millipore), and signal detection was carried out by dot-blotting serial dilution of the DIG-labeled product. Only mRNA samples producing significant signals compared with controls (DIG-labeled RNA) in this test labeling were used in subsequent fluorescent probe synthesis. Preparation of fluorescent DNA probe was performed as follows: 650 ng embryonic mRNA and spiking RNA (1 ng GA1, 200 pg GA4, 100 pg GAI, 20 pg SM2) were mixed with 0.5 μg oligo(dT) primer in a final volume of 19 μL. Spiking control RNAs were generated by in vitro transcription (RiboMAX RNA production System, Promega) from constructs containing these genes with an artificially added poly(A) tail. The RNA–primer mixture was incubated at 65°C for 5 min and then chilled on ice and added to 23 μL of labeling mixture (final concentration: 1X Superscript II buffer; 10 mM DTT; 500 μm each of dATP, dTTP, and dGTP; 200 μm dCTP; 60 μM Cy3– or Cy5-dCTP [Amersham Pharmacia]; 1 μL RNAsin [Promega]; 2 μL Superscript II). This reaction was incubated at 42°C for 1 h; after that, an additional 2 μL of Superscript II was added and incubation was continued for a further 1 h. At the end of labeling reaction, 5 μL 0.5 M EDTA and 10 μL 1M NaOH were added and incubation was continued at 65°C for 1 h, followed by 25 μL Tris-Cl (pH 7.5) to neutralize the mixture. The labeled cDNA was purified using microcon-YM30 (Amicon). Prior to the final concentration step, 1 μL (of the 50- to 80-μL washed probe retained in the column) was serially diluted and dotted on a coated glass slide for fluorescence detection, using a ScanArray 5000 laser scanner (GSI Lumonics). Only labeled probes producing significant signal in this quality control test were used for slide hybridization.
Array Hybridization
Cy3-labeled test cDNA (e.g., E2) and Cy5-labeled reference cDNA (E0) (or their reciprocally labeled pairs) were coprecipitated in a final mixture of 3 μg calf thymus, 10–40 μg tRNA, 10 μg oligo(dA)(40–60), 2.5 M NH4OAc, and 2.5 volume ethanol washed with 70% ethanol. The final dried pellet was resuspended in 40–50 μL 0.3% SDS/3.5 × SSC. The probe solution was denatured for 2 min at 100°C, cooled to 37°C for 30 min, and applied to the array with a 22 × 45-mm coverslip (Sigma); then it was placed in a sealed, humidified chamber (TeleChem International). Hybridization was carried out in a 65°C water bath for 16 h; after that, the slides were washed with five changes of buffers in the order 2 × SSC/0.1%SDS, 1 × SSC/0.1%SDS, 1 × SSC, 0.2 × SSC, and 0.05 × SSC (10 min each at room temperature), and then spun dry (10 min at 500 rpm).
Data Processing and Analysis
Using a ScanArray 5000 laser scanner, hybridized arrays were first scanned with ‘quickscan’ to determine the array area as well as the appropriate laser power and photo multiplier (PMT) value without causing photo-bleaching. On determining those parameters, the slides were scanned (5 μm) sequentially for Cy5—first followed by Cy3. The two images obtained from each fluorophore were superimposed manually for analysis using the software QuantArray (GSI Lumonics). Normalization of the intensity values from the two channels was performed using the method of lowness (R program) as described (http://www.stat.berkeley.edu/users/terry/zarray/Html/papersindex.html, Yang et al. 2001). In most cases, ratios presented here (Supplementary Table 8) represent the average from four independent hybridizations (reciprocal plus duplicate). Only in a few cases, average ratios were obtained from two or three available hybridization data. Hierarchical clustering and K-means clustering analysis was performed as described (http://ep.ebi.ac.uk/EP/EPCLUST/). Statistics and data processing were performed on Microsoft Excel and Access programs. SVM is constructed based on the SVM Toolbox (http://theoval.sys.uea.ac.uk/∼gcc/svm/toolbox) developed by Gavin Cawley.
Northern Blot Analysis
Ten micrograms of total RNA was separated on a formaldehyde gel and then transferred to a nylon membrane (Hybond N+, Amersham). Probes for candidate genes were DIG-labeled (Roche Molecular Biochemicals) through PCR amplification using vector primer pairs T3/T7, respectively. Hybridization was performed as described previously (Lee et al. 2002).
In Situ Hybridization
Samples of 24-h embryos were pretreated and fixed as described (Jowett 1997). To generate probes, we PCR amplified inserts of candidate genes using the vector outer primer pairs M13 forward/reverse, and the obtained PCR product (Roche Molecular Biochemicals) was used as a template for in vitro transcription (T3/T7 polymerase, NEB) to generate DIG-labeled (Roche Molecular Biochemicals) sense and anti-sense probes. In situ hybridization was performed as described (Jowett 1997).
WEB SITE REFERENCES
ftp://ftp.ncbi.nlm.nih.gov/blast/db/nr; Non-redundant (NCBI).
ftp://ftp.ncbi.nlm.nih.gov/repository/Unigene/Dr.seq.uniq; National Centre for Biotechnology Information (NCBI).
http://ep.ebi.ac.uk/EP/EPCLUST/; Expression Profiler of European Bioinformatics Institute (EBI) for hierarchy clustering and K-means clustering.
http://theoval.sys.uea.ac.uk/∼gcc/svm/toolbox; SVM Toolbox (by Gavin Cawley).
http://www.mrc-lmb.cam.ac.uk/pubseq/manual/gap4_unix_toc.html; MRC gap4 program in Staden Package for sequence alignment.
http://www.mrc-lmb.cam.ac.uk/pubseq/manual/pregap4_unix_toc.html; MRC pregap4 program in Staden Package for sequence editing.
http://www.tigr.org/software/assembler; Tigr-Assembler.
http://zfish.wustl.edu/; Washington University–Zebrafish Genome Resources Project
Acknowledgments
We thank Dr. Steve Johnson for his useful discussion on EST sequencing during his visit in Singapore. We thank Dr. Robert Schaffer, Matthew Larson, and Dr. Ellen Wisman (Michigan State University) for helpful advice on techniques in microarray. We thank Dr. Wai Ming Kong and Dr. Keng Wah Choo (Nanyang Polytechnic, Singapore) for helping with data normalization. We thank Dave Oh in the IMA sequencing unit for performing part of the EST sequencing. This work is financially supported by the Agency for Science, Technology and Research (A*STAR) in Singapore.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL pengjr@imcb.a-star.edu.sg; FAX 65-68727007.
E-MAIL zilong@imcb.a-star.edu.sg; FAX 65-68727007.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.885403.
REFERENCES
- 1.Amsterdam A., Burges, S., Golling, G., Chen, W., Sun, Z., Townsend, K., Farrington, S., Haldi, M., and Hopkins, N. 1999. A large-scale insertional mutagenesis screen in zebrafish. Genes & Dev. 13: 2713-2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ando H., Furuta, T., Tsien, R.Y., and Okamoto, H. 2001. Photo-mediated gene activation using caged RNA/DNA in zebrafish embryos. Nat. Genet. 28: 317-325. [DOI] [PubMed] [Google Scholar]
- 3.Barresi M.J.F., Stickney, H.L., and Devoto, S.H. 2000. The zebrafish slow-muscle-omitted gene product is required for Hedgehog signal transduction and the development of slow muscle identity. Development 127: 2189-2199. [DOI] [PubMed] [Google Scholar]
- 4.Brown M.P.S., Grundy, W.N., Lin, D., Cristianini, N., Sugnet, C.W., Furey, T.S., Ares, M., Jr., and Haussler, D. 2000. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc. Natl. Acad. Sci. 97: 262-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho R.J., Campbell, M.J., Winzeler, E.A., Steinmetz, L., Conway, A., Wodicka, L., Wolfsberg, T.G., Gabrielian, A.E., Landsman, D., Lockhart, D.J., et al. 1998. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol. Cell 2: 65-73. [DOI] [PubMed] [Google Scholar]
- 6.Coimbra R.S., Weil, D., Brottier, P., Blanchard, S., Levi, M., Hardelin, J-P., Weissenbach, J., and Petit, C. 2002. A subtracted cDNA library from the zebrafish (Danio rerio) embryonic inner ear. Genome Res. 12: 1007-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Didier D.K., Schiffenbauer, J., Woulfe, S.L., Zacheis, M., and Schwartz, B.D. 1988. Characterization of the cDNA encoding a protein binding to the major histocompatibility complex class II Y box. Proc. Natl. Acad. Sci. 85: 7322-7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dooley K. and Zon, L.I. 2000. Zebrafish: A model system for the study of human disease. Curr. Opin. Genet. Dev. 10: 252-256. [DOI] [PubMed] [Google Scholar]
- 9.Driever W., Solnica-Krezel, L., Schier, A.F., Neuhauss, S.C.F., Malicki, J., Stemple, D.L., Stainier, D.Y., Zwartkruis, F., Abdelilah, S., Rangini, Z., et al. 1996. A genetic screen for mutations affecting embryogenesis in zebrafish. Development 123: 37-46. [DOI] [PubMed] [Google Scholar]
- 10.Eisen M., Spellman, P., Brown, P., and Botstein, D. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. 95: 14863-14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furlong E.E.M., Anderson, E.C., Null, B., White, K.P., and Scott, M.P. 2001. Patterns of gene expression during Drosophila mesoderm development. Science 293: 1629-1633. [DOI] [PubMed] [Google Scholar]
- 12.Gai X.X., Lipson, K.E., and Prystowsky, M.B. 1992. Unusual DNA binding characteristics of an in vitro translation product of the CCAAT binding protein mYB-1. Nucleic Acids Res. 20: 601-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gates M.A., Kim, L., Egan, E.S., Cardozo, T., Sirotkin, H.I., Dougan, S.T., Laskari, D., Abagyan, R., Schier, A.F., and Talbot, W.S. 1999. A genetic linkage map for zebrafish: Comparative analysis of genes and expressed sequences. Genome Res. 9: 334-347. [PubMed] [Google Scholar]
- 14.Golling G., Amsterdam, A., Sun, Z., Antonelli, M., Maldonado, E., Chen, W., Burgess, S., Haldi, M., Artzt, K., Farrington, S., et al. 2002. Insertional mutagenesis in zebrafish rapidly identifies genes essential for early vertebrate development. Nat. Genet. 31: 135-140. [DOI] [PubMed] [Google Scholar]
- 15.Grant C.E. and Deeley, R.G. 1993. Cloning and characterization of the chicken YB-1: Regulation of expression in liver. Mol. Cell. Biol. 13: 4186-4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haffter P., Granato, M., Brand, M., Mullins, M.C., Hammerschmidt, M., Kane, D.A., Odenthal, J., van Eeden, F.J.M., Jiang, Y.J., Heisenberg, C.P., et al. 1996. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development 123: 1-36. [DOI] [PubMed] [Google Scholar]
- 17.Halpern M.E., Ho, R.K., Walker, C., and Kimmel, C.B. 1993. Induction of muscle pioneers and floor plate is distinguished by the zebrafish no tail mutation. Cell 75: 99-111. [PubMed] [Google Scholar]
- 18.Jowett T., 1997. Tissue in situ hybridization: Methods in animal development. John Wiley & Sons, New York. NY.
- 19.Karlstrom R.O., Talbot, W.S., and Schier, A.F. 1999. Comparative synteny cloning of zebrafish you-too: Mutations in the hedgehog target gli2 affect ventral forebrain patterning. Genes & Dev. 13: 388-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimmel C.B., Ballard, W.W., Kimmel, S.R., Ullmann, B., and Schilling, T.F. 1995. Stages of embryonic development of the zebrafish. Dev. Dyn. 203: 253-310. [DOI] [PubMed] [Google Scholar]
- 21.Knapik E.W., Goodman, A., Ekker, M., Chevrette, M., Delgado, J., Neuhauss, S., Shimoda, N., Driever, W., Fishman, M.C., and Jacob, H.J. 1998. A microsatellite genetic linkage map for zebrafish (Danio rerio). Nat. Genet. 18: 338-343. [DOI] [PubMed] [Google Scholar]
- 22.Ko M.S.H. 1990. An “equalized cDNA library” by the reassociation of short double stranded cDNAs. Nucleic Acids Res. 18: 5705-5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee S.C., Cheng, H., King, K.E., Wang, W., He, Y., Hussian, A., Lo, J., Harberd, N.P., and Peng, J.R. 2002. Gibberellin regulates Arabidopsis seed germination via RGL2, a GAI/RGA-like gene whose expression is up-regulated following imbibition. Genes & Dev. 16: 646-658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y-X., Farrell, M.J., Liu, R-P., Mohanty, N., and Kirby, M.L. 2000. Double-stranded RNA injection produces null phenotype in zebrafish. Dev. Biol. 217: 394-405. [DOI] [PubMed] [Google Scholar]
- 25.Makabe K.W., Kawashima, T., Kawashima, S., Minokawa, T., Adachi, A., Kawamura, H., Ishikawa, H., Yasuda, R., Yamamoto, H., Kondoh, K., et al. 2001. Large-scale cDNA analysis of the maternal genetic information in the egg of Halocynthia roretzi for a gene expression catalog of ascidian development. Development 128: 2555-2567. [DOI] [PubMed] [Google Scholar]
- 26.McDonald M.J. and Rosbash, M. 2001. Microarray analysis and organization of circadian gene expression in Drosophila. Cell 107: 567-578. [DOI] [PubMed] [Google Scholar]
- 27.Nasevicius A. and Ekker, S.C. 2000. Effective targeted gene “knockdown” in zebrafish. Nat. Genet. 26: 216-220. [DOI] [PubMed] [Google Scholar]
- 28.Patanjali S.R., Parimoo, S., and Weissman, S.M. 1991. Construction of a uniform abundance (normalized) cDNA library. Proc. Natl. Acad. Sci. 88: 1943-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Postlethwait J.H., Yan, Y.L., Gates, M.A., Horne, S., Amores, A., Brownlie, A., Donovan, A., Egan, E.S., Force, A., Gong, Z., et al. 1998. Vertebrate genome evolution and the zebrafish gene map. Nat. Genet. 18: 345-349. [DOI] [PubMed] [Google Scholar]
- 30.Roy S., Wolff, C., and Ingham, P.W. 2001. The u-boot mutation identifies a Hedgehog-regulated myogenic switch for fiber-type diversification in the zebrafish embryo. Genes & Dev. 15: 1563-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schulte-Merker S., van Eeden, F.J., Halpern, M.E., Kimmel, C.B., and Nusslein-Volhard, C. 1994. no tail (ntl) is the zebrafish homologue of the mouse T (Brachyury) gene. Development 120: 1009-1015. [DOI] [PubMed] [Google Scholar]
- 32.Shimoda N., Knapik, E.W., Ziniti, J., Sim, C., Yamada, E., Kaplan, S., Jackson, D., de Sauvage, F., Jacob, H., and Fishman, M.C. 1999. Zebrafish genetic map with 2000 microsatellite markers. Genomics 58: 219-232. [DOI] [PubMed] [Google Scholar]
- 33.Streisinger G., Walker, C., Dower, N., Knauber, D., and Singer, F. 1981. Production of clones of homozygous diploid zebrafish (Brachydanio rerio). Nature 291: 293-296. [DOI] [PubMed] [Google Scholar]
- 34.Streisinger G., Singer, F., Walker, C., Knauber, D., and Dower, N. 1986. Segregation analyses and gene-centromere distances in zebrafish. Genetics 112: 311-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Talbot W.S. and Hopkins, N. 2000. Zebrafish mutations and functional analysis of the vertebrate genome. Genes & Dev. 14: 755-762. [PubMed] [Google Scholar]
- 36.Weinberg E.S., Allende, M.L., Kelly, C.S., Abdelhamid, A., Andermann, P., Doerre, G., Grunwald, D.J., and Riggleman, B. 1996. Developmental regulation of zebrafish MyoD in wild-type, no tail, and spadetail embryos. Development 122: 271-280. [DOI] [PubMed] [Google Scholar]
- 37.Xie Y.F., Chen, X., and Wagner, T.E. 1997. A ribozyme-mediated, gene “knock-down” strategy for the identification of gene function in zebrafish. Proc. Natl. Acad. Sci. 94: 13777-13781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Y-F., He, J-Y., Wang, X-K., Lim, T-M., and Gong, Z-Y. 2000. Asynchronous activation of 10 muscle-specific protein (MSP) genes during zebrafish somitogenesis. Dev. Dyn. 219: 201-215. [DOI] [PubMed] [Google Scholar]
- 39.Yang Y.H., Dudoit, S., Luu, P., and Speed, T.P., 2001. Normalization for cDNA microarray data. SPIE BiOS 2001, San Jose, CA. http://www.stat.berkeley.edu/users/terry/zarray/Html/papersindex.html.
- 40.Zhang J., Talbot, W.S., and Schier, A.F. 1998. Positional cloning identifies zebrafish one-eyed pinhead as a permissive EGF-related ligand required during gastrulation. Cell 92: 241-251. [DOI] [PubMed] [Google Scholar]