

Abstract
The capability to conditionally inactivate gene function is essential for understanding the molecular basis of development. In gene and mRNA targeting approaches, protein products can perdure, complicating genetic analysis. Current methods for selective protein degradation require drug treatment or take hours for protein removal, limiting their utility in studying rapid developmental processes in vivo. Here, we repurpose an endogenous protein degradation system to rapidly remove targeted C. elegans proteins. We show that upon expression of the E3 ubiquitin ligase substrate-recognition subunit ZIF-1, proteins tagged with the ZF1 zinc-finger domain can be quickly degraded in all somatic cell types examined with temporal and spatial control. We demonstrate that genes can be engineered to become conditional loss-of-function alleles by introducing sequences encoding the ZF1 tag into endogenous loci. Finally, we use ZF1 tagging to establish the site of cdc-42 gene function during a cell invasion event. ZF1 tagging provides a powerful new tool for the analysis of dynamic developmental events.
Keywords: C. elegans, Genetic tool, Mosaic, Protein degradation
INTRODUCTION
Conditional gene inactivation is an essential tool for elucidating the molecular mechanisms of organismal development. Genes tagged with recombinogenic sequences can be deleted conditionally by expressing site-specific recombinases such as Cre or Flp (del Valle Rodriguez et al., 2012; Jones et al., 2005). Alternatively, RNA gene products can be removed using strategies such as RNA interference (RNAi), which induces the degradation of targeted mRNAs (Fire et al., 1998). However, neither conditional gene deletion nor RNAi removes protein gene products, which can perdure and mask phenotypes until they decay. Protein persistence is a particular problem when conditionally inactivating genes in embryos. In C. elegans, Drosophila and zebrafish, significant levels of maternally derived mRNA and proteins persist through key developmental stages, as evidenced by the numerous maternal-effect mutations that have been identified in these species (Abrams and Mullins, 2009; Perrimon and Gans, 1983; Wood et al., 1980). Maternal gene products also contribute to early mammalian development, likely to a greater extent than currently appreciated (Li et al., 2010).
Several methods have been developed to cleave or degrade targeted proteins directly. Strategies include introducing temperature-sensitive protease cleavage sites, or using heterologous adaptor proteins, drugs or light to induce the ubiquitin-mediated degradation of a tagged target protein (Banaszynski et al., 2006; Bonger et al., 2011; Nishimura et al., 2009; Raina and Crews, 2010; Renicke et al., 2013). For example, using a procedure named deGradFP, proteins fused to green fluorescent protein (GFP) can be depleted by expressing a fusion protein consisting of an anti-GFP antibody coupled to an F-box domain; the fusion protein recruits GFP-tagged protein to SKP1-CUL1-F-box (SCF) E3 ubiquitin ligase complexes, marking it for ubiquitylation and proteasome-mediated degradation (Caussinus et al., 2012). deGradFP has been shown to degrade target proteins over a period of 2-3 hours (Caussinus et al., 2012), comparable with the kinetics of most other protein degradation strategies (Bonger et al., 2011; Taxis et al., 2009; Zhou et al., 2000). However, many developmental events occur on a much faster timescale. In the C. elegans embryo, for example, multiple rounds of cell division, many cell fate specification events, and morphogenetic processes occur within the first 2 hours of development (Goldstein, 1992; Lee and Goldstein, 2003; Nance and Priess, 2002; Priess and Thomson, 1987), and most embryonic cells are born within 7 hours (Sulston et al., 1983). Even during the relatively prolonged 2-day larval phase of development, crucial morphogenetic and signaling events, such as uterine-vulval specification and morphogenesis, occur within short time periods (Kimble, 1981; Sharma-Kishore et al., 1999; Sherwood and Sternberg, 2003; Wang and Sternberg, 2000). An inducible protein degradation system has been described that can deplete proteins in less than 1 hour, but this method requires the addition of auxin, which may not be permeable to many living embryos and organisms (Holland et al., 2012; Nishimura et al., 2009). Therefore, existing protein-degradation strategies are poorly suited to study rapid developmental events in vivo.
Here, we repurpose an endogenous C. elegans protein degradation mechanism to remove heterologous proteins that are tagged with a small zinc-finger domain called ZF1. We show that ZF1 tag-mediated degradation of heterologous target proteins occurs within 30-45 min, functions in all somatic cell types examined and can recapitulate loss-of-function phenotypes. Using genome editing, we convert a wild-type allele into a conditional loss-of-function allele by inserting sequences encoding the ZF1 tag into the endogenous locus. Finally, using ZF1 tagging we demonstrate a cell-autonomous role in anchor cell invasion for the Rho GTPase CDC-42, a protein that is essential at earlier developmental stages.
RESULTS
zif-1 expression reactivates ZF1-mediated degradation
The 36 amino acid PIE-1 C-C-C-H type zinc-finger domain (ZF1) targets endogenous PIE-1 protein for degradation in somatic cells of the early embryo, thereby helping to restrict PIE-1 to the single germ-line precursor cell (Reese et al., 2000). We and others showed previously that fusing the ZF1 domain to heterologous cytosolic or transmembrane proteins is sufficient to trigger their rapid degradation within early embryonic somatic cells (Achilleos et al., 2010; Anderson et al., 2008; Chan and Nance, 2013; Chihara and Nance, 2012; Nance et al., 2003; Reese et al., 2000; Totong et al., 2007; Wehman et al., 2011), mimicking loss-of-function phenotypes. For example, degradation of the RhoGAP PAC-1 produces cell polarity defects in somatic cells of the early embryo that are identical to those of pac-1 null mutants (Anderson et al., 2008), and degradation of the RhoGEF ECT-2 causes cytokinesis defects, similar to ect-2(RNAi) early embryos (Chan and Nance, 2013). ZF1-mediated degradation is thought to occur when ZIF-1, a maternally expressed SOCS-box adaptor protein that binds to ZF1 domains, recruits ZF1-containing protein to an ECS (Elongin-C, Cul2, SOCS-box family) E3 ubiquitin ligase complex for subsequent proteasome-mediated destruction (DeRenzo et al., 2003) (Fig. 1A). ZF1-tagged proteins are not degraded at later stages of embryogenesis or in larvae (Achilleos et al., 2010; Chan and Nance, 2013; Chihara and Nance, 2012; Nance et al., 2003; Totong et al., 2007), presumably because ZIF-1 or components of the ECS E3 ubiquitin ligase complex are absent or inactive at these stages.
Fig. 1.
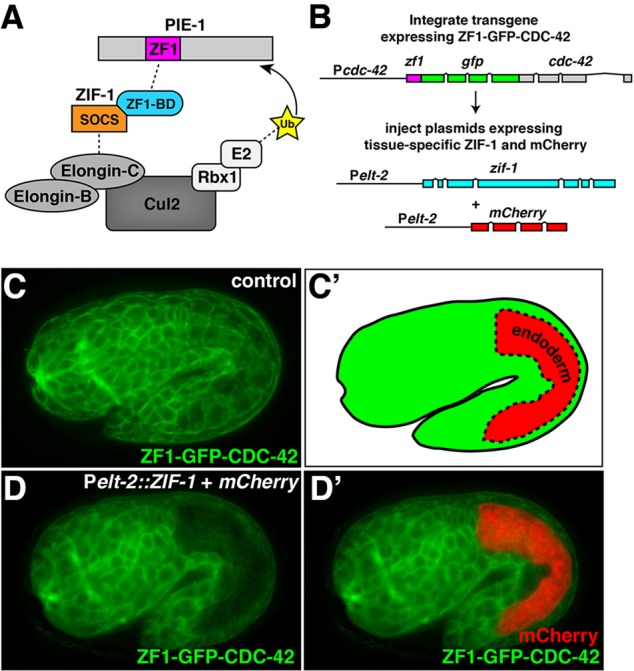
ZF1-tagged protein degradation in endodermal cells. (A) Schematic model of ZIF-1 recruiting the ZF1-containing protein PIE-1 to an ECS E3 ligase complex for ubiquitylation (Ub) and subsequent degradation. (B) Strategy for testing effectiveness of ZF1 tagging at later developmental stages. (C,C′) ZF1-GFP-CDC-42 expression in a 1.75-fold stage embryo; the position of the endodermal cells is indicated in C′; expression was detected in endodermal cells, in addition to all other somatic cells (n=14). (D,D′) ZF1-GFP-CDC-42 in a 1.5-fold embryo containing Pelt-2::ZIF-1+mCherry; ZF1-GFP-CDC-42 has degraded specifically in endodermal cells (n=22/22), where mCherry is expressed (D′). Whole embryos are ∼50 µm in length.
The ECS E3 ligase components elongin C (elc-1), cul-2 and rbx-1 are expressed throughout development, while zif-1 expression is enriched in or limited to the germline and early embryo (Levin et al., 2012). This suggested that the absence of ZIF-1 might limit the activity of the degradation system later in development. To test this, we expressed ZIF-1 at later developmental stages and monitored the degradation of an integrated transgene that expresses the ubiquitous Rho GTPase CDC-42 in somatic tissues, fused at its N-terminus with the ZF1 tag and GFP (Fig. 1B). ZF1-GFP-CDC-42 appeared in somatic cells throughout the embryo several hours after fertilization and was enriched at the plasma membrane (Fig. 1C). To determine whether reintroducing ZIF-1 would degrade ZF1-GFP-CDC-42, we first expressed ZIF-1 from the elt-2 promoter (Pelt-2::ZIF-1), which drives expression specifically in endodermal cells (Fig. 1C′, red region) from the middle stages of embryogenesis onwards (Fukushige et al., 1998). Because previous attempts to tag ZIF-1 fluorescently did not produce visible fusion protein (DeRenzo et al., 2003), we monitored ZIF-1 expression indirectly by co-expressing mCherry from the same promoter (Fig. 1B). Pelt-2::ZIF-1 expression from a transgenic array, which did not appear to cause any toxicity, triggered a dramatic elimination of ZF1-GFP-CDC-42 specifically in endodermal cells (Fig. 1D,D′). ZF1-GFP-CDC-42 depletion occurred before mCherry expression was evident, presumably because mCherry requires additional time to fold. We induced a similar depletion of ZF1-GFP-CDC-42 in larval neuronal tissues by using the neuron-specific rab-3 promoter (Mahoney et al., 2006) to express ZIF-1 (supplementary material Fig. S1A-B′). Thus, reintroducing the adaptor protein ZIF-1 is sufficient to trigger the elimination of ZF1-tagged proteins in at least a subset of embryonic and larval somatic tissues.
ZF1-tagged proteins can be degraded in most or all somatic cells
We next asked which embryonic and larval tissues, in addition to endoderm and neurons, are competent for ZF1-mediated degradation. For these experiments, we used the cdc-42 promoter to express ZIF-1 ubiquitously and monitored expression from the transgene indirectly using a Pcdc-42::mCherry reporter. Pcdc-42::ZIF-1 (together with Pcdc-42::mCherry) was expressed from a transgenic array, which is inherited by a subset of progeny and at low frequency is lost during cell division to yield mosaic animals. Controls, which did not inherit the transgenic array containing Pcdc-42::ZIF-1, expressed ZF1-GFP-CDC-42 in most or all somatic tissues of embryos and larvae (Fig. 2A,C′). By contrast, we failed to detect ZF1-GFP-CDC-42 expression in embryos or larvae that inherited the Pcdc-42::ZIF-1 transgenic array (Fig. 2B,C′). In a small number of animals, ZF1-GFP-CDC-42 was visible in a few cells, although the identity of these cells was not reproducible; these animals are likely genetic mosaics that lost the transgenic array in cells with persistent ZF1-GFP-CDC-42 expression. We conclude that most or all somatic cells in the animal are capable of degrading ZF1-tagged proteins to visibly undetectable levels upon reintroduction of ZIF-1. These findings suggest that other components of the ECS E3 ubiquitin ligase complex are expressed ubiquitously, and that ZIF-1 is the limiting factor needed to degrade ZF1-tagged proteins.
Fig. 2.

ZIF-1-mediated degradation in all somatic tissues. (A) Control 1.5-fold embryo expressing ZF1-GFP-CDC-42 in all somatic cells (n=22). (B,B′) ZF1-GFP-CDC-42 in an embryo containing Pcdc-42::ZIF-1+mCherry, which induces ZF1-GFP-CDC-42 degradation (n=41/41). (C-C″) DIC (C), GFP (C′) and GFP/mCherry (C″) channels showing larvae expressing ZF1-GFP-CDC-42. Control larvae express ZF1-GFP-CDC-42 in most or all somatic cells (n=37). In larvae also containing Pcdc-42::ZIF-1+mCherry (arrow), ZF1-GFP-CDC-42 degrades (n=14/14). (D) Western analysis of ZF1-GFP-CDC-42 levels in worms lacking (left two lanes) or expressing (right two lanes) ZIF-1 protein from two different Pcdc-42::ZIF-1+mCherry transgene insertions (xnIs520 and xnIs521). The top and bottom blots were probed with anti-GFP antibody. The leftmost lane is a wild-type (N2) control. Scale bar: 100 µm. Whole embryos are ∼50 µm in length.
To determine the extent of ZF1-GFP-CDC-42 degradation by ZIF-1 using a more sensitive assay, we performed western analysis, comparing ZF1-GFP-CDC-42 levels in embryos with and without ubiquitously expressed Pcdc-42::ZIF-1 (Fig. 2D). To generate transgene insertions that express ZIF-1 ubiquitously without the mosaicism that is characteristic of transgenic arrays, we integrated a high-copy transgenic array containing Pcdc-42::ZIF-1 and Pcdc-42::mCherry. Neither of the two independent transgene insertions isolated, xnIs520 and xnIs521, caused embryonic lethality [dead eggs/total progeny: wild type, 12/568 (2.1%); xnIs520: 2/511 (0.4%); xnIs521: 3/405 (0.7%)] or other obvious developmental defects, demonstrating that ubiquitous ZIF-1 expression is not deleterious to development. ZF1-GFP-CDC-42 was easily detected in adult worms containing Pcdc-42::ZF1-GFP-CDC-42 alone. By contrast, expression was nearly eliminated in worms that also expressed Pcdc-42::ZIF-1 from either transgene insertion (Fig. 2D). We conclude that ectopically expressing ZIF-1 can degrade ZF1-tagged proteins to undetectable levels and does not cause lethality or developmental defects.
ZF1-tagged proteins degrade rapidly
In early embryos, ZF1-mediated degradation occurs exclusively in somatic cells (Reese et al., 2000), which are born through a series of four asymmetric divisions, beginning with the cleavage of the zygote; at each division, one daughter retains the germ-line precursor cell fate, whereas the other differentiates into a somatic cell. Maternally expressed ZF1-tagged proteins such as PAR-3 and PAR-6 begin to degrade visibly from the daughters of the first somatic cell during the four-cell stage, and degradation in this lineage is usually complete one cell cycle later (Nance et al., 2003). Therefore, endogenous ZIF-1 in early embryonic somatic cells can target degradation of ZF1-tagged proteins within a period of ∼30-45 min. To determine whether the kinetics of ZF1-mediated degradation are similar when ZIF-1 is supplied exogenously at later stages, we expressed ZIF-1 and mCherry from a heat-shock promoter and monitored the degradation of ZF1-GFP-CDC-42 in embryos using time-lapse microscopy. Following a 15 min heat shock to induce ZIF-1 expression, ZF1-GFP-CDC-42 (normalized to controls) degraded sharply: 50% remained at 21 min, 8% remained at 30 min and <1% remained at 40 min (Fig. 3; supplementary material Movie 1). Thus, ZF1-tagged proteins can be degraded rapidly throughout development.
Fig. 3.

Time-lapse analysis of induced ZF1-mediated degradation. (A-F) Frames from time-lapse movies of embryos expressing ZF1-GFP-CDC-42 and either lacking (A-C) or containing (D-F) Phsp::ZIF-1+mCherry. ZF1-GFP-CDC-42 expression persists in embryos lacking Phsp::ZIF-1+mCherry, whereas ZF1-GFP-CDC-42 degrades and mCherry is expressed in embryos containing Phsp::ZIF-1+mCherry. (G) GFP fluorescence levels from regions of interest (boxed region) in embryos containing Phsp::ZIF-1+mCherry relative to controls captured on the same slide. Dashed lines indicate 50% and 90% degradation. Error bars represent s.e.m. from three independent experiments. Whole embryos are ∼50 µm in length.
Endogenous genes can be engineered into conditional loss-of-function alleles by inserting ZF1-coding sequences
To determine whether depleting proteins by ZF1-tagging phenocopies loss-of-function mutations in the corresponding gene, we compared embryos with compromised maternal and zygotic function of the sec-5 gene with embryos where SEC-5 protein was removed by ZF1-tagging. sec-5 encodes a component of the exocyst complex and is an essential gene expressed throughout development (Armenti et al., 2014; Dupuy et al., 2007; Frische et al., 2007). Owing to maternal contribution, sec-5 mutants homozygous for the nonsense allele pk2358 are viable when obtained from a self-fertilized heterozygous mother (Frische et al., 2007). However, all progeny produced by homozygous sec-5(pk2358) mutants (sec-5 maternal-zygotic mutant embryos) die (Frische et al., 2007) (Table 1). To determine whether depleting SEC-5 by ZF1-tagging causes a similar lethal phenotype, we used CRISPR/Cas9 genome editing (Dickinson et al., 2013) to insert sequences encoding the ZF1 tag and YFP (as well as the unc-119 transformation marker) into the endogenous sec-5 locus, creating the sec-5(xn51[sec-5-zf1-yfp + unc-119(+)]) knock-in allele. SEC-5-ZF1-YFP was expressed maternally, degraded rapidly in early embryonic somatic cells and subsequently reappeared during the middle stages of embryogenesis as a result of zygotic expression (supplementary material Fig. S2A-C). Moreover, the ZF1 and YFP tags did not interfere with SEC-5 function, as homozygous sec-5(xn51) worms were viable and fertile (Table 1). To degrade SEC-5-ZF1-YFP, we crossed the Pcdc-42::ZIF-1 transgenic array into the sec-5(xn51) knock-in background. All progeny of genotype sec-5(xn51); Pcdc-42::ZIF-1 lacked detectable SEC-5-ZF1-YFP expression (supplementary material Fig. S2D,E) and died (Table 1), like sec-5(pk2358) maternal-zygotic mutants. We conclude that ZF1-tagging can recapitulate loss-of-function phenotypes at later stages of development, and that ZF1-coding sequences can be engineered directly into an endogenous locus to produce a conditional loss-of-function allele. ZF1-coding sequences are likely to be similarly effective when inserted into various regions of the coding sequence, as SEC-5-ZF1-YFP degraded at a rate comparable with ZF1-GFP-CDC-42 when we expressed ZIF-1 from a heat-shock promoter (50% remaining after 31 min; supplementary material Fig. S3).
Table 1.
Comparison of sec-5 mutation and depletion of SEC-5 by ZF1-tagging

ZF1-mediated degradation of CDC-42 reveals a cell-autonomous role for CDC-42 in cell invasion
To establish the utility of ZF1 tagging for investigating gene function, we examined whether this approach could determine the site of action of CDC-42 during anchor cell invasion. Anchor cell invasion is a visually and experimentally tractable model for understanding invasion through the basement membrane: the dense and highly cross-linked barrier that surrounds most tissues. In the L3 larval stage, the uterine anchor cell invades through the underlying basement membrane and contacts the central (primary) vulval precursor cells to initiate uterine-vulval connection (Sherwood and Sternberg, 2003) (supplementary material Fig. S4). Anchor cell invasion is regulated cell-autonomously by genes that promote invasion in the anchor cell, and non-autonomously by a pro-invasive signal from the primary vulval cells (Matus et al., 2010). A previous whole-body RNAi screen identified cdc-42 as a gene required for anchor cell invasion (Matus et al., 2010) (supplementary material Fig. S4). ZF1-GFP-CDC-42 is expressed broadly throughout the gonad and vulval cells, suggesting it could function in the anchor cell, the vulval precursor cells or both (Fig. 4A-A‴). Anchor cell invasion was blocked in 34% of homozygous cdc-42(gk388) mutants from maternally rescued embryos (n=50), and invasion was fully restored upon expression of ZF1-GFP-CDC-42 (100% invasion, n=50). To determine the site of action of CDC-42, we used cell-specific expression of ZIF-1. We employed the 5′ cis-regulatory element of the egl-17 gene to direct ZIF-1 expression in the primary vulval precursor cells and used a 5′cis-regulatory element of the cdh-3 gene to drive anchor cell-specific expression of ZIF-1 (Kirouac and Sternberg, 2003). When ZF1-GFP-CDC-42 was depleted by expressing ZIF-1 specifically in the primary vulval precursor cells (Pegl-17::ZIF-1 + mCherry) in homozygous cdc-42(gk388) mutants, anchor cell invasion through the basement membrane was not affected, even though no ZF1-GFP-CDC-42 was detectable in the primary vulval precursor cells (100% invasion, n=20; Fig. 4B-B‴). Expression of Pcdh-3::ZIF-1 + mCherry in homozygous cdc-42(gk388) mutants depleted ZF1-GFP-CDC-42 specifically in the anchor cell and resulted in 40% blocked invasion (n=20; Fig. 4C-C‴), similar to cdc-42(gk388) mutants alone. Together, these findings indicate that CDC-42 functions in the anchor cell to regulate its invasion through basement membrane. Therefore, tissue-specific depletion of proteins by ZF1-tagging can be used to determine the larval stage site of action of genes, such as cdc-42 (Gotta et al., 2001; Kay and Hunter, 2001), that are required earlier in development.
Fig. 4.

CDC-42 functions within the anchor cell to regulate invasion. (A-A‴) A DIC image (A), ZF1-GFP-CDC-42 expression in homozygous cdc-42(gk388) (A′), mCherry fluorescence of worm lacking mCherry construct (A″) and a schematic diagram (A‴). The anchor cell (arrowhead, labeled AC in A‴) has breached the basement membrane (arrow, interruption in phase-dense line in the DIC image; see black line labeled BM in schematic) and contacted the central primary vulval precursor cells (bracket) (n=50/50 normal invasion). (B-B‴) Expression of ZIF-1 in the primary vulval cells (B″, Pegl-17::ZIF-1+mCherry) led to loss of detectable ZF1-GFP-CDC-42 (B′), but did not affect anchor cell invasion (n=20/20 invaded normally). (C-C‴) Expression of ZIF-1 in the anchor cell (C″, Pcdh-3::ZIF-1+mCherry) led to loss of detectable ZF1-GFP-CDC-42 (C′, there is complete loss of fluorescence within the anchor cell; fluorescence outlining the anchor cell is from neighboring cell membranes) and resulted in an invasion defect (non-invaded basement membrane indicated by intact phase-dense line in DIC image; n=8/20 blocked invasion; P<0.005, Fisher's exact test). Scale bar: 5 µm.
DISCUSSION
Assessing gene function within a specific tissue or during a precise developmental stage remains challenging for genes with essential roles earlier in development. C. elegans geneticists have circumvented these difficulties using strategies such as mosaic analysis and tissue-specific RNAi. However, these approaches can only reveal full loss-of-function phenotypes once pre-existing protein in the targeted cell type decays. Furthermore, neither technique is always feasible or desirable. For example, in many cases identifying genetic mosaics requires considerable effort (Yochem and Herman, 2003), and RNAi works poorly in some tissues and is often incompletely effective (Asikainen et al., 2005; Fire et al., 1998; Kamath et al., 2001). Here, we introduce ZF1 tagging as a novel approach to rapidly and acutely inactivate gene function at the protein level with spatial and temporal resolution. ZF1 tagging provides a powerful genetic tool with which to study dynamic developmental events, such as those that occur within the rapidly dividing embryo, where perduring maternal protein can mask phenotypes. In addition, ZF1 tagging should greatly facilitate the analysis of essential genes in specific tissues or cells in larvae or adults – a role we have demonstrated here for the essential cdc-42 gene in anchor cell invasion. Given the difficulty in obtaining effective RNAi knockdown in many neuronal cell types (Asikainen et al., 2005), ZF1-tagging should prove particularly useful in the study of essential neuronal genes in adult behaviors.
Most proteins should be amenable to analysis by ZF1 tagging. For example, we have shown previously that both cytoplasmic (Anderson et al., 2008; Chan and Nance, 2013; Nance et al., 2003) and transmembrane proteins (Chihara and Nance, 2012; Wehman et al., 2011) are effectively degraded. However, a few types of proteins would be difficult to analyze, including those essential for cell viability or division; these proteins would be removed in the early embryo by endogenous ZIF-1 protein, causing cell death or division defects. However, the window in which endogenous ZIF-1 degrades proteins is small, and transient loss of many important proteins (like CDC-42 and SEC-5) can be tolerated. Moreover, maternal contribution of the wild-type protein could be used to bypass this window, permitting study of the essential gene at later stages.
We have shown that ZF1-tagged proteins can be degraded in all examined embryonic and larval somatic cell types, and by altering the promoter used to express ZIF-1, we have demonstrated both spatial (tissue-specific promoter) and temporal (heat-shock promoter) control of targeted protein destruction. By combining ZF1 tagging with existing conditional gene expression technologies, such as the FLP-FRT system and tissue-specific heat-shock methods (Davis et al., 2008; Voutev and Hubbard, 2008; Bacaj and Shaham, 2007; Churgin et al., 2013), it should be feasible to achieve simultaneous spatial and temporal control of gene product function. Using recently developed genome editing techniques, we have shown that sequences encoding the ZF1 tag can be inserted directly into endogenous loci, greatly facilitating genetic analysis by ZF1-tagging and obviating the need for a loss-of-function mutation. We envision that this approach will be feasible for most genes, especially given the small size of the ZF1 tag (36 amino acids), which is unlikely to interfere with the function of most tagged proteins. Moreover, the ZF1 tag itself appears robust, as we have used it to tag proteins at the N-terminus (ZF1-GFP-CDC-42) or C-terminus (SEC-5-ZF1-YFP) (see also Anderson et al., 2008; Chihara and Nance, 2012; Nance et al., 2003).
In comparison with other inducible targeted protein degradation strategies that do not require drug administration, ZF1-tagging occurs more rapidly (see Introduction) – removing protein within 30-45 min. This speed of protein degradation is particularly important given the fast rate of development in C. elegans. We speculate that the rapid rate with which ZF1-tagged proteins degrade might have evolved to match the fast pace of C. elegans embryogenesis – cell cycles are short in the early embryo and endogenous ZIF-1 must quickly clear away the C-C-C-H zinc-finger domain germline proteins from each somatic cell lineage soon after it is born. Although the SOCS-box protein ZIF-1 does not share homology with other proteins outside of related nematode species (DeRenzo et al., 2003), components of the ECS E3 ubiquitin ligase complex are highly conserved and can also associate with SOCS-box proteins (Linossi and Nicholson, 2012), raising the possibility that ZF1 tagging could be adapted in the future to function in other species.
MATERIALS AND METHODS
Strains
Strains used in this study are listed in Table 2. Worms were cultured on NGM plates at 20°C unless specified otherwise.
Table 2.
C. elegans strains

Transgene construction
The Pcdc-42::ZF1-GFP-CDC-42 plasmid was constructed by modifying a cdc-42 genomic subclone containing 2775 bp 5′ of the ATG and 780 bp 3′ of the stop codon. An ApaI site was introduced immediately 3′ of the ATG, and zf1-gfp, fused as described (Nance et al., 2003), was ligated into the ApaI site. The unc-119 genomic sequence was inserted into the vector backbone NotI site via partial digestion and ligation, as described previously (Nance et al., 2003).
Pelt-2::ZIF-1, Pelt-2::mCherry, Prab-3::ZIF-1, Prab-3::mCherry, Pcdc-42::ZIF-1 and Pcdc-42::mCherry were generated by Gibson end-joining. All promoters were amplified from genomic DNA. The cdc-42 promoter was identical to that used to generate Pcdc-42::ZF1-GFP-CDC-42 (see above). The elt-2 promoter contains 5001 bp upstream of the start codon (Fukushige et al., 1998). The rab-3 promoter contains 1206 bp upstream of the start codon (Mahoney et al., 2006). The complete zif-1 gene was amplified from genomic DNA. The mCherry-coding sequence contains synthetic introns and was amplified from pBALU4 (lacking galK sequences; a gift from Oliver Hobert, Columbia University Medical Center, NY, USA) (Tursun et al., 2009). A 703 bp region from the unc-54 3′ UTR was amplified from genomic DNA and recombined downstream of zif-1 or mCherry. For zif-1 expression from Phsp-16.41, the hsp-16.41 promoter was obtained from genomic DNA, similar to plasmid pCD6.09AP (a gift from Geraldine Seydoux, Johns Hopkins University School of Medicine, MD, USA) (Hao et al., 2006). mCherry was co-expressed in the same operon by inserting SL2 trans-splice acceptor sequences (244 bp intergenic sequence between gpd-2 stop codon and gpd-3 start site) between the zif-1 stop codon and the mCherry start codon (Tursun et al., 2009), creating Phsp-16.41::ZIF-1-SL2-mCherry. For all constructs above, fragments were recombined into vector pJN566, which is derived from MosSCI vector pCFJ150 (Frøkjær-Jensen et al., 2008) modified to include a PmeI restriction site adjacent to the unc-119-coding region (using primers 5′-PmeI-GGCCTAGTTCTTAGACATTCTC-3′ and 5′-PmeI-CACTGGCCGTCGTTTTACAC-3′). Prior to end-joining, pJN566 was linearized by digestion with PmeI.
Plasmids for CRISPR/Cas9 genomic editing of sec-5 were constructed as described previously (Dickinson et al., 2013), with modifications noted below. The homologous guide RNA sequence (5′-GATATCAGTCTGTTTC-GTAA-3′) from plasmid pDD122 was replaced with the sequence (5′-GGATTATCGGCTGTGTTGTA-3′), which was designed to direct cleavage near the sec-5 C-terminus (plasmid pSA121). The homologous repair plasmid (pSA122) was constructed using Gibson end-joining and the following DNA segments in order: 1658 bp downstream of sec-5 stop codon (including a point mutation in the predicted guide RNA cut site) as homology arm #1; zf1-yfp with unc-119 (see below); and the 3′-terminal 1551 bp of sec-5 genomic sequence as homology arm #2. zf1-yfp with unc-119 flanked by LoxP sites was amplified from plasmid pJN601, which contains LoxP-flanked unc-119 inserted in reverse orientation into a synthetic intron within yfp; the presence of unc-119 does not prevent YFP-tagged protein expression following genome insertion, and vice versa. The vector backbone for each construct was PmeI-linearized pJN566, as above.
Constructs for anchor cell and primary vulval-specific expression of zif-1 (Pegl-17::ZIF-1-SL2-mCherry and Pcdh-3::ZIF-1-SL2-mCherry) were built by PCR fusion. The egl-17 promoter was amplified from pPD107.94/mk84-148 and the cdh-3 promoter was amplified from pPD104.97/mk62-63 (Hagedorn et al., 2013; Ihara et al., 2011). Both promoters were amplified using primers for the pPD104.97 backbone (forward outer primer, 5′-CAGCTATGACCATGATTACGC-3′; reverse primer, 5′-TTTTTCTGAGCTCGGTACCCTC-3′). ZIF-1-SL2-mCherry was amplified from pSA120: forward primer (with overhang for promoter fusion), 5′-gagggtaccgagctagaaaaaATGAGTGAGTGTTCC-GCG-3′; reverse outer primer, 5′-CCGTACGTCTCGAGTGTA-3′. The promoters were fused by PCR to ZIF-1-SL2-mCherry (forward nested primer, 5′-CACTCACAACGATGGATACGC-3′; reverse nested primer 5′-AAACAGTTATGTTTGGTATATTGGG-3′).
Worm transformation and transgene integration
xnIs23 and xnIs39 [Pcdc-42::ZF1-GFP-CDC-42] were created by biolistic transformation of unc-119(ed3) worms, as described previously (Praitis et al., 2001). xnIs23 is silenced in the germ line and xnIs39 is expressed in the germ line. All extrachromosomal arrays were obtained by microinjecting plasmid DNA into the germ line (Mello et al., 1991), using the co-transformation markers indicated in Table 2.
xnEx377 [Pcdc-42::ZIF-1, Pcdc-42::mCherry] was integrated using Trioxsalen (Sigma) and UV irradiation. A mixed population of washed transgenic worms was incubated in 600 ml of 33.3 ng/ml Trioxsalen in DMSO in the dark for 15 min. Worms were dripped onto an unseeded NGM agar plate and, after the solution had soaked in, the agar plate was irradiated with 360 µJ of UV light in a Stratalinker. NA22 bacterial food was dripped onto the worms and, after 5 h in darkness, 20 L4 stage transgenic worms were picked to each of 20 peptone plates (10 cm) seeded with NA22 bacteria. F1 adults were bleached to collect eggs, which were plated 200 per plate onto 70 NGM plates (6 cm). Seven-hundred and twenty transgenic F2 were picked into individual wells of 24-well plates, and those with an F3 brood containing only worms expressing mCherry were saved. Two transgene insertions, xnIs520 and xnIs521, were isolated and outcrossed twice.
sec-5(xn51[sec-5-zf1-yfp + unc-119(+)]) was generated by microinjecting the guide RNA plasmid pSA121 (which also contains Cas9) and the homologous repair template pSA122 [which also contains the unc-119(+) transformation marker] with a plasmid encoding excretory cell-specific mCherry (pSA086) into unc-119(ed3) mutant worms. pSA086 served as a visible marker for successful transgenic array formation, and was generated by Gibson end-joining: PmeI-linearized vector pJN566, 2853 bp 5′ of the pgp-12 start site (Zhao et al., 2005), mCherry (as above) and unc-54 3′UTR (as above) were end-joined. Non-Unc transformants expressing mCherry in the excretory cell were isolated in the F1 generation, and successfully edited F2 non-Unc animals were identified by the absence of excretory cell mCherry expression in the F2 generation.
Microscopy and image acquisition
Differential interference contrast (DIC) and fluorescence images were collected using a Zeiss AxioImager, 63×1.4 NA objective, an Axiocam MRM camera and AxioVision software. Fluorescence images of fixed embryos were deconvolved using AxioVision software, except for images showing SEC-5-ZF1-YFP depletion; embryos shown for this experiment were taken with the same exposure settings and images were not deconvolved. Images were cropped in ImageJ (NIH), and control and mutant images processed similarly using Photoshop (CS6, Adobe), with no γ adjustments and level adjustments across all pixels. For all live-imaging experiments, embryos and larvae were mounted onto 4% agarose pads made in M9 or water.
Images in Fig. 4 and supplementary material Fig. S4 were acquired using an EM-CCD (Hamamatsu Photonics) and a spinning disk confocal microscope (CSU-10, Yokogawa; AxioImager, Carl Zeiss) with a Plan-APOCHROMAT 100×/1.4 oil DIC objective controlled by Micromanager (Edelstein et al., 2010) or iVision software (Biovision Technologies). Acquired images were processed using ImageJ 1.40 g and Photoshop (CS3 Extended, Adobe).
Western analysis of ZF1-GFP-CDC-42 degradation
Two-hundred synchronized young adults of the indicated genotype were washed five times in M9 buffer, resuspended in 50 µl of LDS sample buffer (Invitrogen) containing 100 mM DTT and boiled 10 min. Proteins in 10 µl lysate were separated and analyzed by western analysis. Primary antibodies used were mouse anti-α-tubulin (Sigma, 1:30,000) and mouse anti-GFP (Roche, 1:1000). HRP-conjugated secondary antibodies (sheep anti-mouse IgG, Amersham, 1:10,000) and the ECL Prime kit (Amersham) were used for detection. The experiment was repeated in triplicate using biological replicates, and a representative experiment is shown.
Heat-shock expression of ZIF-1 and analysis of protein degradation kinetics
FT1547 or FT1553 embryos at the bean or comma stage were mounted under a coverslip on 4% agarose pads and placed on a pre-warmed metal plate at 34°C for 15 min. Embryos were not selected for absence or presence of transgenic arrays, so that both control (lacking array) and experimental (carrying array) progeny were heat-shocked on the same slide. Following incubation, embryos were immediately transferred to the microscope for imaging. Fluorescence movies were obtained using a Zeiss AxioImager and a 40×1.3NA objective, and exposure times were consistent throughout each experimental condition (10 ms for ZF1-GFP-CDC-42 and 150 ms for SEC-5-ZF1-YFP). GFP (or YFP) and mCherry signals were captured in a single central z-plane every 5 min. Control and array-bearing embryos were captured in the same field of view to control for experimental conditions and to ensure identical image collection. To measure fluorescence intensity at each time point, regions of interest (ROIs) were drawn around a representative area in the center of each embryo (for ZF1-GFP-CDC-42) or a line overlying the apical surface of the pharynx (for SEC-5-ZF1-YFP). In each experiment, at least three embryos were measured for each condition (control and array bearing) and the experiment was performed three times. To measure degradation, percent reduction in fluorescence intensity was calculated over time. Fluorescence intensity in mutant embryos was normalized to control embryos to correct for minor reduction in signal due to photobleaching. For SEC-5-ZF1-YFP, given the longer exposure required to collect the YFP signal, embryo autofluorescence (from N2 control embryos imaged similarly) was subtracted at each time point to correct for background. Error bars represent s.e.m., and were calculated from the average percentage change of array-bearing embryos compared with controls at each time point over three experiments.
Phenotypic comparison of sec-5 mutants with animals depleted of ZF1-tagged SEC-5
Hermaphrodites were allowed to self-fertilize on seeded NGM agar plates at 20°C for 8 h and eggs laid on the plate were counted. Eggs remaining unhatched after 16 h were scored as dead. Larvae were counted 32 h later when wild-type worms should be in the L4 stage. The number of dead larvae was calculated as the number of larvae present subtracted from the number of eggs that hatched. Lethality was expressed as the sum of dead eggs and dead larvae. sec-5(pk2358) maternal-zygotic mutants were obtained from balanced sec-5(pk2358)/mIn1 hermaphrodites. Because egg-laying is defective in most sec-5(pk2358) hermaphrodites, eggs were obtained by dissecting hermaphrodites in egg buffer and liberated eggs were moved to a seeded NGM agar plate by mouth pipet. Eggs of genotype sec-5(xn51[sec-5-zf1-yfp + unc-119(+)]); xnEx377 (Pcdc-42::ZIF-1 + mCherry) were obtained by crossing sec-5(xn51) hermaphrodites with sec-5(xn51)/mIn1; xnEx377 males. Successfully mated hermaphrodites were allowed to lay eggs on an NGM agar plate for ∼8 h, then removed. Two days later, young adult progeny carrying the xnEx377 transgenic array were identified by mCherry expression and scored for the presence or absence of the GFP-marked mIn1 balancer. As no animals lacking mIn1 and containing xnEx377 survived, the number of animals of this genotype was deduced from the number of progeny containing mIn1 and xnEx377.
Embryo immunostaining
Embryos were placed onto poly-L-lysine coated slides, flash-frozen on dry ice and freeze-cracked by quick removal of cover slips. Freeze-cracked slides were fixed in methanol for 20 min and 4% paraformaldehyde with salts for 5 min, as described previously (Anderson et al., 2008). Slides were incubated with rabbit anti-GFP (1:2000, Abcam), washed, incubated in Alexa-488 conjugated anti-rabbit secondary antibodies, incubated in DAPI, washed and mounted in DABCO (Sigma) as described previously (Anderson et al., 2008).
Anchor cell invasion and RNAi targeting of cdc-42
Anchor cell invasion was scored as previously described using DIC microscopy (Sherwood et al., 2005). Briefly, animals were scored for invasion at the P6.p four-cell stage when basement membrane invasion is completed in wild-type animals. Anchor cells were scored as ‘normal’ invasion if there was a visible breach in the phase-dense line at the P6.p four-cell stage and as ‘blocked’ invasion if the phase-dense line remained intact (see supplementary material Fig. S4). RNA interference (RNAi) targeting cdc-42 or an empty vector control was delivered by RNAi feeding to synchronized L1-arrested worms as previously described (Matus et al., 2010). For anchor cell and primary vulval specific targeting of ZF1-tagged CDC-42, extrachromosomal lines with cell type-specific zif-1 expression were crossed into the cdc-42(gk388)/mIn1 [dpy-10(e128) mIs14]; xnIs23 [Pcdc-42::ZF1-GFP-CDC-42] background, and homozygous cdc-42 mutant progeny were examined. Maternal contribution of untagged cdc-42 from the heterozygous mothers was sufficient to bypass the requirement for CDC-42 during the one-cell stage in cdc-42(gk388) homozygous progeny (as xnIs23 is silenced maternally).
Supplementary Material
Acknowledgements
We thank Geraldine Seydoux and Oliver Hobert for plasmids; Fried Zwartkruis for the sec-5(pk2358) strain; Justin Yeh for help with western analysis; Shai Shaham and Lena Kutscher for the transgene insertion protocol; and Ryan Cinalli, Niels Ringstad, Matthias Stadtfeld and members of the Nance lab for critical comments on the manuscript.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
All authors designed the experiments; S.T.A., L.L.L. and J.N. performed the experiments; and all authors interpreted the data and wrote the manuscript.
Funding
This work was funded by grants from the National Institutes of Health to S.T.A. [F30DK093197], D.R.S. [R01GM079320, R01GM100083] and J.N. [R01GM098492, R01GM078341]. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.115048/-/DC1
References
- Abrams E. W. and Mullins M. C. (2009). Early zebrafish development: it's in the maternal genes. Curr. Opin. Genet. Dev. 19, 396-403 10.1016/j.gde.2009.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achilleos A., Wehman A. M. and Nance J. (2010). PAR-3 mediates the initial clustering and apical localization of junction and polarity proteins during C. elegans intestinal epithelial cell polarization. Development 137, 1833-1842 10.1242/dev.047647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D. C., Gill J. S., Cinalli R. M. and Nance J. (2008). Polarization of the C. elegans embryo by RhoGAP-mediated exclusion of PAR-6 from cell contacts. Science 320, 1771-1774 10.1126/science.1156063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenti S. T., Chan E. and Nance J. (2014). Polarized exocyst-mediated vesicle fusion directs intracellular lumenogenesis within the C. elegans excretory cell. Dev. Biol. 394, 110-121 10.1016/j.ydbio.2014.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asikainen S., Vartiainen S., Lakso M., Nass R. and Wong G. (2005). Selective sensitivity of Caenorhabditis elegans neurons to RNA interference. Neuroreport 16, 1995-1999 10.1097/00001756-200512190-00005 [DOI] [PubMed] [Google Scholar]
- Bacaj T. and Shaham S. (2007). Temporal control of cell-specific transgene expression in Caenorhabditis elegans. Genetics 176, 2651-2655 10.1534/genetics.107.074369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski L. A., Chen L.-C., Maynard-Smith L. A., Ooi A. G. and Wandless T. J. (2006). A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995-1004 10.1016/j.cell.2006.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonger K. M., Chen L.-C., Liu C. W. and Wandless T. J. (2011). Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nat. Chem. Biol. 7, 531-537 10.1038/nchembio.598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caussinus E., Kanca O. and Affolter M. (2012). Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat. Struct. Mol. Biol. 19, 117-121 10.1038/nsmb.2180 [DOI] [PubMed] [Google Scholar]
- Chan E. and Nance J. (2013). Mechanisms of CDC-42 activation during contact-induced cell polarization. J. Cell Sci. 126, 1692-1702 10.1242/jcs.124594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chihara D. and Nance J. (2012). An E-cadherin-mediated hitchhiking mechanism for C. elegans germ cell internalization during gastrulation. Development 139, 2547-2556 10.1242/dev.079863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churgin M. A., He L., Murray J. I. and Fang-Yen C. (2013). Efficient single-cell transgene induction in Caenorhabditis elegans using a pulsed infrared laser. G3 3, 1827-1832 10.1534/g3.113.007682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. W., Morton J. J., Carroll D. and Jorgensen E. M. (2008). Gene activation using FLP recombinase in C. elegans. PLoS Genet. 4, e1000028 10.1371/journal.pgen.1000028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Valle Rodríguez A., Didiano D. and Desplan C. (2012). Power tools for gene expression and clonal analysis in Drosophila. Nat. Methods 9, 47-55 10.1038/nmeth.1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRenzo C., Reese K. J. and Seydoux G. (2003). Exclusion of germ plasm proteins from somatic lineages by cullin-dependent degradation. Nature 424, 685-689 10.1038/nature01887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson D. J., Ward J. D., Reiner D. J. and Goldstein B. (2013). Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat. Methods 10, 1028-1034 10.1038/nmeth.2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuy D., Bertin N., Hidalgo C. A., Venkatesan K., Tu D., Lee D., Rosenberg J., Svrzikapa N., Blanc A., Carnec A. et al. (2007). Genome-scale analysis of in vivo spatiotemporal promoter activity in Caenorhabditis elegans. Nat. Biotechnol. 25, 663-668 10.1038/nbt1305 [DOI] [PubMed] [Google Scholar]
- Edelstein A., Amodaj N., Hoover K., Vale R. and Stuurman N. (2010). Computer control of microscopes using microManager. Curr. Protoc. Mol. Biol. Chapter 14, Unit 14.20. 10.1002/0471142727.mb1420s92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A., Xu S., Montgomery M. K., Kostas S. A., Driver S. E. and Mello C. C. (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806-811 10.1038/35888 [DOI] [PubMed] [Google Scholar]
- Frische E. W., Pellis-van Berkel W., van Haaften G., Cuppen E., Plasterk R. H. A., Tijsterman M., Bos J. L. and Zwartkruis F. J. T. (2007). RAP-1 and the RAL-1/exocyst pathway coordinate hypodermal cell organization in Caenorhabditis elegans. EMBO J. 26, 5083-5092 10.1038/sj.emboj.7601922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjær-Jensen C., Davis M. W., Hopkins C. E., Newman B. J., Thummel J. M., Olesen S.-P., Grunnet M. and Jorgensen E. M. (2008). Single-copy insertion of transgenes in Caenorhabditis elegans. Nat. Genet. 40, 1375-1383 10.1038/ng.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushige T., Hawkins M. G. and McGhee J. D. (1998). The GATA-factor elt-2 is essential for formation of the Caenorhabditis elegans intestine. Dev. Biol. 198, 286-302. [PubMed] [Google Scholar]
- Goldstein B. (1992). Induction of gut in Caenorhabditis elegans embryos. Nature 357, 255-257 10.1038/357255a0 [DOI] [PubMed] [Google Scholar]
- Gotta M., Abraham M. C. and Ahringer J. (2001). CDC-42 controls early cell polarity and spindle orientation in C. elegans. Curr. Biol. 11, 482-488 10.1016/S0960-9822(01)00142-7 [DOI] [PubMed] [Google Scholar]
- Hagedorn E. J., Ziel J. W., Morrissey M. A., Linden L. M., Wang Z., Chi Q., Johnson S. A. and Sherwood D. R. (2013). The netrin receptor DCC focuses invadopodia-driven basement membrane transmigration in vivo. J. Cell Biol. 201, 903-913 10.1083/jcb.201301091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y., Boyd L. and Seydoux G. (2006). Stabilization of cell polarity by the C. elegans RING protein PAR-2. Dev. Cell 10, 199-208 10.1016/j.devcel.2005.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland A. J., Fachinetti D., Han J. S. and Cleveland D. W. (2012). Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc. Natl. Acad. Sci. USA 109, E3350-E3357 10.1073/pnas.1216880109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara S., Hagedorn E. J., Morrissey M. A., Chi Q., Motegi F., Kramer J. M. and Sherwood D. R. (2011). Basement membrane sliding and targeted adhesion remodels tissue boundaries during uterine-vulval attachment in Caenorhabditis elegans. Nat. Cell Biol. 13, 641-651 10.1038/ncb2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J. R., Shelton K. D. and Magnuson M. A. (2005). Strategies for the use of site-specific recombinases in genome engineering. Methods Mol. Med. 103, 245-257. [DOI] [PubMed] [Google Scholar]
- Kamath R. S., Martinez-Campos M., Zipperlen P., Fraser A. G. and Ahringer J. (2001). Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2, RESEARCH0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay A. J. and Hunter C. P. (2001). CDC-42 regulates PAR protein localization and function to control cellular and embryonic polarity in C. elegans. Curr. Biol. 11, 474-481 10.1016/S0960-9822(01)00141-5 [DOI] [PubMed] [Google Scholar]
- Kimble J. (1981). Alterations in cell lineage following laser ablation of cells in the somatic gonad of Caenorhabditis elegans. Dev. Biol. 87, 286-300 10.1016/0012-1606(81)90152-4 [DOI] [PubMed] [Google Scholar]
- Kirouac M. and Sternberg P. W. (2003). cis-Regulatory control of three cell fate-specific genes in vulval organogenesis of Caenorhabditis elegans and C. briggsae. Dev. Biol. 257, 85-103 10.1016/S0012-1606(03)00032-0 [DOI] [PubMed] [Google Scholar]
- Lee J.-Y. and Goldstein B. (2003). Mechanisms of cell positioning during C. elegans gastrulation. Development 130, 307-320 10.1242/dev.00211 [DOI] [PubMed] [Google Scholar]
- Levin M., Hashimshony T., Wagner F. and Yanai I. (2012). Developmental milestones punctuate gene expression in the Caenorhabditis embryo. Dev. Cell 22, 1101-1108 10.1016/j.devcel.2012.04.004 [DOI] [PubMed] [Google Scholar]
- Li L., Zheng P. and Dean J. (2010). Maternal control of early mouse development. Development 137, 859-870 10.1242/dev.039487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linossi E. M. and Nicholson S. E. (2012). The SOCS box-adapting proteins for ubiquitination and proteasomal degradation. IUBMB Life 64, 316-323 10.1002/iub.1011 [DOI] [PubMed] [Google Scholar]
- Mahoney T. R., Liu Q., Itoh T., Luo S., Hadwiger G., Vincent R., Wang Z.-W., Fukuda M. and Nonet M. L. (2006). Regulation of synaptic transmission by RAB-3 and RAB-27 in Caenorhabditis elegans. Mol. Biol. Cell 17, 2617-2625 10.1091/mbc.E05-12-1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus D. Q., Li X.-Y., Durbin S., Agarwal D., Chi Q., Weiss S. J. and Sherwood D. R. (2010). In vivo identification of regulators of cell invasion across basement membranes. Sci. Signal. 3, ra35 10.1126/scisignal.2000654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello C. C., Kramer J. M., Stinchcomb D. and Ambros V. (1991). Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959-3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance J. and Priess J. R. (2002). Cell polarity and gastrulation in C. elegans. Development 129, 387-397. [DOI] [PubMed] [Google Scholar]
- Nance J., Munro E. M. and Priess J. R. (2003). C. elegans PAR-3 and PAR-6 are required for apicobasal asymmetries associated with cell adhesion and gastrulation. Development 130, 5339-5350 10.1242/dev.00735 [DOI] [PubMed] [Google Scholar]
- Nishimura K., Fukagawa T., Takisawa H., Kakimoto T. and Kanemaki M. (2009). An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 6, 917-922 10.1038/nmeth.1401 [DOI] [PubMed] [Google Scholar]
- Perrimon N. and Gans M. (1983). Clonal analysis of the tissue specificity of recessive female-sterile mutations of Drosophila melanogaster using a dominant female-sterile mutation Fs(1)K1237. Dev. Biol. 100, 365-373 10.1016/0012-1606(83)90231-2 [DOI] [PubMed] [Google Scholar]
- Praitis V., Casey E., Collar D. and Austin J. (2001). Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics 157, 1217-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priess J. R. and Thomson J. N. (1987). Cellular interactions in early C. elegans embryos. Cell 48, 241-250 10.1016/0092-8674(87)90427-2 [DOI] [PubMed] [Google Scholar]
- Raina K. and Crews C. M. (2010). Chemical inducers of targeted protein degradation. J. Biol. Chem. 285, 11057-11060 10.1074/jbc.R109.078105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese K. J., Dunn M. A., Waddle J. A. and Seydoux G. (2000). Asymmetric segregation of PIE-1 in C. elegans is mediated by two complementary mechanisms that act through separate PIE-1 protein domains. Mol. Cell 6, 445-455 10.1016/S1097-2765(00)00043-5 [DOI] [PubMed] [Google Scholar]
- Renicke C., Schuster D., Usherenko S., Essen L.-O. and Taxis C. (2013). A LOV2 domain-based optogenetic tool to control protein degradation and cellular function. Chem. Biol. 20, 619-626 10.1016/j.chembiol.2013.03.005 [DOI] [PubMed] [Google Scholar]
- Sharma-Kishore R., White J. G., Southgate E. and Podbilewicz B. (1999). Formation of the vulva in Caenorhabditis elegans: a paradigm for organogenesis. Development 126, 691-699. [DOI] [PubMed] [Google Scholar]
- Sherwood D. R. and Sternberg P. W. (2003). Anchor cell invasion into the vulval epithelium in C. elegans. Dev. Cell 5, 21-31 10.1016/S1534-5807(03)00168-0 [DOI] [PubMed] [Google Scholar]
- Sherwood D. R., Butler J. A., Kramer J. M. and Sternberg P. W. (2005). FOS-1 promotes basement-membrane removal during anchor-cell invasion in C. elegans. Cell 121, 951-962 10.1016/j.cell.2005.03.031 [DOI] [PubMed] [Google Scholar]
- Sulston J. E., Schierenberg E., White J. G. and Thomson J. N. (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64-119 10.1016/0012-1606(83)90201-4 [DOI] [PubMed] [Google Scholar]
- Taxis C., Stier G., Spadaccini R. and Knop M. (2009). Efficient protein depletion by genetically controlled deprotection of a dormant N-degron. Mol. Syst. Biol. 5, 267 10.1038/msb.2009.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totong R., Achilleos A. and Nance J. (2007). PAR-6 is required for junction formation but not apicobasal polarization in C. elegans embryonic epithelial cells. Development 134, 1259-1268 10.1242/dev.02833 [DOI] [PubMed] [Google Scholar]
- Tursun B., Cochella L., Carrera I. and Hobert O. (2009). A toolkit and robust pipeline for the generation of fosmid-based reporter genes in C. elegans. PLoS ONE 4, e4625 10.1371/journal.pone.0004625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voutev R. and Hubbard E. J. A. (2008). A “FLP-Out” system for controlled gene expression in Caenorhabditis elegans. Genetics 180, 103-119 10.1534/genetics.108.090274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. and Sternberg P. W. (2000). Patterning of the C. elegans 1° vulval lineage by RAS and Wnt pathways. Development 127, 5047-5058. [DOI] [PubMed] [Google Scholar]
- Wehman A. M., Poggioli C., Schweinsberg P., Grant B. D. and Nance J. (2011). The P4-ATPase TAT-5 inhibits the budding of extracellular vesicles in C. elegans embryos. Curr. Biol. 21, 1951-1959 10.1016/j.cub.2011.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood W. B., Hecht R., Carr S., Vanderslice R., Wolf N. and Hirsh D. (1980). Parental effects and phenotypic characterization of mutations that affect early development in Caenorhabditis elegans. Dev. Biol. 74, 446-469 10.1016/0012-1606(80)90445-5 [DOI] [PubMed] [Google Scholar]
- Yochem J. and Herman R. K. (2003). Investigating C. elegans development through mosaic analysis. Development 130, 4761-4768 10.1242/dev.00701 [DOI] [PubMed] [Google Scholar]
- Zhao Z., Fang L., Chen N., Johnsen R. C., Stein L. and Baillie D. L. (2005). Distinct regulatory elements mediate similar expression patterns in the excretory cell of Caenorhabditis elegans. J. Biol. Chem. 280, 38787-38794 10.1074/jbc.M505701200 [DOI] [PubMed] [Google Scholar]
- Zhou P., Bogacki R., McReynolds L. and Howley P. M. (2000). Harnessing the ubiquitination machinery to target the degradation of specific cellular proteins. Mol. Cell 6, 751-756 10.1016/S1097-2765(00)00074-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.