

Abstract
Non-transfusion-dependent thalassaemia (NTDT) refers to all thalassaemia disease phenotypes that do not require regular blood transfusions for survival. Thalassaemia disorders were traditionally concentrated along the tropical belt stretching from sub-Saharan Africa through the Mediterranean region and the Middle East to South and South-East Asia, but global migration has led to increased incidence in North America and Northern Europe. Transfusionists may be familiar with β-thalassaemia major because of the lifelong transfusions needed by these patients. Although patients with NTDT do not require regular transfusions for survival, they may require transfusions in some instances such as pregnancy, infection or growth failure. The complications associated with NTDT can be severe if not properly managed, and many are directly related to chronic anaemia. Awareness of NTDT is important, and this review will outline the factors that should be taken into consideration when deciding whether to initiate and properly plan for transfusion therapy in these patients in terms of transfusion interval and duration of treatment.
Keywords: haemoglobin E/β thalassaemia, haemoglobin H disease, iron chelation, non-transfusion-dependent thalassaemia, red cell transfusion, β-thalassaemia intermedia
Introduction
Thalassaemia describes all the inherited genetic abnormalities that affect the synthesis of α- or β-globin chains and, consequently, normal erythropoiesis and the oxygen-carrying capacity of blood by haemoglobin. This condition is inherited as an autosomal recessive disorder and can be classified into two main categories: α- and β-thalassaemia. Patients with thalassaemia disease commonly develop chronic haemolytic anaemia and ineffective erythropoiesis due to imbalanced globin synthesis. The severity of anaemia is linked to the number of genetic aberrations, the specific combination of affected genes, other genetic and environmental modifiers and physiological stressors 1–5. For instance, patients with β-thalassaemia major are transfusion dependent, which is the extreme end of transfusion requirement in thalassaemia syndromes 6. Approximately 23 000 children are born every year with the transfusion-dependent form of β-thalassaemia or β-thalassaemia major, while the number with non-transfusion-dependent β-thalassaemia is less well defined 7–9. Due to a wide range of transfusion requirements between transfusion-dependent and asymptomatic thalassaemia carriers, the term non-transfusion-dependent thalassaemia (NTDT) was introduced to include β-thalassaemia intermedia (β-TI) and patients with β-thalassaemia and β-globin variants in particular HbE (Hb E/β thalassaemia) that do not require regular blood transfusions for survival 10–14. In addition, most patients with α-thalassaemia syndrome such as haemoglobin H (HbH) disease are asymptomatic and may only require transfusion therapy when they experience a sudden increase in red cell demand, such as during pregnancy or intercurrent infection 10.
Therefore the most common types of NTDT are β-TI, α-thalassaemia (mainly HbH disease) and HbE/β-thalassaemia. There are also a number of less common NTDT thalassaemias that will not be dealt with here 15–19.
β-thalassaemia intermedia results from inheritance of a β-thalassaemia genotype that is milder than the transfusion-dependent disease β-thalassaemia major. β-thalassaemia is prevalent worldwide but is most common in South-East Asia and the Eastern Mediterranean.
HbE/β-thalassaemia, which results from the co-inheritance of the haemoglobin structural variant known as HbE and many different β-thalassaemia alleles, is highly prevalent in Southeast Asia, the Indian subcontinent and the southern part of mainland China 20. More than 19 000 children a year are born with HbE/β-thalassaemia, around half with transfusion-dependent disease and half with NTDT 7,8.
Haemoglobin H disease, which is caused by the inactivation of three α-globin genes, is highly prevalent in Southeast Asia and Africa. In some regions, such as the West Pacific Islands, incidence of carriers (having one α-globin gene deletion) may be as high as 80–90% 10.
The clinical severity of patients with NTDT ranges widely because a diversity of phenotypes is included in this classification and because patients with the same phenotype can have very different symptoms. The extent of chronic anaemia that NTDT patients experience, rather than their specific thalassaemia phenotypes, determines the severity of clinical complications 2,11,12,21.
Non-transfusion-dependent thalassaemia patients can generally be described as having mild to moderately severe chronic anaemia. The state of anaemia in patients with NTDT can rapidly change when they experience a physiological stressor, such as infection, or it can become more severe over time for various reasons, including the development of splenomegaly. In addition, a low Hb level can hamper physiological processes such as growth and development, and chronic anaemia initiates a cascade of compensatory mechanisms that lead to diverse clinical complications 22–25.
Transfusionists and haematologists may be very familiar with β-thalassaemia major because of the necessary lifelong transfusions in these patients. However, awareness of NTDT and the special considerations relevant to these patients is important. Although NTDT is less severe than transfusion-dependent thalassaemia (TDT), the associated complications can be severe, and quality of life may actually be more impaired in patients with NTDT 26. Moreover, in the light of increasing evidence that many NTDT complications are directly related to chronic anaemia, and with the availability of oral iron chelators, it may be time to revisit the guidelines for transfusion therapy in patients with NTDT.
This review will focus on the factors that should be taken into consideration when deciding whether to transfuse in patients with β-TI, α-thalassaemia and HbE/β-thalassaemia.
Clinical characteristics of NTDT: recognition
Irrespective of the underlying genetic mutation(s), thalassaemia patients who require regular blood transfusions for life support are considered transfusion dependent and those who require occasional transfusions to overcome a period of severe/debilitating anaemia are considered non-transfusion dependent 22,25,27,28. Although the division between transfusion dependent and non-transfusion dependent is theoretically relatively simple, classifying a patient as either transfusion dependent or non-transfusion dependent in a clinical setting can be much more complex. Table1 shows the clinical and haematological characteristics that can be used to differentiate between β-thalassaemia major (transfusion dependent) and β-TI (non-transfusion-dependent) 29. This guideline can be applied to other types of thalassaemia including HbE/β-thalassaemia or HbH disease. Patients with NTDT can present with severe anaemia for several reasons, including acute infection. In the absence of a diagnosis or a long medical history, it is not possible to predict what baseline Hb levels would be in the absence of infection or how well a patient manages with ‘usual’ baseline Hb levels. Studies have shown that some patients with NTDT function well even if they have very low Hb levels 30,31.
Table 1.
Differential characteristics of β-thalassaemia major (β-TM) and β-thalassaemia intermedia (β-TI) 29
| β-TM more likely | β-TI more likely | |
|---|---|---|
| Clinical | ||
| Presentation (years) | <2 | >2 |
| Hb levels (g/dl) | 6–7 | 8–10 |
| Liver/spleen enlargement | Severe | Moderate to severe |
| Haematological | ||
| Fetal Hb (HbF) (%) | >50 | 10–50 but may be up to 100 |
| HbA2 (%) | <4 | >4 |
Modified from Ref. 29.
The findings of several studies and trials have identified characteristics of both patients with TDT and patients with NTDT that can be used as a guide for clinical decisions regarding the transfusion need of a patient 22,25,27,28. Patients with TDT usually present in the first year of life with symptoms of severe anaemia (Hb <7 g/dl), pallor, lethargy, failure to thrive, bone and skeletal deformities, marked splenomegaly and hepatomegaly. Patients with NTDT, in contrast, usually present when they are older than 2 years of age and often have moderate to severe spleen/liver enlargement. Patients with moderately severe anaemia (Hb 8–9 g/dl), a state often associated with β-TI, often present between 2 and 6 years of age, but thalassaemia patients with milder anaemia (Hb >9 g/dl) can present much later, sometimes only in adulthood 6,27.
Patients with NTDT are usually diagnosed because of chronic anaemia rather than the complications that may arise from chronic anaemia, although these complications can be severe if diagnosis and appropriate management are delayed 22,25,27,28,32.
Complications of NTDT and indications for transfusion therapy
On initial presentation and diagnosis of NTDT, it is not recommended to initiate transfusion therapy immediately. A patient may present with severe anaemia that is exacerbated by infection and acute haemolysis and therefore be placed on lifelong transfusion therapy unnecessarily. One study found that 37 out of 84 patients with HbE/β-thalassaemia could be withdrawn from a previous regular blood transfusion regimen without any deleterious effects, suggesting that many of these HbE/β-thalassaemias were in fact NTDT but had received inappropriate regular blood transfusion 33. It is important to monitor the patient for several months after diagnosis of a NTDT thalassaemia. Assessment of transfusion requirement should be based on factors such as activity, growth and development, skeletal changes and disease complications, rather than on Hb levels alone 22. Several studies in HbE/β-thalassaemia found that there was only 1·8–2·6 g/dl difference in Hb levels between the mildest and most severe groups, highlighting the limitations of Hb level as a decision criterion for initiating transfusion therapy 33,34. In addition, some children with HbE/β-thalassaemia are able to adapt to lower Hb levels and function well without transfusions 30,31. A low baseline Hb might be well compensated in most conditions, but become de-compensated in several clinical circumstances or even normal situations such as pregnancy or growth spurt. Therefore, the patient should be regularly monitored for any signs and symptoms of disease complications. It is important to note that all complications seen in NTDT are also characteristic of TDT, however with adequate blood transfusion, many of these complications have become less common in patients with TDT. The profiles of complications seen most often in NTDT vs. TDT are shown in Fig.1. If the decision is taken to initiate transfusion therapy, the patient should be monitored closely and the regimen adjusted according to the patient's needs. Therapeutic goals may be reached with temporary transfusion therapy, thus avoiding the need for lifelong regular transfusions 35. Earlier initiation of occasional transfusions may be beneficial in preventing the development of disease complications and alloimmunization 36.
Fig 1.
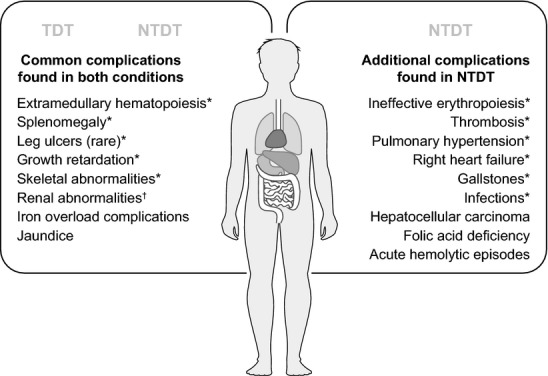
Common complications of TDT and NTDT. *Preventable by blood transfusion; †Probably preventable by blood transfusion. NTDT, Non-transfusion-dependent thalassaemia.
Table2 outlines the current indications for transfusion in each NTDT subtype, although the phenotypic variation of NTDT should be taken into account and decisions made on a case-by-case basis. It is important to note that the clinical phenotype of HbE/β-thalassaemia can be divided into mild, moderate and severe. At the mild end of the spectrum, patients generally require no transfusional intervention, and at the severe end, patients should be managed as TDT. Moderate HbE/β-thalassaemia is clinically more similar to β-TI and therefore falls into the category of NTDT.
Table 2.
Current indications for transfusion therapy in NTDT subtypes
| NTDT subtype | Indications for transfusion |
|---|---|
| HbH disease 11,94,95 | Infections exacerbating anaemia |
| Growth retardation | |
| HbE/β-thalassaemia 6 | |
| Mild | Generally none |
| Moderate | As per β-TI (see below) |
| Severe | Transfusions required for survival, as per β-thalassaemia major |
| β-TI 6,22 | Hb <5 g/dl |
| Failure to thrive secondary to anaemia | |
| Emergence of bone deformities | |
| Tendency to thrombosis | |
| Presence of leg ulcers | |
| Development of pulmonary hypertension | |
| Poor growth and development | |
| Splenic enlargement | |
| Pregnancy | |
| Infection | |
| Cardiovascular disease | |
HbH, haemoglobin H; NTDT, Non-transfusion-dependent thalassaemia; β-TI, β-thalassaemia intermedia.
Ineffective erythropoiesis
The body's response to chronic anaemia is increased erythropoietin (EPO) production, but because of the ineffective erythropoiesis characteristic of NTDT, the erythroid marrow is unable to respond properly to the EPO signal, resulting in proliferation of erythroid precursors which fail to differentiate and die prematurely 37. This causes erythroid marrow expansion, which is associated with osteoporosis and bone deformities, as well as hepatosplenomegaly due to build-up of erythroid precursors in the spleen and liver 38. Ineffective erythropoiesis also leads to the formation of extramedullary haemopoietic pseudotumours, which are essentially masses of haemopoietic tissue. These can occur almost anywhere in the body, but the most concerning are those with paraspinal involvement (around 11% of cases), which can have debilitating consequences due to neuronal compression 39.
The degree of ineffective erythropoiesis in NTDT can vary from one patient to another and is directly associated with morbidity, as it is the underlying dysfunction causing many of the severe clinical complications. Even though patients with NTDT are not reliant on transfusions for survival, transfusion therapy supplies normal erythrocytes and reduces the bone marrow expansion caused by ineffective erythropoiesis 40–42. Transfusion therapy can therefore be beneficial in reducing the risk of complications that are associated with ineffective erythropoiesis. Table3 shows the suppressive effects of transfusion therapy on erythroid activity in patients with β-thalassaemia major with differing pretransfusion Hb levels. In addition, it should be noted that the prevalence of extramedullary pseudotumours is <4% in transfusion-dependent patients, compared with around 20% in patients with NTDT, and transfusion has been shown to be protective against the development of these masses 23. Transfusion therapy has also been successfully used to treat extramedullary pseudotumours, even in the most severe cases with paraspinal involvement 39.
Table 3.
Suppression of erythroid activity by transfusion therapy 42
| Pretransfusion Hb level (g/dl) | Erythroid activity after transfusion |
|---|---|
| 8·6–9 | 2–6 times normal |
| 9–10 | 1–4 times normal |
| 10–11 | 1–2 times normal |
Splenectomy may be considered as an alternative to transfusion therapy to alleviate splenomegaly and increase Hb levels. However, splenectomized patients with NTDT are at higher risk of venous thromboembolism, pulmonary hypertension (PHT), leg ulcers, silent cerebral infarcts and serious infections such as meningitis and sepsis than non-splenectomized patients 23,43–45. Splenectomized patients with NTDT have higher rates of organ iron loading than non-splenectomized patients 46–49. For these reasons, splenectomy is only recommended in circumstances in which transfusion and iron chelation are not available, where hypersplenism results in severe anaemia, leucopenia or thrombocytopenia with recurrent infection and bleeding, or if the spleen exceeds 20 cm (largest dimension) and is at risk of rupture 22,32,35.
Increased intestinal iron absorption
Ineffective erythropoiesis, accompanied by anaemia and hypoxia, results in suppression of hepcidin production, increased intestinal iron absorption and release of iron from the reticuloendothelial system in patients with NTDT 50. This leads to iron accumulation, particularly in the liver, and as a result, patients with NTDT commonly experience liver disease such as fibrosis and cirrhosis and may be at risk of hepatocellular carcinoma (HCC) 49,51,52. This is in contrast with the situation in regularly transfused patients with β-thalassaemia major, who do not have low hepcidin levels. In 211 TI patients with risk factors for HCC, 2·8% developed HCC over 9 years of follow up 52. In patients with NTDT, intestinal absorption of iron can be as much as 3–4 mg/day or 1000 mg/year 35. Transfusion therapy may help to reduce intestinal iron absorption and thus reduce the risk of liver disease and other complications. However, patients with NTDT who receive blood transfusion also require appropriate and adequate iron chelation therapy; otherwise, the effect of transfusional iron overload would outweigh the benefit of reducing intestinal iron absorption.
Vascular complications
Thalassaemia is associated with a hypercoagulable state caused by aggregation of platelets and red-blood-cells (RBCs), increased thrombin production and vasoconstriction 53. As a result, patients are at higher risk of thromboembolic events, and this risk may be up to fourfold higher in patients with β-TI compared with regularly transfused patients with β-thalassaemia major 44. In 584 patients with β-TI enrolled in the OPTIMAL CARE study, splenectomy, older age (over 35 years) and high serum ferritin (≥1000 μg/l) were significantly associated with increased risk of thrombosis, whereas transfusion therapy significantly decreased the risk by more than 70% 23. Patients with β-TI are also at increased risk of silent brain infarcts, with one study finding that 18 out of 30 splenectomized patients had evidence of asymptomatic white matter lesions by magnetic resonance imaging (MRI) 54. Older patients and those not previously transfused had the highest incidence and number of lesions. One possible mechanism by which transfusion therapy may protect against thrombotic and vascular events is the reduced proportion of circulating RBCs with membrane damage that contribute to the hypercoagulable state 55.
Pulmonary hypertension is a severe and common clinical complication in β-TI and HbE/β-thalassaemia 56–58, with an incidence of more than 50% 59–61. However, this complication is rarely reported in HbH disease 62,63. Factors thought to be involved in the development of PHT in patients with NTDT include chronic anaemia, iron overload, splenectomy and hypercoagulability 32,64–67. Importantly, PHT is a leading cause of right-sided heart failure in patients with NTDT 59,68. Response to symptomatic and supportive treatment is poor, and in patients with PHT class III–IV, survival is usually only 2–3 years 69. Transfusion therapy has been shown to ameliorate PHT in cases of sickle cell disease 70 and in splenectomized patients with HbE/β-thalassaemia 70,71, suggesting that transfusion therapy can be considered as a potentially life-saving management strategy for severe PHT in patients with NTDT. However, at present, there is no clear guideline on when to start blood transfusion to prevent potential, long-term development of PHT. It is recommended that particular NTDT types with aforementioned risk factors be regularly evaluated by echocardiography to early detect signs of PHT such as increased tricuspid regurgitation velocity (TR jet), and such patients should be candidates for regular blood transfusion therapy 35.
Patients with NTDT are at risk of developing leg ulcers, particularly as they get older 23,70,72, but transfusion therapy may protect against this complication. Risk factors for the development of leg ulcers include splenectomy and iron overload, which may contribute to lesions becoming chronic 23,51,70,73.
Impaired growth and development
One of the potential consequences of chronic anaemia in children with NTDT is growth retardation and pubertal delay 2,23,74,75. At the severe end of the spectrum, children with Hb levels of 5–6 g/dl develop skeletal deformities 26. A study of the effects of different transfusion regimens on growth in 12 patients with β-thalassaemia major and 36 patients with HbE/β-thalassaemia found that children of both sexes who received regular transfusions during the first 10 years grew normally. In patients who were transfused infrequently, both males and females became increasingly retarded in their linear growth 76. Hence, transfusion therapy, along with adequate iron chelation and hormone therapy, may be beneficial in helping to ensure normal development in some paediatric patients with NTDT.
Endocrine complications
Endocrine problems are well characterized in patients with TDT as a consequence of transfusion-related iron overload. However, the prevalence of adrenal insufficiency in children with HbE/β-thalassaemia was recently found to be 50% for transfusion-dependent patients and 53·5% for non-transfusion-dependent patients 77. It was also recently shown that vitamin D deficiency was significantly more common in children with non-transfusion-dependent HbE/β-thalassaemia than those with transfusion dependency (33·3% vs. 12·2%, P = 0·01) 78. A total of 23% of the OPTIMAL CARE study cohort had osteoporosis, a common complication in β-TI, secondary to bone marrow expansion and vitamin D deficiency 23,79. Transfusion therapy, along with appropriate iron chelation, may be beneficial in paediatric patients to minimize endocrine problems and the development of osteoporosis later in life. However, in a study of 50 paediatric patients (aged 8–13 years) with HbE/β-thalassaemia, osteopenia was common (>50% of patients) even in those receiving adequate transfusion and iron chelation therapy, suggesting that there are other factors influencing the development of this complication apart from anaemia and bone marrow expansion 80.
Transfusion considerations
Transfusion therapy has been shown to be protective against complications such as thrombosis, extramedullary haematopoiesis, PHT, heart failure, cholelithiasis and leg ulcers, but the accompanying increased risk of endocrinopathy secondary to iron overload should be taken into account when making the decision to transfuse a patient with NTDT 23. In addition, the risks of alloimmunization should be considered.
Blood selection and alloimmunization
Alloimmunization is more frequent in patients with thalassaemia than the general population, with reported rates of 4–37% (compared with 1–4% in the general population) 81–87. Alloimmunization rates in patients with thalassaemia appear to vary according to the homogeneity of donor and recipient populations. For example, alloimmunization rates are relatively lower in patients with thalassaemia in Greece and Italy 81,82, whereas a study in the US of Asian patients with thalassaemia, where only 5% of local blood donors were Asian, reported an alloimmunization rate of 20·8% 83. The risk of alloimmunization is 1–1·6% per unit of blood and is highest in older, transfusion-naïve and splenectomized patients, although it is not known why this is the case 81,84,85. It may be that RBC membranes are altered and antigenic exposure increased in splenectomized patients, causing increased immunomodulation and increased risk of alloimmunization 84. The risk of alloimmunization is also increased during pregnancy 35. Reports from Western countries suggest that, in patients with thalassaemia, alloimmunization usually involves C, E and Kell antibodies 81,82,84,86,87. Though some thalassaemia treatment centres routinely perform extended antigen matching, it is not yet clear whether this significantly reduces alloimmunization rates 81,83,87. A recent study examined clinical and transfusion records for 10 years of follow up in 37 patients with thalassaemia intermedia 88. In 2009, the institution had implemented full Rh (C,c,E,e), Kell (K, Kpa, Kpb), Kidd (Jka, Jkb), Duffy (Fya, Fyb), S and s antigen matching for this group of patients. Of the whole cohort, seven patients were alloimmunized, with these antibodies developing prior to 2009, or prior to referral 88. Since 2009, a total of 335 units were transfused in this group of patients, with only one antibody developing after necessary transfusion with non-phenotyped blood. No autoantibodies were found in this study 88. Strategies to reduce the risk of alloimmunization should be employed where possible. Initiation of transfusion at an early age is thought to reduce the risk of alloimmunization, and fully ABO, Rh (D, C, c, E, e) and Kell matched blood should be used 35,81. The risk of autoimmunization should also be considered. In a recent study in 407 patients with thalassaemia, autoantibodies occurred in 6·5% 86. Older age and splenectomy were associated with higher risk of autoimmunization, and 84% of patients with autoantibodies were also alloimmunized 86. Further studies to identify predictive markers for susceptibility to alloimmunization and autoimmunization would be of value to reduce this risk in patients with NTDT. Other complications of blood transfusion including exposure to pathogens and transfusion reactions pose a challenge in the management of patients with thalassaemia. Around half of transfused patients with thalassaemia experience transfusion reactions, and although most are mild or moderate, anaphylaxis can occur in rare cases 86.
Iron overload
Iron overload may occur in patients with NTDT because of both increased intestinal iron absorption and subsequent transfusion therapy; therefore, there is a need to monitor iron burden, irrespective of transfusion. Iron overload associated with transfusion therapy develops more quickly than that caused by the underlying disease, but both can be managed with appropriate chelation therapy. It has been observed that serum ferritin levels can underestimate iron load in patients with β-TI, compared with patients with thalassaemia major. Therefore, relying on this measurement may expose patients with NTDT to high iron levels and delay necessary chelation therapy 89–92. Similar findings were reported in the THALASSA trial of deferasirox in patients with NTDT including HbE/β-thalassaemia and α-thalassaemia 93.
Instead, measurement of liver iron concentration (LIC) by MRI is the recommended method where possible, and chelation therapy should be initiated if LIC reaches ≥5 mg Fe/g dry weight 35,50. If LIC measurement is not possible, a serum ferritin level of ≥800 mg Fe/g dry weight can be used as an indicator for chelation 35. The benefits of transfusion with iron chelation in patients with NTDT was highlighted in the OPTIMAL CARE study, which found that treatment with both transfusion and iron chelation therapy was associated with fewer complications (including endocrinopathy) than either treatment alone 23. Iron overload in patients with NTDT undergoing frequent blood transfusions for any length of time can be managed similarly to patients with transfusion-dependent β-thalassaemia major 35.
Conclusion
Management of NTDT can be complex because of the diverse phenotypes and clinical spectrum of disease. These patients are not reliant on transfusions for survival, but transfusion therapy may convey substantial clinical benefits for some patients if administered properly. Transfusion therapy has been shown to be protective against complications such as thrombosis, extramedullary haematopoiesis, PHT, heart failure, cholelithiasis and leg ulcers in patients with NTDT. In addition, transfusion therapy may help to reduce intestinal iron absorption and thus reduce the risk of serious liver disease. Transfusion strategy should be tailored to the individual patient, and careful monitoring of clinical parameters is essential. Risk of infection and alloimmunization should be considered, and fully ABO, Rh (D, C, c, E, e) and Kell matched blood should be used. Transfusion therapy should be accompanied by the use of an appropriate iron chelator, and iron load should be carefully monitored. Some patients may require frequent transfusions, but it is important to taper or withdraw transfusions when clinical goals have been met to avoid enforcing a lifelong transfusion regimen unnecessarily. Until recently, there have been no formal guidelines on the management of NTDT, but following the position paper in 2012, the Thalassaemia International Federation (TIF) NTDT guidelines are now available on the TIF website 35.
Acknowledgments
We thank Holly Gilbert-Jones for medical editorial assistance with this manuscript. Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals. The authors are fully responsible for the content and editorial decisions for this manuscript.
Conflict of interests
V Viprakasit received research grant support and lecture fees from Novartis Pharmaceuticals and research grant support from GPO−L−ONE, Thailand, and National Institute of Health (NIH); AT Taher received research funding and honoraria from Novartis Pharmaceuticals.
References
- Fucharoen S, Winichagoon P. Hemoglobinopathies in Southeast Asia. Hemoglobin. 1987;11:65–88. doi: 10.3109/03630268709036587. [DOI] [PubMed] [Google Scholar]
- Fucharoen S, Ketvichit P, Pootrakul P, et al. Clinical manifestation of beta-thalassemia/hemoglobin E disease. J Pediatr Hematol Oncol. 2000;22:552–557. doi: 10.1097/00043426-200011000-00022. [DOI] [PubMed] [Google Scholar]
- Fucharoen S, Winichagoon P. New updating into hemoglobinopathies. Int J Lab Hematol. 2012 doi: 10.1111/j.1751-553X.2012.01446.x. ; doi: 10.1111/j.1751-553X.2012.01446.x [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Olivieri NF. The beta-thalassemias. N Engl J Med. 1999;341:99–109. doi: 10.1056/NEJM199907083410207. [DOI] [PubMed] [Google Scholar]
- Sripichai O, Makarasara W, Munkongdee T, et al. A scoring system for the classification of beta-thalassemia/Hb E disease severity. Am J Hematol. 2008;83:482–484. doi: 10.1002/ajh.21130. [DOI] [PubMed] [Google Scholar]
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010;5:11. doi: 10.1186/1750-1172-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86:480–487. doi: 10.2471/BLT.06.036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115:4331–4336. doi: 10.1182/blood-2010-01-251348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherall DJ, Clegg JB. The Thalassaemia Syndromes. 4th edn. Oxford, UK: Blackwell Science; 2001. [Google Scholar]
- Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010;5:13. doi: 10.1186/1750-1172-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal A, Goldrich ML, Haines DA, et al. Heterogeneity of hemoglobin H disease in childhood. N Engl J Med. 2011;364:710–718. doi: 10.1056/NEJMoa1010174. [DOI] [PubMed] [Google Scholar]
- Vichinsky E. Hemoglobin e syndromes. Hematology Am Soc Hematol Educ Program. 2007:79–83. doi: 10.1182/asheducation-2007.1.79. [DOI] [PubMed] [Google Scholar]
- Viprakasit V, Tanphaichitr VS, Pung-Amritt P, et al. Clinical phenotypes and molecular characterization of Hb H-Pakse disease. Haematologica. 2002;87:117–125. [PubMed] [Google Scholar]
- Weatherall DJ. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl 1):S3–S6. doi: 10.1016/S0268-960X(12)70003-6. [DOI] [PubMed] [Google Scholar]
- Lorey F, Cunningham G, Vichinsky EP, et al. Universal newborn screening for Hb H disease in California. Genet Test. 2001;5:93–100. doi: 10.1089/109065701753145538. [DOI] [PubMed] [Google Scholar]
- Michlitsch J, Azimi M, Hoppe C, et al. Newborn screening for hemoglobinopathies in California. Pediatr Blood Cancer. 2009;52:486–490. doi: 10.1002/pbc.21883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanpakit K, Viprakasit V. Variable genotype-phenotype correlations in patients with a rare non-deletional α-thalassemia; Hb Pak Num Po (HBA1:c.396_397insT) J Pediatr Hematol Oncol. 2014;36:e185–e189. doi: 10.1097/MPH.0000000000000016. [DOI] [PubMed] [Google Scholar]
- Viprakasit V. 2013. Alpha thalassemia syndromes: from clinical and molecular diagnosis to bedside management. EHA 18 Education Book.
- Viprakasit V, Ekwattanakit S, Chalaow N, et al. Clinical presentation and molecular identification of four uncommon α-globin variants in Thailand: initiation codon mutation of α2-globin gene (HBA2:c.1delA), donor splice site mutation of α1-globin gene (IVSI-1, HBA1:c.95+1G>A), Hemoglobin Queens Park/Chao Pra Ya (HBA1:c.98T>A) and Hemoglobin Westmead (HBA2:c.369C>G) Acta Haematol. 2013;131:88–94. doi: 10.1159/000353119. [DOI] [PubMed] [Google Scholar]
- Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79:704–712. [PMC free article] [PubMed] [Google Scholar]
- Laosombat V, Viprakasit V, Chotsampancharoen T, et al. Clinical features and molecular analysis in Thai patients with HbH disease. Ann Hematol. 2009;88:1185–1192. doi: 10.1007/s00277-009-0743-5. [DOI] [PubMed] [Google Scholar]
- Taher AT, Musallam KM, Cappellini MD, et al. Optimal management of beta thalassaemia intermedia. Br J Haematol. 2011;152:512–523. doi: 10.1111/j.1365-2141.2010.08486.x. [DOI] [PubMed] [Google Scholar]
- Taher AT, Musallam KM, Karimi M, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood. 2010;115:1886–1892. doi: 10.1182/blood-2009-09-243154. [DOI] [PubMed] [Google Scholar]
- Taher AT, Musallam KM, Karimi M, et al. Splenectomy and thrombosis: the case of thalassemia intermedia. J Thromb Haemost. 2010;8:2152–2158. doi: 10.1111/j.1538-7836.2010.03940.x. [DOI] [PubMed] [Google Scholar]
- Taher AT, Musallam KM, Cappellini MD. Thalassaemia intermedia: an update. Mediterr J Hematol Infect Dis. 2009;1:e2009004. doi: 10.4084/MJHID.2009.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musallam KM, Khoury B, Abi-Habib R, et al. Health-related quality of life in adults with transfusion-independent thalassaemia intermedia compared to regularly transfused thalassaemia major: new insights. Eur J Haematol. 2011;87:73–79. doi: 10.1111/j.1600-0609.2011.01623.x. [DOI] [PubMed] [Google Scholar]
- Taher AT, Musallam KM, Karimi M, et al. Contemporary approaches to treatment of beta-thalassemia intermedia. Blood Rev. 2012;26(Suppl. 1):S24–S27. doi: 10.1016/S0268-960X(12)70008-5. [DOI] [PubMed] [Google Scholar]
- Taher AT, Cappellini MD, Musallam KM. Recent advances and treatment challenges in patients with non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl. 1):S1–S2. doi: 10.1016/S0268-960X(12)00028-8. [DOI] [PubMed] [Google Scholar]
- Cappellini MD, Cohen AR, Eleftheriou A, et al. Guidelines for the Clinical Management of Thalassaemia. 2nd edn revised. Nicosia, Cyprus: Thalassaemia International Federation; 2008. [PubMed] [Google Scholar]
- Allen A, Fisher C, Premawardhena A, et al. Adaptation to anemia in hemoglobin E-ss thalassemia. Blood. 2010;116:5368–5370. doi: 10.1182/blood-2010-06-289488. [DOI] [PubMed] [Google Scholar]
- O'Donnell A, Premawardhena A, Arambepola M, et al. Age-related changes in adaptation to severe anemia in childhood in developing countries. Proc Natl Acad Sci USA. 2007;104:9440–9444. doi: 10.1073/pnas.0703424104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musallam KM, Taher AT, Rachmilewitz EA. Beta-thalassemia intermedia: a clinical perspective. Cold Spring Harb Perspect Med. 2012;2:a013482. doi: 10.1101/cshperspect.a013482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premawardhena A, Fisher CA, Olivieri NF, et al. Haemoglobin E beta thalassaemia in Sri Lanka. Lancet. 2005;366:1467–1470. doi: 10.1016/S0140-6736(05)67396-5. [DOI] [PubMed] [Google Scholar]
- Viprakasit V, Tanphaichitr VS, Chinchang W, et al. Evaluation of alpha hemoglobin stabilizing protein (AHSP) as a genetic modifier in patients with beta thalassemia. Blood. 2004;103:3296–3299. doi: 10.1182/blood-2003-11-3957. [DOI] [PubMed] [Google Scholar]
- Taher A, Vichinsky E, Musallam KM, et al. Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT) Nicosia, Cyprus: Thalassaemia International Federation; 2013. TIF Publication No. 19. [PubMed] [Google Scholar]
- Eder AF, Chambers LA. Noninfectious complications of blood transfusion. Arch Pathol Lab Med. 2007;131:708–718. doi: 10.5858/2007-131-708-NCOBT. [DOI] [PubMed] [Google Scholar]
- Rivella S. The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl. 1):S12–S15. doi: 10.1016/S0268-960X(12)70005-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musallam KM, Rivella S, Vichinsky E, et al. Non-transfusion-dependent thalassemias. Haematologica. 2013;98:833–844. doi: 10.3324/haematol.2012.066845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haidar R, Mhaidli H, Taher AT. Paraspinal extramedullary hematopoiesis in patients with thalassemia intermedia. Eur Spine J. 2010;19:871–878. doi: 10.1007/s00586-010-1357-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzola M, Pootrakul P, Huebers HA, et al. Erythroid marrow function in anemic patients. Blood. 1987;69:296–301. [PubMed] [Google Scholar]
- Cazzola M, Finch CA. Evaluation of erythroid marrow function in anemic patients. Haematologica. 1987;72:195–200. [PubMed] [Google Scholar]
- Cazzola M, De Stefano P, Ponchio L, et al. Relationship between transfusion regimen and suppression of erythropoiesis in beta-thalassaemia major. Br J Haematol. 1995;89:473–478. doi: 10.1111/j.1365-2141.1995.tb08351.x. [DOI] [PubMed] [Google Scholar]
- Musallam KM, Nasreddine W, Beydoun A, et al. Brain positron emission tomography in splenectomized adults with beta-thalassemia intermedia: uncovering yet another covert abnormality. Ann Hematol. 2012;91:235–241. doi: 10.1007/s00277-011-1291-3. [DOI] [PubMed] [Google Scholar]
- Taher A, Isma'eel H, Mehio G, et al. Prevalence of thromboembolic events among 8,860 patients with thalassaemia major and intermedia in the Mediterranean area and Iran. Thromb Haemost. 2006;96:488–491. [PubMed] [Google Scholar]
- Vichinsky E. Advances in the treatment of alpha-thalassemia. Blood Rev. 2012;26(Suppl 1):S31–S34. doi: 10.1016/S0268-960X(12)70010-3. [DOI] [PubMed] [Google Scholar]
- Anuwatanakulchai M, Pootrakul P, Thuvasethakul P, et al. Non-transferrin plasma iron in beta-thalassaemia/Hb E and haemoglobin H diseases. Scand J Haematol. 1984;32:153–158. doi: 10.1111/j.1600-0609.1984.tb02171.x. [DOI] [PubMed] [Google Scholar]
- Pootrakul P, Rugkiatsakul R, Wasi P. Increased transferrin iron saturation in splenectomized thalassaemic patients. Br J Haematol. 1980;46:143–145. doi: 10.1111/j.1365-2141.1980.tb05946.x. [DOI] [PubMed] [Google Scholar]
- Pootrakul P, Vongsmasa V, La-ongpanich P, et al. Serum ferritin levels in thalassemias and the effect of splenectomy. Acta Haematol. 1981;66:244–250. doi: 10.1159/000207129. [DOI] [PubMed] [Google Scholar]
- Taher A, Hershko C, Cappellini MD. Iron overload in thalassaemia intermedia: reassessment of iron chelation strategies. Br J Haematol. 2009;147:634–640. doi: 10.1111/j.1365-2141.2009.07848.x. [DOI] [PubMed] [Google Scholar]
- Musallam KM, Cappellini MD, Wood JC, et al. Iron overload in non-transfusion-dependent thalassemia: a clinical perspective. Blood Rev. 2012;26(Suppl 1):S16–S19. doi: 10.1016/S0268-960X(12)70006-1. [DOI] [PubMed] [Google Scholar]
- Musallam KM, Cappellini MD, Wood JC, et al. Elevated liver iron concentration is a marker of increased morbidity in patients with beta thalassemia intermedia. Haematologica. 2011;96:1605–1612. doi: 10.3324/haematol.2011.047852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restivo Pantalone G, Renda D, Valenza F, et al. Hepatocellular carcinoma in patients with thalassaemia syndromes: clinical characteristics and outcome in a long term single centre experience. Br J Haematol. 2010;150:245–247. doi: 10.1111/j.1365-2141.2010.08180.x. [DOI] [PubMed] [Google Scholar]
- Cappellini MD, Musallam KM, Poggiali E, et al. Hypercoagulability in non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl. 1):S20–S23. doi: 10.1016/S0268-960X(12)70007-3. [DOI] [PubMed] [Google Scholar]
- Taher AT, Musallam KM, Nasreddine W, et al. Asymptomatic brain magnetic resonance imaging abnormalities in splenectomized adults with thalassemia intermedia. J Thromb Haemost. 2010;8:54–59. doi: 10.1111/j.1538-7836.2009.03651.x. [DOI] [PubMed] [Google Scholar]
- Chen S, Eldor A, Barshtein G, et al. Enhanced aggregability of red blood cells of beta-thalassemia major patients. Am J Physiol. 1996;270:H1951–H1956. doi: 10.1152/ajpheart.1996.270.6.H1951. [DOI] [PubMed] [Google Scholar]
- Sonakul D, Pacharee P, Laohapand T, et al. Pulmonary artery obstruction in thalassaemia. Southeast Asian J Trop Med Public Health. 1980;11:516–523. [PubMed] [Google Scholar]
- Sonakul D, Suwanagool P, Sirivaidyapong P, et al. Distribution of pulmonary thromboembolic lesions in thalassemic patients. Birth Defects Orig Artic Ser. 1987;23:375–384. [PubMed] [Google Scholar]
- Sonakul D, Fucharoen S. Pulmonary thromboembolism in thalassemic patients. Southeast Asian J Trop Med Public Health. 1992;23(Suppl 2):25–28. [PubMed] [Google Scholar]
- Aessopos A, Farmakis D, Karagiorga M, et al. Cardiac involvement in thalassemia intermedia: a multicenter study. Blood. 2001;97:3411–3416. doi: 10.1182/blood.v97.11.3411. [DOI] [PubMed] [Google Scholar]
- Teawtrakul N, Ungprasert P, Pussadhamma B, et al. Effect of genotype on pulmonary hypertension in patients with thalassemia. Blood. 2013;122 doi: 10.1111/ejh.12261. :abst 3456. [DOI] [PubMed] [Google Scholar]
- Chuncharunee S, Atichartakarn V, Udomsubpayakul U, et al. Features associated with pulmonary hypertension in splenectomized patients with hemoglobin E/β-thalassemia disease. Blood. 2013;122 :abst 3457. [Google Scholar]
- Chueamuangphan N, Wongtheptien W, Nawarawong W, et al. Clinical indicators for pulmonary arterial hypertension in thalassemia. J Med Assoc Thai. 2012;95:16–21. [PubMed] [Google Scholar]
- Yin X, Zhang X, Wu Z, et al. Pulmonary hypertension risk in patients with haemoglobin H disease: low incidence and absence of correlation with splenectomy. Acta Haematol. 2013;130:153–159. doi: 10.1159/000347177. [DOI] [PubMed] [Google Scholar]
- Isma'eel H, Chafic AH, Rassi FE, et al. Relation between iron-overload indices, cardiac echo-Doppler, and biochemical markers in thalassemia intermedia. Am J Cardiol. 2008;102:363–367. doi: 10.1016/j.amjcard.2008.03.066. [DOI] [PubMed] [Google Scholar]
- Morris CR, Vichinsky EP. Pulmonary hypertension in thalassemia. Ann N Y Acad Sci. 2010;1202:205–213. doi: 10.1111/j.1749-6632.2010.05580.x. [DOI] [PubMed] [Google Scholar]
- Phrommintikul A, Sukonthasarn A, Kanjanavanit R, et al. Splenectomy: a strong risk factor for pulmonary hypertension in patients with thalassaemia. Heart. 2006;92:1467–1472. doi: 10.1136/hrt.2005.079970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer ST, Kuypers FA, Styles L, et al. Pulmonary hypertension in thalassemia: association with platelet activation and hypercoagulable state. Am J Hematol. 2006;81:670–675. doi: 10.1002/ajh.20640. [DOI] [PubMed] [Google Scholar]
- Aessopos A, Stamatelos G, Skoumas V, et al. Pulmonary hypertension and right heart failure in patients with beta-thalassemia intermedia. Chest. 1995;107:50–53. doi: 10.1378/chest.107.1.50. [DOI] [PubMed] [Google Scholar]
- Gibbs JSR, Higenbottam TW. Recommendations on the management of pulmonary hypertension in clinical practice. Heart. 2001;86:i1–i13. [PMC free article] [PubMed] [Google Scholar]
- Claster S, Hammer M, Hagar W, et al. Treatment of pulmonary hypertension in sickle cell disease with transfusion. Blood. 1999;94(Suppl 1) ):abst 420a. [Google Scholar]
- Atichartakarn V, Chuncharunee S, Chandanamattha P, et al. Correction of hypercoagulability and amelioration of pulmonary arterial hypertension by chronic blood transfusion in an asplenic hemoglobin E/beta-thalassemia patient. Blood. 2004;103:2844–2846. doi: 10.1182/blood-2003-09-3094. [DOI] [PubMed] [Google Scholar]
- Taher AT, Musallam KM, El-Beshlawy A, et al. Age-related complications in treatment-naive patients with thalassaemia intermedia. Br J Haematol. 2010;150:486–489. doi: 10.1111/j.1365-2141.2010.08220.x. [DOI] [PubMed] [Google Scholar]
- Levin C, Koren A. Healing of refractory leg ulcer in a patient with thalassemia intermedia and hypercoagulability after 14 years of unresponsive therapy. Isr Med Assoc J. 2011;13:316–318. [PubMed] [Google Scholar]
- Phadke SR, Agarwal S. Phenotype score to grade the severity of thalassemia intermedia. Indian J Pediatr. 2003;70:477–481. doi: 10.1007/BF02723137. [DOI] [PubMed] [Google Scholar]
- Taher A, Isma'eel H, Cappellini MD. Thalassemia intermedia: revisited. Blood Cells Mol Dis. 2006;37:12–20. doi: 10.1016/j.bcmd.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Viprakasit V, Tanphaichitr VS, Mahasandana C, et al. Linear growth in homozygous beta-thalassemia and beta-thalassemia/hemoglobin E patients under different treatment regimens. J Med Assoc Thai. 2001;84:929–941. [PubMed] [Google Scholar]
- Nakavachara P, Viprakasit V. Adrenal insufficiency is prevalent in HbE/beta-thalassaemia paediatric patients irrespective of their clinical severity and transfusion requirement. Clin Endocrinol (Oxf) 2013;79:776–783. doi: 10.1111/cen.12235. [DOI] [PubMed] [Google Scholar]
- Nakavachara P, Viprakasit V. Children with hemoglobin E/beta-thalassemia have a high risk of being vitamin D deficient even if they get abundant sun exposure: a study from Thailand. Pediatr Blood Cancer. 2013;60:1683–1688. doi: 10.1002/pbc.24614. [DOI] [PubMed] [Google Scholar]
- Napoli N, Carmina E, Bucchieri S, et al. Low serum levels of 25-hydroxy vitamin D in adults affected by thalassemia major or intermedia. Bone. 2006;38:888–892. doi: 10.1016/j.bone.2005.11.018. [DOI] [PubMed] [Google Scholar]
- Viprakasit V, Sawathiparnich P, Sangpraypanm T, et al. Osteopenia is commonly present in prepubertal children with severe HbE/βthalassemia despite adequate transfusion and iron chelation therapy. Blood. 2007;110 :abst 3830. [Google Scholar]
- Spanos T, Karageorga M, Ladis V, et al. Red cell alloantibodies in patients with thalassemia. Vox Sang. 1990;58:50–55. doi: 10.1111/j.1423-0410.1990.tb02055.x. [DOI] [PubMed] [Google Scholar]
- Sirchia G, Zanella A, Parravicini A, et al. Red cell alloantibodies in thalassemia major. Results of an Italian cooperative study. Transfusion. 1985;25:110–112. doi: 10.1046/j.1537-2995.1985.25285169198.x. [DOI] [PubMed] [Google Scholar]
- Singer ST, Wu V, Mignacca R, et al. Alloimmunization and erythrocyte autoimmunization in transfusion-dependent thalassemia patients of predominantly asian descent. Blood. 2000;96:3369–3373. [PubMed] [Google Scholar]
- Chou ST, Liem RI, Thompson AA. Challenges of alloimmunization in patients with haemoglobinopathies. Br J Haematol. 2012;159:394–404. doi: 10.1111/bjh.12061. [DOI] [PubMed] [Google Scholar]
- Kosaryan M, Mahdavi MR, Roshan P, et al. Prevalence of alloimmunisation in patients with beta thalassaemia major. Blood Transfus. 2012;10:396–397. doi: 10.2450/2012.0072-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vichinsky E, Neumayr L, Trimble S, et al. Transfusion complications in thalassemia patients: a report from the Centers for Disease Control and Prevention. Transfusion. 2014;54:972–981. doi: 10.1111/trf.12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AA, Cunningham MJ, Singer ST, et al. Red cell alloimmunization in a diverse population of transfused patients with thalassaemia. Br J Haematol. 2011;153:121–128. doi: 10.1111/j.1365-2141.2011.08576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Riyami AZ, Al-Mahrooqi S, Al-Hinai S, et al. Transfusion therapy and alloimmunization in thalassemia intermedia: a 10 year experience at a tertiary care university hospital. Transfus Apher Sci. 2014 doi: 10.1016/j.transci.2014.04.009. http://dx.doi.org/10.1016/j.transci.2014.04.009 (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Taher A, El RF, Isma'eel H, et al. Correlation of liver iron concentration determined by R2 magnetic resonance imaging with serum ferritin in patients with thalassemia intermedia. Haematologica. 2008;93:1584–1586. doi: 10.3324/haematol.13098. [DOI] [PubMed] [Google Scholar]
- Taher AT, Musallam KM, Wood JC, et al. Magnetic resonance evaluation of hepatic and myocardial iron deposition in transfusion-independent thalassemia intermedia compared to regularly transfused thalassemia major patients. Am J Hematol. 2010;85:288–290. doi: 10.1002/ajh.21626. [DOI] [PubMed] [Google Scholar]
- Taher AT, Viprakasit V, Musallam KM, et al. Treating iron overload in patients with non-transfusion-dependent thalassemia. Am J Hematol. 2013;88:409–415. doi: 10.1002/ajh.23405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tony S, Daar S, Elshinawy M, et al. T2* MRI in regularly transfused children with thalassemia intermedia: serum ferritin does not reflect liver iron stores. Pediatr Hematol Oncol. 2012;29:579–584. doi: 10.3109/08880018.2012.708891. [DOI] [PubMed] [Google Scholar]
- Taher AT, Porter J, Viprakasit V, et al. Deferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood. 2012;120:970–977. doi: 10.1182/blood-2012-02-412692. [DOI] [PubMed] [Google Scholar]
- Cohen AR, Galanello R, Pennell DJ, et al. Thalassemia. Hematology Am Soc Hematol Educ Program. 2004:14–34. doi: 10.1182/asheducation-2004.1.14. [DOI] [PubMed] [Google Scholar]
- Origa R, Sollaino MC, Giagu N, et al. Clinical and molecular analysis of haemoglobin H disease in Sardinia: haematological, obstetric and cardiac aspects in patients with different genotypes. Br J Haematol. 2007;136:326–332. doi: 10.1111/j.1365-2141.2006.06423.x. [DOI] [PubMed] [Google Scholar]