

Abstract
Our earlier work showed that knockout of hematopoietic prostaglandin D synthase (HPGDS, an enzyme that produces prostaglandin D2) caused more adenomas in ApcMin/+ mice. Conversely, highly expressed transgenic HPGDS allowed fewer tumors. Prostaglandin D2 (PGD2) binds to the prostaglandin D2 receptor known as PTGDR (or DP1). PGD2 metabolites bind to peroxisome proliferator-activated receptor γ (PPARG). We hypothesized that Ptgdr or Pparg knockouts may raise numbers of tumors, if these receptors take part in tumor suppression by PGD2. To assess, we produced ApcMin/+ mice with and without Ptgdr knockouts (147 mice). In separate experiments, we produced ApcMin/+ mice expressing transgenic lipocalin-type prostaglandin D synthase (PTGDS), with and without heterozygous Pparg knockouts (104 mice). Homozygous Ptgdr knockouts raised total numbers of tumors by 30–40% at 6 and 14 weeks. Colon tumors were not affected. Heterozygous Pparg knockouts alone did not affect tumor numbers in ApcMin/+ mice. As mentioned above, our Pparg knockout assessment also included mice with highly expressed PTGDS transgenes. ApcMin/+ mice with transgenic PTGDS had fewer large adenomas (63% of control) and lower levels of v-myc avian myelocytomatosis viral oncogene homolog (MYC) mRNA in the colon. Heterozygous Pparg knockouts appeared to blunt the tumor-suppressing effect of transgenic PTGDS. However, tumor suppression by PGD2 was more clearly mediated by receptor PTGDR in our experiments. The suppression mechanism did not appear to involve changes in microvessel density or slower proliferation of tumor cells. The data support a role for PGD2 signals acting through PTGDR in suppression of intestinal tumors.
Keywords: Adenomatous polyposis coli, gastrointestinal neoplasms, PPAR gamma, prostaglandin D2 receptor, prostaglandin D2 synthases
Introduction
Prostaglandin studies in intestinal neoplasia usually focus on prostaglandin E2 (PGE2), a pro-tumorigenic compound 1–4. In an opposite effect, knockout of the gene for hematopoietic prostaglandin D synthase (HPGDS) caused more adenomas in ApcMin/+ mice. Moreover, high HPGDS production from transgenes allowed fewer 5. Prostaglandin D2 (PGD2) and PGE2 are both made from PGH2, so Hpgds knockouts could have shunted conversion of PGH2 to PGE2. Likewise, HPGDS transgenes could have drawn prostaglandin synthesis away from PGE2.
Lewis lung cancer cells implanted onto the backs of mice lacking the PGD2 receptor (PTGDR, also known as DP1), grew faster than tumors implanted onto wild-type mice 6. Furthermore, the PTGDR agonist, BW245C, reduced tumor growth. These results support a role for PGD2 itself.
Tumor suppression by PGD2 might also occur through inhibition of inflammatory genes by molecules that bind PGD2 metabolites. For example, PGD2 metabolites bind to peroxisome proliferator-activated receptor γ (PPARG). Such binding can induce conjugation of small ubiquitin-related modifier-1 (SUMO-1) to PPARG. SUMOylation is thought to increase PPARG binding to nuclear receptor corepressor complexes, causing transrepression of inflammatory genes 7. Additionally, 15-deoxy-Δ12,14-PGJ2 may down-regulate inflammatory genes, through covalent binding to nuclear factor-κB or IκB kinase 8.
Here, we show that knockouts of Ptgdr increased tumor numbers in ApcMin/+ mice, indicating that PGD2 and PTGDR act to suppress tumors. PPARG had smaller effects in our experiments.
Material and Methods
Mice
The protocol and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Los Angeles Biomedical Research Institute. C57BL/6, FVB/N, and ApcMin/+ (C57BL/6; no. 002020) mice came from Jackson (Bar Harbor, ME), as did mice carrying the Cre transgene controlled by the adenovirus EIIa promoter [Tg(EIIa-Cre) C5379Lmgd/J; FVB/N strain; no. 003314] 9. Mice in which exon 2 of the Pparg gene is flanked by loxP sites were from F. Gonzalez (Ppargflox/flox FVB/N mice).
To produce ApcMin/+ mice with heterozygous Ptgdr knockouts, we crossed male ApcMin/+ mice with female homozygous Ptgdr knockout mice 10. Male ApcMin/+ mice with heterozygous or homozygous Ptgdr knockouts were then bred with female homozygous Ptgdr knockout mice to produce ApcMin/+ mice with homozygous Ptgdr knockouts (all 100% C57BL/6).
Our PTGDS transgenic mice (line B20; FVB/N) overexpress human PTGDS in all tissues 11. Reported basal brain levels of PGD2 were 1.5-fold higher than wild-type levels and rose fivefold upon stimulation. PGE2 levels did not change. The mice had more eosinophilia in a bronchial asthma model, compared to HPGDS transgenic mice 12.
To generate heterozygotic Pparg knockout mice, we crossed Ppargflox/flox mice with Tg(EIIa-Cre) mice and identified heterozygotes lacking exon 2 of the Pparg gene (Pparg+/− mice; all FVB/N) 13. We then crossed female Pparg+/− FVB/N mice with male ApcMin/+ C57BL/6 mice to produce ApcMin/+ Pparg+/−, ApcMin/+, and Pparg+/− mice, all on an F1 mixed background. Similarly, we bred PTGDS transgenic FVB/N males with C57BL/6 females to produce transgenic mice on an F1 C57BL/6 × FVB/N background. We intercrossed these various offspring to obtain additional mice with desired genotypes. Fifteen of the 104 mice used were C57BL/6 × FVB/N F1 mice, and 89 were from matings of F1 mice or mice in later generations (all 50% C57BL/6).
Intestinal histopathology and definitions of tumor sizes
Adenomas were counted histologically at 6 or 14 weeks, without knowing genotypes 5. We used 24 Swiss roll sections spaced 150 μm apart for PTGDS transgenic mice, Pparg knockout mice, and their controls. We used 10 Swiss roll sections (250 μm apart) for Ptgdr knockout mice and their controls. Tumors sizes were gauged by the number of sections spanned. Small tumors were defined as those seen in only 1 section. Large tumors were those with profiles in multiple sections. Mitotic figures were identified as described 14.
Statistical analyses of tumor data
Tumor data were analyzed by nonparametric methods (Kruskal–Wallis and Mann–Whitney), because numbers of tumors per mouse did not follow a Gaussian distribution. We analyzed total, small, large, and colon tumors. We also calculated ratios of the geometric mean number of tumors in genetically modified mice to the geometric mean number in controls. Ratios were estimated from differences in logarithm-transformed tumor numbers. For the colon, we added 0.5 to all numbers of tumors before taking logarithms, to handle zero values. Data from 6- and 14-week-old mice were analyzed separately. These statistical methods were also used to reanalyze tumor data from ApcMin/+ mice with transgenic HPGDS (and controls) from earlier work 5.
Immunohistochemistry
Antibodies used were: mouse monoclonal anti-human PTGDS 15; rabbit polyclonal anti-human HPGDS; mouse monoclonal anti-rat proliferating cell nuclear antigen (PCNA); and rat monoclonal anti-mouse CD31. Staining for HPGDS, PCNA, and CD31 was done on slides from ApcMin/+ mice with transgenic HPGDS from earlier work 5.
In situ hybridization
Digoxigenin-labeled probes were prepared by in vitro transcription from a linearized plasmid vector containing the mouse PTGDR cDNA (DIG RNA labeling kit; Roche; Indianapolis, IN). T7 RNA polymerase was used to make anti-sense probes. SP6 RNA polymerase was used to prepare control sense probes 16.
mRNA analyses by reverse transcription and real-time PCR (RT-PCR)
Primers, probes, and procedures for preparing RNA and determining copy numbers of RNA transcripts are in Supporting Information. Assays for v-myc avian myelocytomatosis viral oncogene homolog (MYC), GAPDH, and vascular endothelial growth factor A (VEGFA) were performed with kits (Applied Biosystems; Grand Island, NY; Mm00487803_m1, Mm99999915_g1, Mm00437304_m1, respectively).
Results
Tumor scoring
We histologically examined >35,000 tumors in Swiss roll sections (Fig.1A–E), including 9837 tumors from 147 mice in Ptgdr knockout experiments, 21,763 tumors from 104 mice in experiments on PTGDS transgenic and Pparg knockout mice, and 3431 tumors reexamined from 39 HPGDS transgenic mice and controls from earlier work 5.
Figure 1.
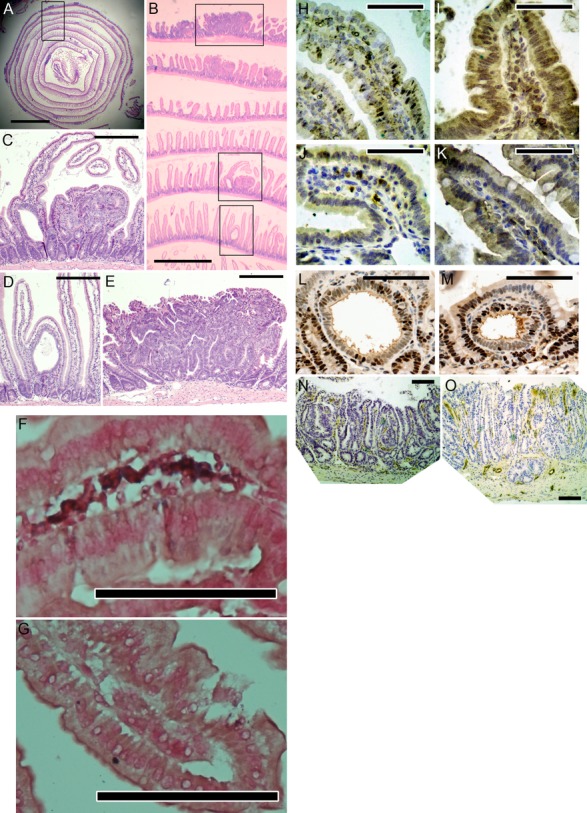
(A–E) Swiss roll section (14 weeks). (A) The box outlines B. Scale bar, 5 mm. (B) The top, middle, and bottom boxes outline E, C, and D, respectively. Scale bar, 1 mm. (C) An early adenoma abutting against a larger adenoma. Scale bar, 200 μm. (D) An early adenoma expanding the villus base. Scale bar, 200 μm. (E) A large adenoma. Scale bar, 200 μm. (F–G) In situ hybridization for PTGDR (12 weeks). (F) Detection of PTGDR mRNA with antisense probes. PTGDR mRNA appears as blue deposits in stromal cells, in a pattern consistent with lymphocytes or monocytes, or both. (G) Sense probes showed no staining (negative control). Counterstained with neutral red. Scale bar, 100 μm. (H–O) Immunohistochemistry (14 weeks). (H–K) High production of human PTGDS and HPGDS in transgenic mice, shown by immunoperoxidase staining (with rabbit polyclonal anti-human PTGDS [H, I] or HPGDS [J, K] antibodies). Staining (brown) occurred in all cell types of the small bowel and colon (epithelium and stroma). Scale bars, 50 μm. (H) Small bowel villi from a wild-type mouse. Antibody labeling is mostly in the cytosol of some epithelial cells, with occasional stromal cell staining. (I) A small bowel villus from a PTGDS transgenic mouse. Antibody binding is heavy throughout the villus, with a cytoplasmic staining pattern. (J) Small bowel villi from a wild-type mouse. HPGDS staining is mainly within the stroma of villi, not epithelial cells. Earlier studies showed these cells to be macrophages and monocytes. (K) Small bowel villi from an HPGDS transgenic mouse, showing heavy antibody staining in all cells (as in I). (L, M) Staining for PCNA in intravillar tumors from ApcMin/+ mice, with and without HPGDS transgenes. There was no consistent difference in staining between mice with and without transgenic HPGDS. Scale bars, 100 μm. (L) An intravillar adenoma from an ApcMin/+ mouse. (M) An intravillar tumor from an HPGDS transgenic ApcMin/+ mouse. (N, O) Staining for microvessels with anti-CD31 antibodies in tumors from ApcMin/+ mice with and without HPGDS transgenes. There was no consistent difference in staining between mice with and without transgenic HPGDS. Scale bars, 100 μm. (N) A tumor from an ApcMin/+ mouse. (O) A tumor from an HPGDS transgenic ApcMin/+ mouse.
The earliest tumors were uniglandular, intravillar lesions with a simple cystic configuration, or intravillar neoplasms, also known as intravillous microadenomas 17,18, dysplastic crypts 19, and cystic crypts (Fig. S1A and S1D) 20,21. More advanced early tumors may have other dysplastic features, such as extension of dysplastic cells (Fig. S1E), multiple lumina (Fig. S1C and G), loss of epithelial cell polarity (Fig. S1G), pseudo-stratification, or crowding (Fig. S1F).
Intravillar tumors progressed by enlarging, forming adjoining cysts (Figs. S1C and S2), or erupting through the villus surface into the bowel lumen (Figs. S1B and S3). Although early tumors arise from crypts 17,22, we found only a few examples of out-pouching of cysts from crypts (Fig. S4). Serial sections from two tumors (75 sections each) showed that early tumors may have no crypt connection (Fig. S5) 21. Examples of early colon tumors are shown in Fig. S6.
Ptgdr knockouts and intestinal tumors
At 6 weeks (Fig.2A), homozygous Ptgdr knockouts raised total numbers of tumors (medians 64 vs. 49.5; P = 0.0086; Table S1A) and numbers of small tumors (medians 58 vs. 42.5; P = 0.0089; Table S1B). Large tumors and colon tumors were not affected by Ptgdr knockouts at 6 weeks.
Figure 2.
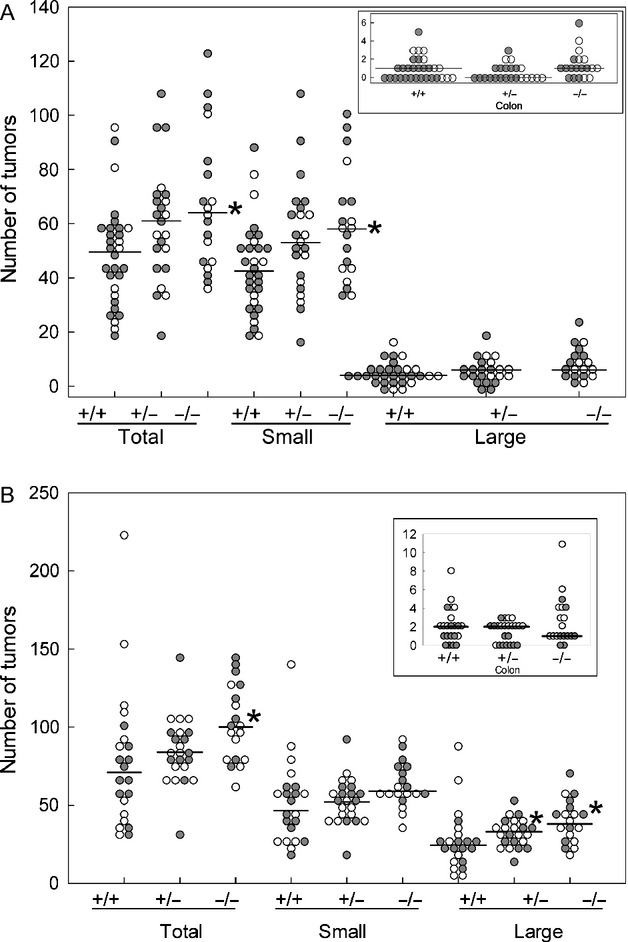
Tumors in ApcMin/+ mice, with and without Ptgdr knockouts (total, small, large, and colon). (A) Tumors at 6 weeks. (B) Tumors at 14 weeks. +/+, control ApcMin/+ mice. +/− and −/−, ApcMin/+ mice with heterozygous and homozygous Ptgdr knockouts, respectively. Filled symbols: females. Open symbols: males. Horizontal bars: medians. *P < 0.025. See Tables S1 and S2 for details.
At 14 weeks (Fig.2B), heterozygous Ptgdr knockouts increased the median number of large tumors (33 vs. 24; P = 0.023; Table S2C). Also at 14 weeks, homozygous Ptgdr knockouts raised median numbers of total tumors (100 vs. 71; P = 0.0060; Table S2A) and large tumors (38 vs. 24; P = 0.0040; Table S2C). Ptgdr knockouts did not affect small or colon tumors at 14 weeks (Table S2B and D).
To obtain data on occurrence of the earliest tumors, we also scored tumors in ten 3-week-old mice: six ApcMin/+ mice (3–8 tumors each); three ApcMin/+ mice with heterozygous Ptgdr knockouts (5–11 tumors each); and one ApcMin/+ mouse with homozygous Ptgdr knockouts (11 tumors). However, data from these 10 mice were not included in statistical analyses, because of the age difference.
In situ hybridization for PTGDR mRNA showed consistent, but weak, staining of inflammatory cells in the mucosal stroma (lymphocytes or monocytes, or both; Fig.1F–G). There was no detectable staining in epithelial cells of crypts or villi. Staining with PTGDR antibodies was not conclusive (not shown).
Expression of transgenic PTGDS
Human PTGDS transgenes were highly expressed in the intestines, as measured RT-PCR. Specifically, we found 1.61 × 105 and 8.13 × 105 copies of human PTGDS transcripts per nanogram of total RNA in two transgenic mice (geometric mean, 3.6 × 105 copies). These values were comparable to levels for HPGDS transgenes in earlier work (7.5 × 105 copies—a 375-fold increase in expression of transgenic HPGDS over endogenous mouse Hpgds) 5. Immunohistochemistry showed heavy staining of transgenic PTGDS in all intestinal cells (Fig.1H–I). Endogenous mouse PTGDS mRNA was not detectable in the colon.
Transgenic PTGDS and large tumors
With 104 ApcMin/+ mice, we scored intestinal tumors in relation to transgenic PTGDS, with and without heterozygous Pparg knockouts. Among mice without Pparg knockouts, only large tumors were reduced in number by transgenic PTGDS (medians were 52 vs. 83 for controls; P = 0.011; Fig.3A–D; Table S3). Tumor suppression was also reflected by the ratio of the geometric mean number of large tumors in PTGDS transgenic mice to the geometric mean number in controls (ratio = 0.56 for large tumors; 95% confidence interval 0.34–0.92; Table S3C). Large tumors were >150–300 μm in diameter, based on the spacing between sections.
Figure 3.

Tumors in ApcMin/+ mice with PTGDS transgenes, with and without heterozygous Pparg knockouts (all 14 weeks). Data are for total (A), small (B), large (C), and colon tumors (D). There were statistically significant reductions in numbers of large tumors (C) in PTGDS transgenic mice without Pparg knockouts. See Table S3 for details. Pparg +/+ indicates no Pparg knockout. Pparg +/− indicates heterozygous Pparg knockout. Symbols are as in Figure2. *P < 0.025.
We measured colon mRNA levels for VEGFA and MYC, relative to endogenous GAPDH transcript levels. PTGDS transgenes lowered median levels of MYC and VEGFA transcripts by 50% in ApcMin/+ mice (Fig. S7).
Heterozygotic Pparg knockouts and transgenic PTGDS
Without PTGDS transgenes, the numbers of tumors in heterozygotic Pparg knockout mice were comparable to numbers in mice without Pparg knockouts (Fig.3; Table S3; see “Control”). Thus, heterozygous Pparg knockouts alone did not increase tumors in ApcMin/+ mice.
On the other hand, ApcMin/+ mice with both transgenic PTGDS and heterozygotic Pparg knockouts had intermediate numbers of large tumors. Specifically, going by medians, there were 52 large tumors in mice with PTGDS transgenes alone, 88 in mice with heterozygotic Pparg knockouts alone, and 70 in mice with both mutations (Table S3C). Similarly, the ratio of the mean number of large tumors in PTGDS transgenic mice to the mean number in controls was 0.56 for mice without heterozygotic Pparg knockouts (95% confidence interval, 0.34–0.92), compared to 0.78 for mice with heterozygotic Pparg knockouts (95% confidence interval, 0.48–1.26).
PTGDS versus HPGDS
As mentioned above, RT-PCR showed similar expression of transgenic PTGDS, compared to transgenic HPGDS (as measured in our previous work) 5. Also, immunohistochemistry showed high levels of PTGDS and HPGDS (Fig.1H–K). Both experiments scored tumors in the same way (24 Swiss roll sections; 150 μm between sections). Therefore, we reanalyzed slides from HPGDS transgenic mice from our first report 5 to directly compare PTGDS to HPGDS (Fig. S8; Table S4). Ratios of the mean total number of tumors in transgenic mice to the mean total in controls were 0.70 for PTGDS, compared to 0.28 for HPGDS (Tables S3A and S4A). Thus, HPGDS may be two times stronger than PTGDS in suppressing tumors.
We assessed tumor cell proliferation in relation to HPGDS transgenes, by use of immunohistochemistry with anti-PCNA antibodies. Again, we used slides from our earlier work on ApcMin/+ mice with HPGDS transgenes 5. We focused on intravillar tumors, because they are fairly uniform in size. There was no difference in PCNA staining in intravillar tumors in HPGDS transgenic versus nontransgenic ApcMin/+ mice (Fig.1L–M). We also counted mitotic figures in all intravillar tumors of 24 HPGDS transgenic mice and 15 ApcMin/+ controls. There were 0.18 mitoses per tumor in HPGDS transgenic ApcMin/+ mice (33 mitotic figures in 182 intravillar tumors), compared to 0.11 mitoses per tumor in non-transgenic ApcMin/+ mice (64 mitotic figures in 587 intravillar tumors). Thus, transgenic HPGDS did not reduce tumor cell proliferation.
Immunohistochemistry with anti-CD31 antibodies showed no consistent difference in microvessel staining between HPGDS transgenic and nontransgenic tumors (Fig.1N–O). Thus, microvessel growth does not appear to explain occurrence of fewer tumors with PGD2.
Tumors in eight mutants with altered PGD2 synthesis or binding
We have now analyzed tumors in eight different ApcMin/+ mouse mutants that have altered PGD2 production or binding, due to knockouts or transgenes. Some experiments used different procedures for cutting sections. For example, we used up to 24 Swiss roll sections for scoring tumors in our first report 5 and in the PTGDS and PPARG experiments shown here. Alternatively, we used 10 sections per Swiss roll in the PTGDR experiments, because reanalysis of earlier data showed that the same conclusions can be reached with 8–10 sections.
To compare data across experiments, we converted the total number of tumors for each mouse to a “multiple of the median” value. Specifically, we divided the total number of tumors for each mouse by the median number of tumors for that mouse's controls. By this analysis, the most tumor-promoting mutations were Hpgds knockouts and homozygous Ptgdr knockouts—raising tumor numbers 40% above control values (Fig.4; all mice were analyzed at 14 weeks). In contrast, HPGDS transgenes were the most tumor-suppressing mutations—reducing tumor numbers to 20–30% of the control value.
Figure 4.
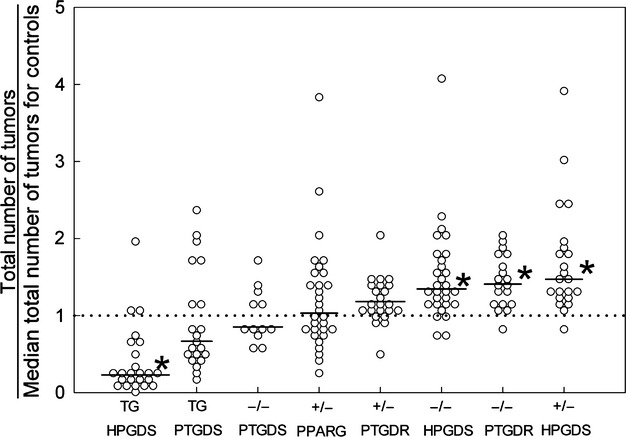
Tumors in ApcMin/+ mice with various mutations that affect PGD2 production or binding (all at 14 weeks). Total numbers of tumors scored for each mouse were divided by the median total number of tumors scored among that mouse's controls. HPGDS transgenes are the most tumor-suppressive mutations, whereas homozygous Ptgdr knockouts and homozygous and heterozygous Hpgds knockouts are the most tumorigenic. The dotted line represents median control values (defined as 1.0). TG, transgenic; +/−, heterozygous knockouts; −/−, homozygous knockouts. Horizontal bars, median values: HPGDS TG, 0.23; PTGDS TG, 0.67; Ptgds −/−, 0.85; Pparg +/−, 1.03; Ptgdr +/−, 1.18; Hpgds −/−, 1.34; Ptgdr −/−, 1.40; Hpgds +/−, 1.47. *P < 0.05.
Female versus male ApcMin/+ mice
To assess female–male differences in tumor numbers at 14 weeks, we used current data and two earlier reports 5,23 (for a total of 61 female and 75 male ApcMin/+ mice; Fig. S9). Males and females had similar numbers of intestinal tumors (ratio of tumors in males vs. females, 0.82; P = 0.069). However, males had more colon tumors (ratio of colon tumors in males vs. females, 1.6; P = 0.0002). Results are consistent with McAlpine et al. 24.
Discussion
PTGDR and intestinal tumors
Homozygous deletion of the gene for PGD2 receptor PTGDR led to 30–40% more intestinal tumors in ApcMin/+ mice. The result supports an interpretation that PTGDR mediates tumor inhibition by PGD2 in these mice. We now have data on eight different ApcMin/+ mouse mutants, each with a different alteration in PGD2 production or binding. Homozygous Ptgdr and homozygous or heterozygous Hpgds knockout mutations are the most pro-tumorigenic. On the other hand, HPGDS transgenes are the most tumor-suppressive mutations—lowering numbers of tumors by 70-80% (Fig.4).
There was no detectable staining of PTGDR mRNA in the epithelium of intestinal crypts or villi. However, PTGDR mRNA was consistently detected in inflammatory cells in the mucosal stroma (Fig.1F–G). Tissue-specific gene knockouts will be needed to more conclusively identify the cells that respond to PGD2.
Mutoh et al. 25 treated homozygous Ptgdr knockout mice with azoxymethane starting at 7 weeks and examined colons at 12 weeks. They did not find more aberrant crypt foci in the colons of knockout mice, compared to controls. Our results are consistent with Mutoh et al., because we did not observe more colon tumors at 6 or 14 weeks with Ptgdr knockouts (Fig.2A–B). However, a role for PTGDR in colon tumor growth is supported by human data. Gustafsson et al. 26 found fivefold lower expression of PTGDR in colorectal cancers, compared to normal tissues (62 tumors and 43 normal tissues, from 62 patients). Galamb et al. 27 showed a trend toward decreased PTGDR expression going from normal tissues, to adenomas, to early cancers, and to advanced cancers.
Comparison of PTGDS and HPGDS effects
Transgenic PTGDS in ApcMin/+ mice reduced numbers of large adenomas (>150–300 μm; Fig.3C; Table S3C). In this way, PTGDS had a tumor blocking effect. However, transgenic PTGDS was less effective than transgenic HPGDS in suppressing tumors (Fig.4). Reasons are unknown. A difference between PTGDS and HPGDS is secretion of PTGDS into body fluids, whereas HPGDS stays in the cytosol 28. We recognize that our comparison between PTGDS and HPGDS is based on only one transgenic mouse line for each mutant. But these lines had comparable numbers of PTGDS or HPGDS mRNA transcripts in the intestines (3.6 × 105 and 7.5 × 105 copies, respectively).
Transgenic PTGDS was associated with lower colon expression of MYC (Fig. S7). MYC is a major part of WNT signaling following Apc loss 29. Moreover, disruption of Myc restores the normal appearance of intestinal crypts in mice with intestine-specific Apc knockouts 30. Thus, lower intestinal levels of MYC may be part of the tumor preventive mechanism of PGD2.
Reduced levels of VEGFA mRNA were also seen in PTGDS transgenic mice (Fig. S7). The finding is consistent with VEGFA effects in Apc mice 31. However, we did not see a decrease in microvessel density in large tumors in ApcMin/+ mice with transgenic HPGDS (Fig.1N–O). Thus, tumor suppression by PGD2 did not appear to involve antiangiogenesis in our experiments 32, at a level detectable by anti-CD31 immunohistochemistry.
Transgenic HPGDS did not reduce PCNA immunostaining in early tumors in ApcMin/+ mice (Fig.1L–M). PCNA is a marker of intestinal cell proliferation and belongs to the family of sliding DNA clamps that bind factors at replication forks 33. Similarly, transgenic HPGDS did not lower numbers of mitotic figures in early tumors. Thus, PGD2 does not appear to suppress tumors by lowering rates of tumor cell division.
A possible explanation is increased tumor cell death with PGD2 and PTGDR, as shown by Lewis lung cancer cells implanted onto the backs of mice 6. Alternatively, PGD2 may prevent tumors by slowing initiation.
PPARG and intestinal tumors
Heterozygous Pparg knockouts alone did not increase the numbers of tumors in our ApcMin/+ mice. The result is consistent with earlier reports 24,34. However, McAlpine et al. 24 found ∼30% more tumors in male ApcMin/+ mice with heterozygous or homozygous intestine-specific Pparg deletions.
In our transgenic mice with PTGDS overproduction and reduced adenoma occurrence, the decrease in numbers of large tumors caused by PTGDS appeared blunted in heterozygous Pparg knockout mice (Fig.3C and Table S3C). Such blunting could be compatible with tumor suppression by PGD2 metabolites bound to PPARG 35, when PGD2 production is increased. A limitation in our experiments with heterozygous Pparg knockouts and PTGDS transgenes was the use of mice with mixed C57BL/6-FVB/N backgrounds (all 50% C57BL/6, but not all F1). However, fairly large numbers of mice were used in the Pparg experiments (104 in total). The 147 mice in the Ptgdr knockout experiments were all 100% C57BL/6.
PGD2 and inflammation
Mechanisms for tumor suppression by PGD2 in the intestines have not been proven, but useful information is available. For example, in the skin 36 and lung 37, PGD2 delays migration of dendritic cells to draining lymph nodes, where T cells are primed. PGD2 also reduces the ability of dendritic cells to stimulate naïve T cells 38,39. In the intestinal mucosa, dendritic cells produce IL-23, to stimulate release of IL-22 by immune cells (innate lymphoid cells 40,41, TH17 cells 42, and TH22 cells 43). In turn, IL-22 induces proliferation of epithelial cells, production of inflammatory mediators, and release of antimicrobial proteins, to guard against invaders 44. This cytokine can be neutralized by IL-22-binding protein, a soluble receptor also made by dendritic cells in the colon. Huber et al. 45 showed that IL-22 gene knockouts allowed fewer tumors in ApcMin/+ mice, whereas knockouts of IL-22-binding protein caused more (in the colon). Further work is needed to determine if these functions of dendritic cells explain tumor suppression by PGD2. Identification of mechanisms involving PGD2 and PTGDR may suggest molecular targets for tumor prevention studies.
Conclusions
By scoring tumors in ApcMin/+ mice histologically at 6 and 14 weeks, we showed that homozygous knockouts of the gene for the PGD2 receptor, PTGDR, raised median numbers of tumors by 30–40%. The results support an interpretation that PGD2 is a tumor-suppressing molecule, acting through PTGDR. Heterozygous Pparg knockouts had smaller effects in our experiments. The observation that PGD2 and PTGDR can affect tumorigenesis may have impact for prevention.
Acknowledgments
Peter D. Christenson provided statistical consultation, supported by the National Center for Advancing Translational Sciences through UCLA CTSI Grant UL1TR000124. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Funding was from the: National Institutes of Health (CA73403, CA91179, CA132184 to H. J. L.; AA08116 to S. W. F.); Spanish Ministry of Science (SAF2011-23933 to E. S.); and Los Angeles Biomedical Research Institute (to H. J. L.).
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Examples of early stage intravillar neoplasms (A and D) and later stage tumors (B, C, E–G) in the small bowel of ApcMin/+ mice. Arrows indicate single, intravillar, neoplastic glands. (A) An early stage intravillar neoplasm with a single neoplastic gland lying at the level of normal glands and displacing them. (B) Profile of an intravillar neoplasm that progressed by erupting upward through the villus surface and opening into the lumen. Serial sections of this tumor are shown in Figure S3. (C) Profile of an intravillar neoplasm that progressed by forming multiple attached cysts. Serial sections of this lesion are shown in Figure S2. (D) Higher magnification view of an early intravillar neoplasm (not the same tumor as shown in A). (E) A later stage intravillar neoplasm consisting of a single neoplastic gland between the stalk of the villus and underlying normal glands, with extension of neoplastic cells (outlined by asterisks) beyond the gland. (F) A later stage intravillar neoplasm in which the glandular cells display crowding and pseudo-stratification. (G) A more complex, later stage intravillar neoplasm, characterized by polarized neoplastic glandular columnar cells (arrow), multiple neoplastic glandular lumina in a cribriform pattern (two examples are indicated by arrowheads), and solid areas of nonpolarized cells (asterisks). Hematoxylin and eosin staining. Scale bars, 100 μm.
Figure S2. Sections of an intravillar tumor that contains multiple adjoining cystic structures. The sections indicate that intravillar neoplasms can progress by forming new cysts that abut existing cysts. The slides of this tumor (beginning with the section represented by the top left image and ending with the section represented by the bottom right image) spanned 48 sections (4 μm each). However, only 20 sections are shown here, to save space. Scale bar, 100 μm.
Figure S3. Sections of an intravillar tumor that erupted through the villus surface. The sections indicate that intravillar neoplasms can progress by expanding toward the top of the villus, erupting through the villus surface, and opening into the bowel lumen. The slides of this tumor (beginning with the section represented by the top left image and ending with the section represented by the bottom right image) spanned 31 sections (4 μm each). However, only 18 sections are shown here, to save space. Scale bar, 100 μm.
Figure S4. Three examples of intravillar tumors that show a connection to a normal crypt. Scale bar, 100 μm.
Figure S5. Sections of an intravillar neoplasm in the small bowel of an ApcMin/+ mouse, showing a uniglandular, intravillar lesion with a simple cystic structure. Although tumors arise from crypt cells, we did not observe a connection between the cystic structure and the crypt for this tumor. Thus, early tumors may become fully enclosed or “sealed off.” All mounted sections containing profiles for this tumor are shown here. Scale bar, 100 μm.
Figure S6. Examples of colon tumors seen at 6 weeks. Tumors at this age are typically small and lie below the mucosal surface. They would be overlooked without histological examination. The inset in A shows a higher magnification view of the tumor. Scale bar, 100 μm (applies to all panels, except the inset).
Figure S7. Lower expression of VEGFA and MYC in the colon of ApcMin/+ mice with PTGDS transgenes (TG) and without (WT). mRNA was prepared from colon tissue, and expression levels for VEGFA and MYC were quantitated relative to endogenous mouse GAPDH. Plotted points are averages of triplicate measurements in different mice. VEGFA expression in PTGDS transgenic mice was approximately 50% of expression in controls (P = 0.022, Mann–Whitney; P = 0.012, t-test). MYC expression was also 50% lower in PTGDS transgenic mice (P = 0.041, Mann–Whitney; P = 0.050, t-test). Filled symbols: females. Open symbols: males. Horizontal bars show medians. *P < 0.05.
Figure S8. Numbers of adenomas in ApcMin/+ mice with HPGDS transgenes (TG) and without (WT). Transgenic HPGDS was associated with statistically significant reductions in the numbers of tumors in all size categories. See Table S4 for median values, ranges, numbers of mice, P-values, and ratios of numbers of tumors in HPGDS transgenic mice to numbers in controls. Filled symbols: females. Open symbols: males. Horizontal bars indicate medians. *P < 0.05.
Figure S9. Tumor development in female and male ApcMin/+ mice at 14 weeks. We combined data from the current experiments with data from two earlier reports 5,23. For each mouse, numbers of tumors (in the entire intestine and in the colon) were normalized to the median number among females in the same experiment. For colon tumors, we added 0.5 to the number of tumors before taking the median. Horizontal bars indicate medians. The dotted horizontal lines indicate 1.0 (which is the median value for females). Males (77 mice) and females (61 mice) tended to have similar numbers of total tumors throughout the intestine (median ratio for males to females = 0.82; P = 0.069; A), but males had roughly 60% more colon tumors, compared to females (median ratio for males to females = 1.6; P = 0.0002; B).
Table S1. Adenomas at 6 weeks in ApcMin/+ mice with Ptgdr knockouts.
Table S2. Adenomas at 14 weeks in ApcMin/+ mice with Ptgdr knockouts.
Table S3. Adenomas in ApcMin/+ mice with PTGDS transgenes, with and without heterozygous Pparg knockouts.
Table S4. Adenomas in ApcMin/+ mice with HPGDS transgenes.
References
- Nakanishi M, Montrose DC, Clark P, Nambiar PR, Belinsky GS, Claffey KP, et al. Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008;68:3251–3259. doi: 10.1158/0008-5472.CAN-07-6100. [DOI] [PubMed] [Google Scholar]
- Wang D. Dubois RN. Eicosanoids and cancer. Nat. Rev. Cancer. 2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Park YY, Kim SW, Lee JS, Wang D. DuBois RN. ANGPTL4 induction by prostaglandin E2 under hypoxic conditions promotes colorectal cancer progression. Cancer Res. 2011;71:7010–7020. doi: 10.1158/0008-5472.CAN-11-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia D, Wang D, Kim SH, Katoh H. DuBois RN. Prostaglandin E2 promotes intestinal tumor growth via DNA methylation. Nat. Med. 2012;18:224–226. doi: 10.1038/nm.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Kanaoka Y, Eguchi N, Aritake K, Grujic S, Materi AM, et al. Hematopoietic prostaglandin D synthase suppresses intestinal adenomas in ApcMin/+ mice. Cancer Res. 2007;67:881–889. doi: 10.1158/0008-5472.CAN-05-3767. [DOI] [PubMed] [Google Scholar]
- Murata T, Lin MI, Aritake K, Matsumoto S, Narumiya S, Ozaki H, et al. Role of prostaglandin D2 receptor DP as a suppressor of tumor hyperpermeability and angiogenesis in vivo. Proc. Natl. Acad. Sci. USA. 2008;105:20009–20014. doi: 10.1073/pnas.0805171105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima T, Koga H. Shimotohno K. Transcriptional activity of peroxisome proliferator-activated receptor γ is modulated by SUMO-1 modification. J. Biol. Chem. 2004;279:29551–29557. doi: 10.1074/jbc.M403866200. [DOI] [PubMed] [Google Scholar]
- Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, et al. 15-Deoxy-Δ12,14-prostaglandin J2 inhibits multiple steps in the NF-κB signaling pathway. Proc. Natl. Acad. Sci. USA. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. USA. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, et al. Prostaglandin D2 as a mediator of allergic asthma. Science. 2000;287:2013–2017. doi: 10.1126/science.287.5460.2013. [DOI] [PubMed] [Google Scholar]
- Pinzar E, Kanaoka Y, Inui T, Eguchi N, Urade Y, Hayaishi O, et al. Prostaglandin D synthase gene is involved in the regulation of non-rapid eye movement sleep. Proc. Natl. Acad. Sci. USA. 2000;97:4903–4907. doi: 10.1073/pnas.090093997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujitani Y, Kanaoka Y, Aritake K, Uodome N, Okazaki-Hatake K. Urade Y. Pronounced eosinophilic lung inflammation and Th2 cytokine release in human lipocalin-type prostaglandin D synthase transgenic mice. J. Immunol. 2002;168:443–449. doi: 10.4049/jimmunol.168.1.443. [DOI] [PubMed] [Google Scholar]
- Nicol CJ, Yoon M, Ward JM, Yamashita M, Fukamachi K, Peters JM, et al. PPARγ influences susceptibility to DMBA-induced mammary, ovarian and skin carcinogenesis. Carcinogenesis. 2004;25:1747–1755. doi: 10.1093/carcin/bgh160. [DOI] [PubMed] [Google Scholar]
- van Diest PJ, Baak JPA, Matze-Cok P, Wisse-Brekelmans ECM, van Galen CM, Kurver PHJ, et al. Reproducibility of mitosis counting in 2,469 breast cancer specimens: results from the Multicenter Morphometric Mammary Carcinoma Project. Hum. Pathol. 1992;23:603–607. doi: 10.1016/0046-8177(92)90313-r. [DOI] [PubMed] [Google Scholar]
- Oda H, Shiina Y, Seiki K, Sato N, Eguchi N. Urade Y. Development and evaluation of a practical ELISA for human urinary lipocalin-type prostaglandin D synthase. Clin. Chem. 2002;48:1445–1453. [PubMed] [Google Scholar]
- Moorman AF, Houweling AC, de Boer PA. Christoffels VM. Sensitive nonradioactive detection of mRNA in tissue sections: novel application of the whole-mount in situ hybridization protocol. J. Histochem. Cytochem. 2001;49:1–8. doi: 10.1177/002215540104900101. [DOI] [PubMed] [Google Scholar]
- Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C. Taketo M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc. Natl. Acad. Sci. USA. 1995;92:4482–4486. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR. Wnt signalling in the mouse intestine. Oncogene. 2006;25:7512–7521. doi: 10.1038/sj.onc.1210065. [DOI] [PubMed] [Google Scholar]
- Oshima H, Oshima M, Kobayashi M, Tsutsumi M. Taketo MM. Morphological and molecular processes of polyp formation in ApcΔ716 knockout mice. Cancer Res. 1997;57:1644–1649. [PubMed] [Google Scholar]
- Shoemaker AR, Haigis KM, Baker SM, Dudley S, Liskay RM. Dove WF. Mlh1 deficiency enhances several phenotypes of ApcMin/+ mice. Oncogene. 2000;19:2774–2779. doi: 10.1038/sj.onc.1203574. [DOI] [PubMed] [Google Scholar]
- Shoemaker AR, Moser AR. Dove WF. N-ethyl-N-nitrosourea treatment of multiple intestinal neoplasia (Min) mice: age-related effects on the formation of intestinal adenomas, cystic crypts, and epidermoid cysts. Cancer Res. 1995;55:4479–4485. [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- Kwong AM, Tippin BL, Materi AM, Buslon VS, French SW. Lin HJ. High dietary niacin may increase prostaglandin formation but does not increase tumor formation in ApcMin/+ mice. Nutr. Cancer. 2011;63:950–959. doi: 10.1080/01635581.2011.590266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlpine CA, Barak Y, Matise I. Cormier RT. Intestinal-specific PPARγ deficiency enhances tumorigenesis in ApcMin/+ mice. Int. J. Cancer. 2006;119:2339–2346. doi: 10.1002/ijc.22115. [DOI] [PubMed] [Google Scholar]
- Mutoh M, Watanabe K, Kitamura T, Shoji Y, Takahashi M, Kawamori T, et al. Involvement of prostaglandin E receptor subtype EP4 in colon carcinogenesis. Cancer Res. 2002;62:28–32. [PubMed] [Google Scholar]
- Gustafsson A, Hansson E, Kressner U, Nordgren S, Andersson M, Lönnroth C, et al. Prostanoid receptor expression in colorectal cancer related to tumor stage, differentiation and progression. Acta Oncol. 2007;46:1107–1112. doi: 10.1080/02841860701403061. [DOI] [PubMed] [Google Scholar]
- Galamb O, Sipos F, Spisák S, Galamb B, Krenács T, Valcz G, et al. Potential biomarkers of colorectal adenoma-dysplasia-carcinoma progression: mRNA expression profiling and in situ protein detection on TMAs reveal 15 sequentially upregulated and 2 downregulated genes. Cell Oncol. 2009;31:19–29. doi: 10.3233/CLO-2009-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urade Y. Hayaishi O. Biochemical, structural, genetic, physiological, and pathophysiological features of lipocalin-type prostaglandin D synthase. Biochim. Biophys. Acta. 2000;1482:259–271. doi: 10.1016/s0167-4838(00)00161-8. [DOI] [PubMed] [Google Scholar]
- Myant K. Sansom OJ. Wnt/Myc interactions in intestinal cancer: partners in crime. Exp. Cell Res. 2011;317:2725–2731. doi: 10.1016/j.yexcr.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446:676–679. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- Goodlad RA, Ryan AJ, Wedge SR, Pyrah IT, Alferez D, Poulsom R, et al. Inhibiting vascular endothelial growth factor receptor-2 signaling reduces tumor burden in the ApcMin/+ mouse model of early intestinal cancer. Carcinogenesis. 2006;27:2133–2139. doi: 10.1093/carcin/bgl113. [DOI] [PubMed] [Google Scholar]
- Murata T, Aritake K, Matsumoto S, Kamauchi S, Nakagawa T, Hori M, et al. Prostaglandin D2 is a mast cell-derived antiangiogenic factor in lung carcinoma. Proc. Natl. Acad. Sci. USA. 2011;108:19802–19807. doi: 10.1073/pnas.1110011108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan GL, Pfander B. Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Girnun GD, Smith WM, Drori S, Sarraf P, Mueller E, Eng C, et al. APC-dependent suppression of colon carcinogenesis by PPARγ. Proc. Natl. Acad. Sci. USA. 2002;99:13771–13776. doi: 10.1073/pnas.162480299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus DS. Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Angeli V, Faveeuw C, Roye O, Fontaine J, Teissier E, Capron A, et al. Role of the parasite-derived prostaglandin D2 in the inhibition of epidermal Langerhans cell migration during schistosomiasis infection. J. Exp. Med. 2001;193:1135–1147. doi: 10.1084/jem.193.10.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhao J, Legge K. Perlman S. Age-related increases in PGD2 expression impair respiratory DC migration, resulting in diminished T cell responses upon respiratory virus infection in mice. J. Clin. Invest. 2011;121:4921–4930. doi: 10.1172/JCI59777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosset P, Pichavant M, Faveeuw C, Bureau F, Tonnel AB. Trottein F. Prostaglandin D2 affects the differentiation and functions of human dendritic cells: impact on the T cell response. Eur. J. Immunol. 2005;35:1491–1500. doi: 10.1002/eji.200425319. [DOI] [PubMed] [Google Scholar]
- Hammad H, Kool M, Soullié T, Narumiya S, Trottein F, Hoogsteden HC, et al. Activation of the D prostanoid 1 receptor suppresses asthma by modulation of lung dendritic cell function and induction of regulatory T cells. J. Exp. Med. 2007;204:357–367. doi: 10.1084/jem.20061196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Briseño CG, Lee JS, Ng D, Nicholas A, Manieri NA, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat. Immunol. 2013;14:937–948. doi: 10.1038/ni.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med. 2013;210:917–931. doi: 10.1084/jem.20122308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg GF, Fouser LA. Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 2011;12:383–390. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- Xie MH, Aggarwal S, Ho WH, Foster J, Zhang Z, Stinson J, et al. Interleukin (IL)-22, a novel human cytokine that signals through the interferon receptor-related proteins CRF2-4 and IL-22R. J. Biol. Chem. 2000;275:31335–31339. doi: 10.1074/jbc.M005304200. [DOI] [PubMed] [Google Scholar]
- Rutz S, Eidenschenk C. Ouyang W. IL-22, not simply a Th17 cytokine. Immunol. Rev. 2013;252:116–132. doi: 10.1111/imr.12027. [DOI] [PubMed] [Google Scholar]
- Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;49:259–263. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Examples of early stage intravillar neoplasms (A and D) and later stage tumors (B, C, E–G) in the small bowel of ApcMin/+ mice. Arrows indicate single, intravillar, neoplastic glands. (A) An early stage intravillar neoplasm with a single neoplastic gland lying at the level of normal glands and displacing them. (B) Profile of an intravillar neoplasm that progressed by erupting upward through the villus surface and opening into the lumen. Serial sections of this tumor are shown in Figure S3. (C) Profile of an intravillar neoplasm that progressed by forming multiple attached cysts. Serial sections of this lesion are shown in Figure S2. (D) Higher magnification view of an early intravillar neoplasm (not the same tumor as shown in A). (E) A later stage intravillar neoplasm consisting of a single neoplastic gland between the stalk of the villus and underlying normal glands, with extension of neoplastic cells (outlined by asterisks) beyond the gland. (F) A later stage intravillar neoplasm in which the glandular cells display crowding and pseudo-stratification. (G) A more complex, later stage intravillar neoplasm, characterized by polarized neoplastic glandular columnar cells (arrow), multiple neoplastic glandular lumina in a cribriform pattern (two examples are indicated by arrowheads), and solid areas of nonpolarized cells (asterisks). Hematoxylin and eosin staining. Scale bars, 100 μm.
Figure S2. Sections of an intravillar tumor that contains multiple adjoining cystic structures. The sections indicate that intravillar neoplasms can progress by forming new cysts that abut existing cysts. The slides of this tumor (beginning with the section represented by the top left image and ending with the section represented by the bottom right image) spanned 48 sections (4 μm each). However, only 20 sections are shown here, to save space. Scale bar, 100 μm.
Figure S3. Sections of an intravillar tumor that erupted through the villus surface. The sections indicate that intravillar neoplasms can progress by expanding toward the top of the villus, erupting through the villus surface, and opening into the bowel lumen. The slides of this tumor (beginning with the section represented by the top left image and ending with the section represented by the bottom right image) spanned 31 sections (4 μm each). However, only 18 sections are shown here, to save space. Scale bar, 100 μm.
Figure S4. Three examples of intravillar tumors that show a connection to a normal crypt. Scale bar, 100 μm.
Figure S5. Sections of an intravillar neoplasm in the small bowel of an ApcMin/+ mouse, showing a uniglandular, intravillar lesion with a simple cystic structure. Although tumors arise from crypt cells, we did not observe a connection between the cystic structure and the crypt for this tumor. Thus, early tumors may become fully enclosed or “sealed off.” All mounted sections containing profiles for this tumor are shown here. Scale bar, 100 μm.
Figure S6. Examples of colon tumors seen at 6 weeks. Tumors at this age are typically small and lie below the mucosal surface. They would be overlooked without histological examination. The inset in A shows a higher magnification view of the tumor. Scale bar, 100 μm (applies to all panels, except the inset).
Figure S7. Lower expression of VEGFA and MYC in the colon of ApcMin/+ mice with PTGDS transgenes (TG) and without (WT). mRNA was prepared from colon tissue, and expression levels for VEGFA and MYC were quantitated relative to endogenous mouse GAPDH. Plotted points are averages of triplicate measurements in different mice. VEGFA expression in PTGDS transgenic mice was approximately 50% of expression in controls (P = 0.022, Mann–Whitney; P = 0.012, t-test). MYC expression was also 50% lower in PTGDS transgenic mice (P = 0.041, Mann–Whitney; P = 0.050, t-test). Filled symbols: females. Open symbols: males. Horizontal bars show medians. *P < 0.05.
Figure S8. Numbers of adenomas in ApcMin/+ mice with HPGDS transgenes (TG) and without (WT). Transgenic HPGDS was associated with statistically significant reductions in the numbers of tumors in all size categories. See Table S4 for median values, ranges, numbers of mice, P-values, and ratios of numbers of tumors in HPGDS transgenic mice to numbers in controls. Filled symbols: females. Open symbols: males. Horizontal bars indicate medians. *P < 0.05.
Figure S9. Tumor development in female and male ApcMin/+ mice at 14 weeks. We combined data from the current experiments with data from two earlier reports 5,23. For each mouse, numbers of tumors (in the entire intestine and in the colon) were normalized to the median number among females in the same experiment. For colon tumors, we added 0.5 to the number of tumors before taking the median. Horizontal bars indicate medians. The dotted horizontal lines indicate 1.0 (which is the median value for females). Males (77 mice) and females (61 mice) tended to have similar numbers of total tumors throughout the intestine (median ratio for males to females = 0.82; P = 0.069; A), but males had roughly 60% more colon tumors, compared to females (median ratio for males to females = 1.6; P = 0.0002; B).
Table S1. Adenomas at 6 weeks in ApcMin/+ mice with Ptgdr knockouts.
Table S2. Adenomas at 14 weeks in ApcMin/+ mice with Ptgdr knockouts.
Table S3. Adenomas in ApcMin/+ mice with PTGDS transgenes, with and without heterozygous Pparg knockouts.
Table S4. Adenomas in ApcMin/+ mice with HPGDS transgenes.