

Abstract
Fibroblast growth factor receptor 3 (FGFR3) is the only gene known to cause achondroplasia (ACH), hypochondroplasia (HCH), and thanatophoric dysplasia types I and II (TD I and TD II). A second, as yet unidentified, gene also causes HCH. In this study, we used sequencing analysis to determine the frequency of FGFR3 mutations for each phenotype in 324 cases from the International Skeletal Dysplasia Registry (ISDR). Our data suggest that there is a considerable overlap of genotype and phenotype between ACH and HCH. Thus, it is important to test for mutations found in either disorder when ACH or HCH is suspected. Only two of 29 cases with HCH did not have an identified mutation in FGFR3, much less than previously reported. We recommend testing other mutations in FGFR3, instead of just the common HCH mutation, p.Asn540Lys. The mutation frequency for TD I and TD II in the largest series of cases to date are also reported. This study provides valuable information on FGFR3 mutation frequency of four skeletal dysplasias for clinical diagnostic laboratories and clinicians.
Keywords: Achondroplasia, FGFR3, hypochondroplasia, mutation frequency, thanatophoric dysplasia
Introduction
Achondroplasia (ACH; MIM:100800) occurs with an estimated prevalence between 1/16,000 and 1/26,000 live births, representing the most common genetic form of human dwarfism (Orioli et al. 1995). Affected patients have short limbs with macrocephaly and characteristic facial features such as frontal bossing and midface hypoplasia. A milder form of ACH is hypochondroplasia (HCH, MIM: 146000). Thanatophoric dysplasia (TD) (MIM: 187600), with an incidence between 1/33,000 and 1/47,000 live births (Waller et al. 2008), is the most common form of neonatal lethal dwarfism. Individuals usually die after birth from respiratory distress secondary to pulmonary hypoplasia. Two phenotypes have been distinguished: the more common form with curved femora (TD I) or the less frequent form with straight femora and a cloverleaf skull (TD II) (Wilcox et al. 1998). Fibroblast growth factor receptor 3 (FGFR3, OMIM# 134934) is the only known gene associated with all four of these skeletal dysplasias as well as severe ACH with developmental delay and acanthosis nigricans (SADDAN), Crouzon syndrome with acanthosis nigricans (MIM: 612247), and Muenke craniosynostosis syndrome (MIM: 602849) (Bellus et al. 1999; Mulliken et al. 1999; Zankl et al. 2008).
FGFR3 is a transmembrane tyrosine kinase receptor that binds fibroblast growth factors. The FGFR3 gene, located on chromosome 4p16.3 (Thompson et al. 1991), consists of 18 coding exons (Keegan et al. 1991, 1993). It was first linked to ACH by Shiang et al. (1994) and Rousseau et al. (1994). Mutations in FGFR3 causing skeletal dysplasia are all inherited in an autosomal dominant pattern, but frequently occur de novo on the paternal allele (Wilkin et al. 1998). The mutations are gain of function causing increased activation of FGFR3 with the severity of the phenotype proportionate to the overactivation of the receptor. FGFR3 overactivation slows endochondral ossification in part through ERK/MAPK (mitogen-activated protein kinases, originally called ERK, extracellular signal-regulated kinases; Foldynova-Trantirkova et al. 2012). Loss of FGFR3 function causes autosomal recessive camptodactyly, tall stature, and hearing loss (CATSHL syndrome; MIM: 610474).
Two mutations in the FGFR3 gene at nucleotide c.1138 (most commonly a G-to-A transition with a less frequent G-to-C transversion) causing a p.Gly380Arg substitution are said to account for the majority of ACH (Shiang et al. 1994; Bellus et al. 1995a,b). Mutations causing HCH are more widespread in FGFR3 with a hotspot in the tyrosine kinase domain at codon 540 in exon 13 (Bellus et al. 1995a,b). Other rare HCH mutations have been reported (Bellus et al. 2000; Heuertz et al. 2006). However, a significant number of HCH cases have no mutations in FGFR3, indicating that other genes may be involved in this phenotype (Flynn and Pauli 2003). Several amino acid substitutions in the extracellular and intracellular domains of the FGFR3 protein have been found in TD I, including p.Arg248Cys, p.Tyr373Cys, and p.Lys650Met (Tavormina et al. 1995; Rousseau et al. 1996a,b,c). In addition, several mutations in the stop codon mutations have also been described (Rousseau et al. 1996a,b,c). Platyspondylic lethal skeletal dysplasia, San Diego type (PLSD-SD) (MIM# 187600), shares many phenotypic features with TD I and is due to the same FGFR3 mutations as TD I (Brodie et al. 1999). For TD II, only the p.Lys650Glu mutation in FGFR3 has been found (Wilcox et al. 1998). In this study, conducted since 1994, we investigated the frequency of mutations in clinically and radiographically diagnosed cases of ACH, HCH, TD I and II from the International Skeletal Dysplasia Registry (ISDR).
Methods
For the last 40 years, samples have been received through the ISDR (http://cedars-sinai.edu/Patients/Programs-and-Services/Skeletal-Dysplasia/). These cases were referred to us by clinicians around the globe and seen in our own clinics. The diagnoses were based on established clinical, radiographic, and for many TD cases, chondro-osseous features. For the purpose of this study, we have combined TD I and PLSD-SD, since they are due to similar mutations in FGFR3 (Brodie et al. 1999). These cases include 93 cases of TDI + PLSD-SD (Tavormina et al. 1995; Brodie et al. 1998, 1999; Kitoh et al. 1998; Wilcox et al. 1998) and 17 cases of TDII (Tavormina et al. 1995; Wilcox et al. 1998) that were previously reported. Almost all cases were referred to the ISDR without prior molecular testing. We excluded cases that had undergone commercial testing and identified a mutation. The study is approved by Cedars-Sinai Medical Center human subjects Institutional Review Board.
Genomic DNA was isolated from samples (whole blood, frozen tissue, or cultured cells) using a commercial kit (Qiagen). All the DNA samples were first tested for the mutations commonly seen for each disorder in FGFR3 (ACH and HCH: p.Gly380Arg and p.Asn540Lys; TD I: p.Arg248Cys, p.Tyr373Cys, p.Ser249Cys, and p.X807 mutations; TD II: p.Lys650Glu) using Sanger sequencing. Sequencing of all the coding exons was performed if no common mutations were found. For primer's sequences see Table S1 (Reference sequence for the FGFR3 gene: NM_000142.4). Polymerase chain reaction (PCR) conditions were as follows: denaturing at 94°C; annealing at 64°C for 5 cycles, 62°C for 5 cycles, and 60°C for 25 cycles; elongating at 72°C; amplification for 30 cycles. PCR products from the more recent cases were directly sequenced using the Big Dye Sequencing kit (Applied Biosystems, Foster City, CA) on an ABIPRISM 3130 Genetic Analyzer and analyzed with the Sequencing 5.2 software package (Applied Biosystems). Older cases were analyzed by prior ABI machines or gel electrophoresis. Radiographs and clinical information from patients carrying FGFR3 mutations uncommon to the phenotype were reexamined after mutation detection to verify the phenotypic assignment was correct.
Results
The frequency of FGFR3 mutations in four skeletal dysplasias is shown in Table1. About 90% of ACH cases had a FGFR3 p.Gly380Arg mutation with the majority due to a c.G1138A substitution. The remainder of the cases had a p.Asn540Lys mutation due to one of two c.1620 substitutions. For HCH, 75.9% of cases had a p.Asn540Lys mutation. p.Gly380Arg and p.Lys650Gln mutations each account for 6.9% of the HCH cases. We found one case with a rare p.Tyr278Cys mutation. Two HCH cases did not have an identified FGFR3 mutation.
Table 1.
Mutation frequency for different skeletal dysplasia phenotypes
| Phenotype | Total No. of cases | Protein amino acid change | cDNA Nucleotide change | No. of cases | Percentage (%) |
|---|---|---|---|---|---|
| ACH | 79 | p.Gly380Arg | c.1138G>A | 58 | 89.9 |
| c.1138G>C | 13 | ||||
| p.Asn540Lys | c.1620C>A | 5 | 10.1 | ||
| c.1620C>G | 3 | ||||
| HCH | 29 | p.Tyr278Cys | c.829A>G | 1 | 3.4 |
| p.Gly380Arg | c.1138G>A | 2 | 6.9 | ||
| p.Asn540Lys | c.1620C>A | 13 | 75.9 | ||
| c.1620C>G | 9 | ||||
| p.Lys650Gln | c.1949A>C | 2 | 6.9 | ||
| No mutation | N/A | 2 | 6.9 | ||
| TD I* (includes PLSD-SD) | 173 | p.Arg248Cys | c.742C>T | 115 | 66.5 |
| p.Ser249Cys | c.746C>G | 11 | 6.4 | ||
| p.Gly370Cys | c.1108G>T | 4 | 2.3 | ||
| p.Tyr373Cys | c.1118A>G | 41 | 23.7 | ||
| p.Lys650Met | c.1949A>T | 2 | 1.2 | ||
| p.X807Arg | c.2419T>A | 2 | 6.9 | ||
| p.X807Arg | c.2419T>C | 2 | |||
| p.X807Gly | c.2419T>G | 3 | |||
| p.X807Ser | c.2420G>C | 1 | |||
| p.X807Leu | c.2420G>T | 1 | |||
| p.X807Trp | c.2421A>G | 3 | |||
| TD II* | 31 | p.Lys650Glu | c.1948A>G | 31 | 100.0 |
Reference sequence for FGFR3: NM_000142.4. Phenotype symbols: ACH, achondroplasia; HCH, hypochondroplasia; TDI, thanatophoric dysplasia I; TDII, thanatophoric dysplasia II.
The p.Arg248Cys mutation caused the majority (66.5%) of the TD I causes, followed by p.Tyr373Cys. The third most common mutation for TD I was at the stop codon p.X807, which was closely followed by the p.Ser249Cys mutation. p.Gly370Cys and p.Lys650Met accounted for a minority of cases, 2.3% and 1.2%, respectively. The p.Lys650Glu mutation was exclusively found in all cases of TD II.
Figure1 shows the radiographs for one of the patients with clinical ACH due to a p.Asn540Lys mutation (R#00-347). He was first seen at our center at 40 months of age. He was brought in by his adoptive parents who knew little of his prior history. Born at 32 weeks, he was diagnosed with ACH in the first year of life. His height was 85 cm (+2 SD on the ACH growth chart) and head circumference 53 cm (95%). He had macrocephaly, midface hypoplasia, rhizomelic shortening of the limbs, and brachydactyly typical for ACH. At his last visit, he was 11.5 years old and his height was 132.8 cm (+2.5 SD on the ACH chart) and his head circumference was 55.9 cm (95%). He had developed genu valgum in addition to the physical features noted previously.
Figure 1.
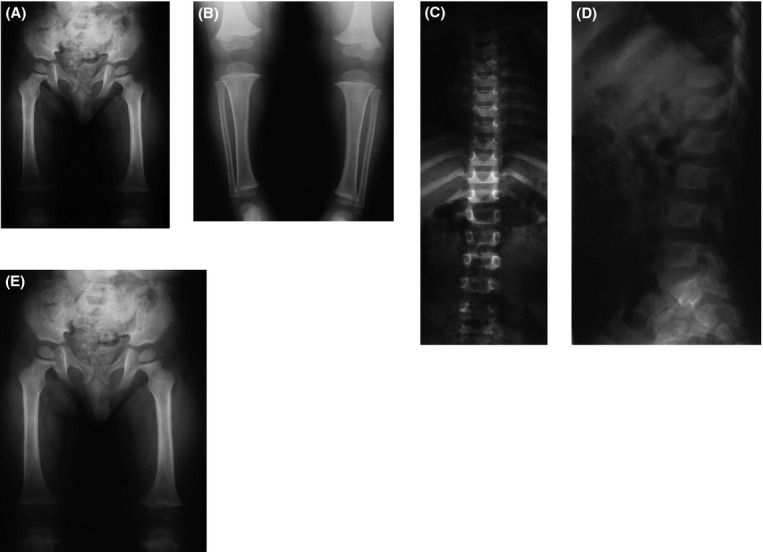
Radiographs of a case of achondroplasia with a p.Asn540Lys mutation (R#00-347). (A) Pelvis and femur, age 3 years. Narrow sacrosciatic notches and femoral shortening. (B) AP lower legs, age 3 years. There is distal overgrowth of the fibulae. (C) AP spine, age 3 years, shows platyspondyly and interpediculate narrowing in the lumbar spine. (D) Lateral spine, age 3 years. Note a narrow spinal canal and a wedge vertebrae at L2. (E) PA, left hand, age 3 years. There is significant brachydactyly and brachymetacarpia.
Radiographic findings, such as narrow spine canal, a wedge vertebrae at L2, (Fig.1D) and significant brachydactyly and brachymetacarpia, support a diagnosis of ACH.
In Figure2, milder radiographic findings such as mild interpediculate narrowing suggest a diagnosis of HCH although this patient (R#92-231A) had a p.Gly380Arg mutation. He was born at term and his growth was in the normal range until age 2. The paternal height was 188 cm and the maternal height was 163 cm. At age 6 years 9 months, he began being followed by an endocrinologist. His height was 105.8 cm (+2.5 SD on the ACH chart). He had rhizomelic shortening of the limbs and mild midface hypoplasia without macrocephaly. At age 10 years 10 months, his height was 125.7 cm (+2.5 SD on the ACH chart). At 12 years 7 months, he was 131.8 cm tall (+2 SD on the ACH chart). He was placed on growth hormone therapy at 12.5 years.
Figure 2.
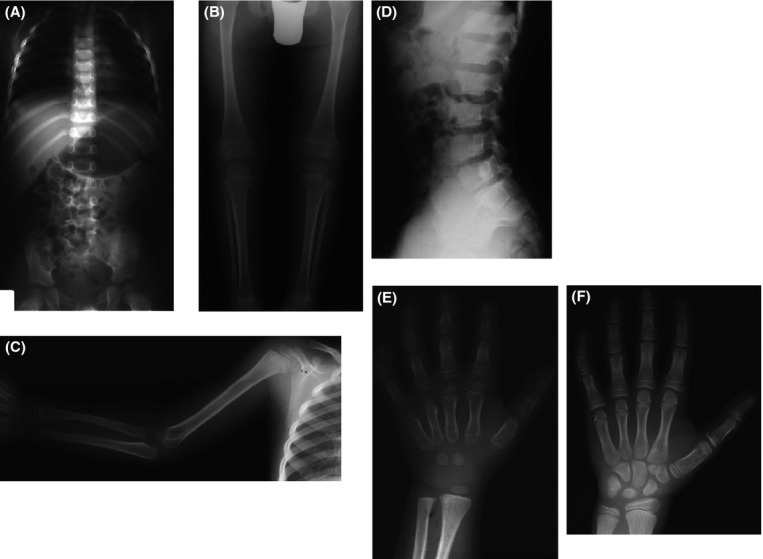
Radiographs of a case of hypochondroplasia with a p.Gly380Arg mutation (R# 92-231A). (A) AP chest and pelvis, age 5 years. The sacrosciatic notch is narrowed, but there is no interpediculate narrowing. (B) AP legs, age 5 years. There is distal fibular overgrowth. (C) AP arm, age 5 years. There is only mild rhizomelic shortening. (D) Lateral lumbar spine, age 12.5 years. There is mild platyspondyly and a narrowed spinal canal. (E) PA left hand, age 5 years. Note the brachydactyly and brachymetacarpia. (F) PA, left hand, age 12.5 years. The brachymetacarpia is less pronounced than at age 5.
Discussion
Overlapping genotype and phenotype of ACH and HCH
Both ACH and HCH share clinical and radiological features including macrocephaly, brachydactyly, metaphyseal flaring, narrowing of the interpediculate distance in the lumbar spine, square iliae, and short femoral necks. The abnormalities seen in HCH are less severe overall than those seen in ACH and HCH patients have less dysmorphic facial features (Matsui et al. 1998). Most cases can be readily distinguished clinically and radiographically. However, there is variability in severity within each group, which sometimes makes it difficult to differentiate severe HCH from mild ACH cases. Based on our large series of cases, the frequency of FGFR3 mutations in ACH and HCH is considerably different than that previously reported. It has been claimed that two common mutations in FGFR3, both resulting in p.Gly380Arg amino acid substitutions, cause over 99% of ACH cases (Shiang et al. 1994; Bellus et al. 1995a,b; Rousseau et al. 1996a,b,c). In this study, 10% of ACH is caused by p.Asn540Lys that is more typically associated with HCH. Interestingly, the p.Gly380Arg mutation was found in about 7% of the clinically diagnosed HCH cases. These data indicate that there is an overlap in the phenotypic spectrum of the p.Gly380Arg and p.Asn540Lys mutations. In our series, we did not identify some of the mutations found in other cohorts, such as p.Asn540Thr (Deutz-Terlouw et al. 1998), p.Asn540Ser (Mortier et al. 2000), p.Ile538Val (Grigelioniene et al. 1998), and p.Asn328Ile (Winterpacht et al. 2000).
In clinical practice, molecular tests are normally used for confirmatory purposes. Most laboratories offer targeted mutation analysis separately for ACH and HCH. Testing for ACH consists of the two point mutations for p.Gly380Arg, whereas testing for HCH is usually limited to p.Asn540Glu. Given the overlapping mutations between these two skeletal dysplasias, we recommend that laboratories should include both p.Gly380Arg and p.Asn540Glu mutations on the test menu when either ACH or HCH is suspected. This study provides data from the first and largest series of cases to support this testing strategy. Until laboratories adopt this practice, clinicians should consider ordering both tests. With this strategy, almost all cases of ACH will have a detected mutation while the detection rate for HCH will be ∼80%.
Incomplete screening may explain the 70% detection rate for FGFR3 mutations in HCH
It has been reported that about 30% of HCH cases do not have a mutation in FGFR3 (Prinos et al. 1995; Bellus et al. 1996; Rousseau et al. 1996a,b,c; Fofanova et al. 1998; Prinster et al. 1998; Ramaswami et al. 1998). We found that p.Asn540Lys, p.Gly380Arg, and p.Lys650Gln mutations in FGFR3 together account for about 90% of the cases. The p.Asn540Lys mutation alone accounts for about 76%, which is in agreement with other studies for HCH (Prinos et al. 1995; Bellus et al. 1996; Rousseau et al. 1996a,b,c; Fofanova et al. 1998; Prinster et al. 1998; Ramaswami et al. 1998). A less common mutation in FGFR3, p.Lys650Gln, was found in 7.4% of HCH cases. This mutation has been reported before with a slightly milder skeletal phenotype than found with the p.Asn540Lys mutation (Bellus et al. 2000). The p.Tyr278Cys mutation, found in one HCH patient, has been reported previously in two patients (Heuertz et al. 2006). These patients had an ACH phenotype at birth, at the age of 6 months, and during the first 2 years of life. By the age of 3.5 years, the phenotype had changed to typical HCH with normal craniofacial features. Our patient is an adult who had HCH clinically and radiographically. It is possible that this patient had an ACH phenotype at younger age and evolved to HCH as he grew older.
The much higher detection rate observed in this study suggests that failing to look for other mutations in FGFR3 such as p.Gly380Arg and p.Lys650Gln maybe the reason for the ∼70% detection rate quoted for HCH. Since all mutations in FGFR3 causing dwarfism are activating mutations, it is not likely that sequencing the coding region will miss a pathogenic mutation. Thus, from our data, genetic locus heterogeneity is to be found in less than 10% of HCH cases. With advanced sequencing technology such as exome sequencing, it is highly possible that the second HCH locus will be identified in the future.
What to expect when testing mutations for TD I and TD II
Although several distinct missense mutations have been described for TD I cases, the most frequent mutations are p.Arg248Cys and p.Tyr373Cys, these two mutations together contributing to about 90% of the cases. TD II patients exclusively have p.Lys650Glu mutation, which agrees with previous studies (Wilcox et al. 1998; Bellus et al. 2000). This information is based on the largest series of TD cases and can help clinical laboratories design a mutation panel for TD, especially for prenatal diagnosis.
Limitations of the study
Some cases (<10%) were referred to the ISDR with known mutations from clinical laboratories. These cases were not analyzed in this study and are excluded from this analysis. We included only cases where we did the molecular analysis. Because commercial testing typically only tests for the common mutations for each phenotype and we sequenced the entire coding region, if necessary, the percentage of rare mutations in a completely unselected population could be slightly lower than we found.
Conclusion
Based on a large number of cases, we report the mutation frequency in FGFR3 for four major skeletal dysplasias. This information can be used to significantly improve analytical sensitivity in a clinical molecular laboratory. When considering a testing strategy, either a mutation panel or reflex testing could be used for ACH and HCH. Panels can be designed to test all these mutations simultaneously. Otherwise, reflex testing can be applied after the common mutation is not detected for a specific phenotype. For example, a p.Asn540Lys mutation should be considered when a p.Gly380Arg mutation is not found in a suspected case of ACH patient and vice versa for HCH. For TD II, p.Lys650Glu is the only mutation that needs to be tested for. In TD I, p.Arg248Cys and p.Tyr373Cys would be tested first followed by less common mutations.
Acknowledgments
We thank the families and referring doctors for participating in this research project. The support from International Skeletal Dysplasia Registry is acknowledged.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Sequences of primer pairs used to amplify FGFR3 coding exons.
References
- Bellus GA, Hefferon TW, Ortiz de Luna RI, Hecht JT, Horton WA, Machado M, et al. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am. J. Hum. Genet. 1995a;56:368–373. [PMC free article] [PubMed] [Google Scholar]
- Bellus GA, McIntosh I, Smith EA, Aylsworth AS, Kaitila I, Horton WA, et al. A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nat. Genet. 1995b;10:357–359. doi: 10.1038/ng0795-357. [DOI] [PubMed] [Google Scholar]
- Bellus GA, McIntosh I, Szabo J, Aylsworth A, Kaitila I. Francomano CA. Hypochondroplasia: molecular analysis of the fibroblast growth factor receptor 3 gene. Ann. N. Y. Acad. Sci. 1996;785:182–187. doi: 10.1111/j.1749-6632.1996.tb56257.x. [DOI] [PubMed] [Google Scholar]
- Bellus GA, Bamshad MJ, Przylepa KA, Dorst J, Lee RR, Hurko O, et al. Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN): phenotypic analysis of a new skeletal dysplasia caused by a Lys650Met mutation in fibroblast growth factor receptor 3. Am. J. Med. Genet. 1999;85:53–65. [PubMed] [Google Scholar]
- Bellus GA, Spector EB, Speiser PW, Weaver CA, Garber AT, Bryke CR, et al. Distinct missense mutations of the FGFR3 lys650 codon modulate receptor kinase activation and the severity of the skeletal dysplasia phenotype. Am. J. Hum. Genet. 2000;67:1411–1421. doi: 10.1086/316892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie SG, Kitoh H, Lipson M, Sifry-Platt M. Wilcox WR. Thanatophoric dysplasia type I with syndactyly. Am. J. Med. Genet. 1998;80:260–262. doi: 10.1002/(sici)1096-8628(19981116)80:3<260::aid-ajmg15>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Brodie SG, Kitoh H, Lachman RS, Nolasco LM, Mekikian PB. Wilcox WR. Platyspondylic lethal skeletal dysplasia, San Diego type, is caused by FGFR3 mutations. Am. J. Med. Genet. 1999;84:476–480. [PubMed] [Google Scholar]
- Deutz-Terlouw PP, Losekoot M, Aalfs CM, Hennekam RC. Bakker E. Asn540Thr substitution in the fibroblast growth factor receptor 3 tyrosine kinase domain causing hypochondroplasia. Hum. Mutat. 1998;(Suppl. 1):S62–S65. doi: 10.1002/humu.1380110122. [DOI] [PubMed] [Google Scholar]
- Flynn MA. Pauli RM. Double heterozygosity in bone growth disorders: four new observations and review. Am. J. Med. Genet. 2003;121A:193–208. doi: 10.1002/ajmg.a.20143. [DOI] [PubMed] [Google Scholar]
- Fofanova OV, Takamura N, Kinoshita E, Meerson EM, Iljina VK, Nechvolodova OL, et al. A missense mutation of C1659 in the fibroblast growth factor receptor 3 gene in Russian patients with hypochondroplasia. Endocr. J. 1998;45:791–795. doi: 10.1507/endocrj.45.791. [DOI] [PubMed] [Google Scholar]
- Foldynova-Trantirkova S, Wilcox WR. Krejci P. Sixteen years and counting: the current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Hum. Mutat. 2012;33:29–41. doi: 10.1002/humu.21636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigelioniene G, Hagenas L, Eklof O, Neumeyer L, Haereid PE. Anvret M. A novel missense mutation Ile538Val in the fibroblast growth factor receptor 3 in hypochondroplasia. Hum. Mutat. 1998;11:333. doi: 10.1002/(SICI)1098-1004(1998)11:4<333::AID-HUMU18>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Heuertz S, Le Merrer M, Zabel B, Wright M, Legeai-Mallet L, Cormier-Daire V, et al. Novel FGFR3 mutations creating cysteine residues in the extracellular domain of the receptor cause achondroplasia or severe forms of hypochondroplasia. Eur. J. Hum. Genet. 2006;14:1240–1247. doi: 10.1038/sj.ejhg.5201700. [DOI] [PubMed] [Google Scholar]
- Keegan K, Johnson DE, Williams LT. Hayman MJ. Isolation of an additional member of the fibroblast growth factor receptor family, FGFR-3. Proc. Natl. Acad. Sci. USA. 1991;88:1095–1099. doi: 10.1073/pnas.88.4.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan K, Rooke L, Hayman M. Spurr NK. The fibroblast growth factor receptor 3 gene (FGFR3) is assigned to human chromosome 4. Cytogenet. Cell Genet. 1993;62:172–175. doi: 10.1159/000133465. [DOI] [PubMed] [Google Scholar]
- Kitoh H, Lachman RS, Brodie SG, Mekikian PB, Rimoin DL. Wilcox WR. Extra pelvic ossification centers in thanatophoric dysplasia and platyspondylic lethal skeletal dysplasia-San Diego type. Pediatr. Radiol. 1998;28:759–763. doi: 10.1007/s002470050461. [DOI] [PubMed] [Google Scholar]
- Matsui Y, Yasui N, Kimura T, Tsumaki N, Kawabata H. Ochi T. Genotype phenotype correlation in achondroplasia and hypochondroplasia. J. Bone Joint Surg. Br. 1998;80B:1052–1056. doi: 10.1302/0301-620x.80b6.9277. [DOI] [PubMed] [Google Scholar]
- Mortier G, Nuytinck L, Craen M, Renard JP, Leroy JG. de Paepe A. Clinical and radiographic features of a family with hypochondroplasia owing to a novel Asn540Ser mutation in the fibroblast growth factor receptor 3 gene. J. Med. Genet. 2000;37:220–224. doi: 10.1136/jmg.37.3.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulliken JB, Steinberger D, Kunze S. Muller U. Molecular diagnosis of bilateral coronal synostosis. Plast. Reconstr. Surg. 1999;104:1603–1615. doi: 10.1097/00006534-199911000-00001. [DOI] [PubMed] [Google Scholar]
- Orioli IM, Castilla EE, Scarano G. Mastroiacovo P. Effect of paternal age in achondroplasia, thanatophoric dysplasia, and osteogenesis imperfecta. Am. J. Med. Genet. 1995;59:209–221. doi: 10.1002/ajmg.1320590218. [DOI] [PubMed] [Google Scholar]
- Prinos P, Costa T, Sommer A, Kilpatrick MW. Tsipouras P. A common FGFR3 gene mutation in hypochondroplasia. Hum. Mol. Genet. 1995;4:2097–2101. doi: 10.1093/hmg/4.11.2097. [DOI] [PubMed] [Google Scholar]
- Prinster C, Carrera P, Del Maschio M, Weber G, Maghnie M, Vigone MC, et al. Comparison of clinical-radiological and molecular findings in hypochondroplasia. Am. J. Med. Genet. 1998;75:109–112. doi: 10.1002/(sici)1096-8628(19980106)75:1<109::aid-ajmg22>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Ramaswami U, Rumsby G, Hindmarsh PC. Brook CG. Genotype and phenotype in hypochondroplasia. J. Pediatr. 1998;133:99–102. doi: 10.1016/s0022-3476(98)70186-6. [DOI] [PubMed] [Google Scholar]
- Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet J-M, Maroteaux P, et al. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371:252–254. doi: 10.1038/371252a0. [DOI] [PubMed] [Google Scholar]
- Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet JM, Maroteaux P, et al. Mutations of the fibroblast growth factor receptor-3 gene in achondroplasia. Horm. Res. 1996a;45:108–110. doi: 10.1159/000184768. [DOI] [PubMed] [Google Scholar]
- Rousseau F, Bonaventure J, Legeai-Mallet L, Schmidt H, Weissenbach J, Maroteaux P, et al. Clinical and genetic heterogeneity of hypochondroplasia. J. Med. Genet. 1996b;33:749–752. doi: 10.1136/jmg.33.9.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F, El Ghouzzi V, Delezoide AL, Legeai-Mallet L, Le Merrer M, Munnich A, et al. Missense FGFR3 mutations create cysteine residues in thanatophoric dwarfism type I (TD1) Hum. Mol. Genet. 1996c;5:509–512. doi: 10.1093/hmg/5.4.509. [DOI] [PubMed] [Google Scholar]
- Shiang R, Thompson LM, Zhu YZ, Church DM, Fielder TJ, Bocian M, et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78:335–342. doi: 10.1016/0092-8674(94)90302-6. [DOI] [PubMed] [Google Scholar]
- Tavormina PL, Shiang R, Thompson LM, Zhu YZ, Wilkin DJ, Lachman RS, et al. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat. Genet. 1995;9:321–328. doi: 10.1038/ng0395-321. [DOI] [PubMed] [Google Scholar]
- Thompson LM, Plummer S, Schalling M, Altherr MR, Gusella JF, Housman DE, et al. A gene encoding a fibroblast growth factor receptor isolated from the Huntington disease gene region of human chromosome 4. Genomics. 1991;11:1133–1142. doi: 10.1016/0888-7543(91)90041-c. [DOI] [PubMed] [Google Scholar]
- Waller DK, Correa A, Vo TM, Wang Y, Hobbs C, Langlois PH, et al. The population-based prevalence of achondroplasia and thanatophoric dysplasia in selected regions of the US. Am. J. Med. Genet. A. 2008;146A:2385–2389. doi: 10.1002/ajmg.a.32485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox WR, Tavormina PL, Krakow D, Kitoh H, Lachman RS, Wasmuth JJ, et al. Molecular, radiologic, and histopathologic correlations in thanatophoric dysplasia. Am. J. Med. Genet. 1998;78:274–281. doi: 10.1002/(sici)1096-8628(19980707)78:3<274::aid-ajmg14>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Wilkin DJ, Szabo JK, Cameron R, Henderson S, Bellus GA, Mack ML, et al. Mutations in fibroblast growth factor receptor 3 in sporadic cases of achondroplasia occur exclusively on the paternally derived chromosome. Am. J. Hum. Genet. 1998;63:711–716. doi: 10.1086/302000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterpacht A, Hilbert K, Stelzer C, Schweikardt T, Decker H, Segerer H, et al. A novel mutation in FGFR-3 disrupts a putative N-glycosylation site and results in hypochondroplasia. Physiol. Genomics. 2000;2:9–12. doi: 10.1152/physiolgenomics.2000.2.1.9. [DOI] [PubMed] [Google Scholar]
- Zankl A, Elaki G, Susman RD, Inglis G, Gardener G, Buckley MF, et al. Prenatal and postnatal presentation of severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) due to the FGFR3 Lys650Met mutation. Am. J. Med. Genet. A. 2008;146:212–218. doi: 10.1002/ajmg.a.32085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequences of primer pairs used to amplify FGFR3 coding exons.