

Abstract
Inherited biallelic mutations of the ATM gene are responsible for the development of ataxia telangiectasia (AT). The objective of the present study was to conduct molecular analysis of the ATM gene in a cohort of 24 Polish patients with ataxia-telangiectasia with aim being to provide an updated mutational spectrum in Polish AT patients. As a result of molecular analysis, the status of recurrent mutation was confirmed and ten new ATM variants were detected. Application of MLPA analysis allowed the detection of large genomic deletion. Previously, this type of mutation had never been seen in our population. Finally, in silico analysis was carried out for newly detected ATM alterations. In addition, functional analysis was performed to evaluate the effects of intronic variants: c.3402+30_3402+32delATC.
Keywords: Ataxia telangiectasia, ATM , Polish population, mutation analysis, sequencing, MLPA
Introduction
Ataxia-telangiectasia (AT, MIM#208900) is a neurodegenerative disorder belonging to primary immunodeficiency diseases and it is associated with DNA repair defects. AT is inherited in an autosomal recessive manner and results from mutations in the ataxia telangiectasia mutated gene (ATM, MIM*607585). Protein encoded by ATM plays an important role in monitoring and maintaining DNA integrity. ATM coordinates cell cycle progression and the cellular response to DNA double-stranded breaks by phosphorylation of several substrates: TP53, BRCA1, CHEK2, and nibrin (Derheimer and Kastan 2010; Keimling et al. 2011). The first symptoms of ataxia-telangiectasia often appear in early childhood, when children begin to walk. The most characteristic manifestations of AT are neurological dysfunction (ataxia) and dilated blood vessels (telangiectasia) in corner of the eyes and in the skin on the ears and cheeks. Neurological manifestations presented by AT patients resulted from cerebellar atrophy (Carlessi et al. 2013). Neurodegeneration of the cerebellum is progressive and is responsible for unsteady gait, poor muscle control, abnormal eye movements and problems with speaking or swallowing (McKinnon 2004). Immunodeficiency is presented by more than half of all patients with AT. Furthermore, the humoral immune system, cellular immune system or both can be affected. Immunoglobulin levels (particularly IgA, IgE, and IgG2) are diminished or absent. Common abnormality of cell-mediated immunity is peripheral lyphmopenia, and especially CD4 T-cells are reduced. Patients with AT suffer from sinopulmonary infections, but opportunistic infections are rare (Lumsden et al. 2004; Staples et al. 2008). One of the biomarkers of AT is an elevated α-fetoprotein (AFP) level in serum. In cytogenetic studies the translocation between 7 and 14 chromosomes is identified in 5–15% cases. Patients with AT have a strong predisposition to malignancy, with an increased risk of leukemia and lymphoma of both B-cell and T-cell origins. In AT patients, the most frequent malignancies are found in the lymphoid system, and T-cell tumors occur more frequently than B-cell tumors. Patients living longer also present with other type of cancers, like ovarian and breast cancer, gastric cancer, melanoma, leiomyomas, and sarcomas (Byrd et al. 1996; Reiman et al. 2011).
Materials and Methods
Patients
26 AT patients from 24 unrelated families were recruited from the department of Immunology and Genetics departments in Poland. The majority of our patients presented typical ataxia-telangiectasia manifestation: immunoglobulin deficiencies involving: IgA, IgE, and IgG, and high levels of alfa-fetoprotein (AFP). Cerebellar ataxia causing uncoordinated movement, swallowing difficulties and dysarthria were observed in our patients. Telangiectasia were detected in only part of the AT patient group. The clinical features of the individual A-T patients were summarized in Table1.
Table 1.
Clinical manifestations, laboratory findings of AT patients
| Patients | Age | Sex | Ataxia (age) | Telangiectasia (age) | Afp | Immunoglobulins |
|---|---|---|---|---|---|---|
| AT01 | 11 | M | − | − | ↓IgG | |
| AT02 | 7 | F | +(1.8) | − | ↓IgA | |
| AT06 | 7 | M | + | + | ↑ | ↓IgA, ↓IgG, ↑IgM |
| AT07 | 5 | F | +(1.8) | − | ↑ | ↓IgA, ↓IgG |
| AT7.1 | 5 | M | +(1.8) | − | ↑ | ↓IgA, ↓IgG |
| AT08 | 6 | F | + | + | ↑ | ↓IgA, ↓IgG2 |
| AT10 | 14 | M | +(1.3) | +(9,5) | ↑ | ↓IgA, ↓IgG2 |
| AT12 | 3 | M | +(1.4) | +(3) | ↓IgG3 | |
| AT13 | 6 | F | +(1.4) | +(3) | ↑ | ↓IgA, ↓IgG2 |
| AT15 | 17 | M | +(1.5) | +(7) | ↑ | ↓IgA |
| AT19 | 2 | M | +(1.2) | − | ↑ | ↓IgG3 |
| AT21 | 9 | F | + | ↑ | ↓IgA | |
| AT23 | 4 | M | +(2.1) | + | ↑ | ↓IgA, ↓IgG |
| AT24 | 16 | M | + | + | ↑ | ↓IgA |
| AT26 | 21 | F | + | Norm | Norm | |
| AT27 | 4 | M | +(1.8) | − | ↑ | ↓IgA |
| AT28 | 9 | M | + | + | ↑ | ↓IgA |
| AT30 | 9 | F | +(2) | +(6) | ↑ | ↓IgA, ↓IgG2, ↓IgG4 |
| AT31 | 13 | M | +(1.3) | +(2) | ↑ | ↓IgA, ↓IgG2 |
| AT33 | 3 | M | +(1) | +(2) | ↑ | ↓IgA |
| AT33.1 | 3 | M | +(1) | +(2) | ↑ | ↓IgA |
| AT34 | 5 | F | +(2) | − | Norm | ↓IgA,↓IgG2, ↓IgG3, ↓IgG4 |
| AT35 | 12 | M | +(1.5) | +(8) | ↑ | ↓IgA, ↓IgG2, |
| AT36 | 4 | M | +(1) | − | ↑ | ↓IgA, ↓IgG2, ↓IgG3, ↓IgG4 |
| AT37 | 8 | F | + | + | ↑ | ↑IgG |
| AT38 | 5 | M | + | + | ↑ | ↓IgG |
F, female; M, male; +, present; –, absent; arrows indicate increase (pointing up)/decrease (pointing down) level of an AFP/immunoglobulin.
Molecular analysis
We performed genomic DNA extraction from peripheral ethylenediamine tetraacetic acid-anticoagulated blood samples using standard phenol-chloroform protocols. The genomic DNA was amplified using a previously reported primers set, flanking all exons and exon/intron boundaries of the ATM gene (Castellvi-Bel et al. 1999). Single-strand conformation polymorphism (SSCP) and heteroduplex (HD) were performed and the products were visualized with silver staining. PCR products with variant migration patterns were sequenced. Multiplex Ligation-dependent Probe Amplification (MLPA) was performed with P041 and P042 kits (MRC Holland, Amsterdam, The Netherlands) in accordance with the manufacturer's instructions. MLPA products were analyzed on an ABI sequencer. Data analysis was performed by exporting the peak areas to a Microsoft Excel file.
We investigated the effect of a c.3402+30_3402+32delATC intronic variant on splicing and expression. Total RNA was obtained from the lymphoblastoid cell line (LCLs). RNA was isolated from two patients carrying c.3402+30_3402+32delATC and from controls by using a QIAmp RNA blood mini kit (Qiagen, Hilden, Germany) and was reverse-transcribed into cDNA using Superscript III Reverse Transcriptase (Invitrogen, Carlsbad, CA). Expression of the ATM gene was measured quantitatively by real-time PCR using KAPA SYBR® FAST One-Step qRT-PCR Kits (KAPA Biosystem, Boston, MA) by gene specific primers and β-actin was used as a reference control. The immunoblotting analysis was performed using an ATM 2C1 monoclonal antibody raised against amino acids 2577-3056 and an antibody against β-actin as an internal loading control (both antibodies; Santa Cruz Biotechnology, Inc., Heidelberg, Germany).
In silico analyses of the ATM variants were performed using the Protein Variation Effect Analyzer (PROVEAN, J. Craig Venter Institute), Align Grantham Variation Grantham Deviation (Align GVGD, International Agency for Research on Cancer, Lyon, France) and Alibaba 2.1 TF Binding Prediction (BIOBASE), which are freely available web-based programs.
The reference sequence for ATM used GenBank NM_000051.3. Mutation numbering uses the A of the ATG initiation codon as +1.
The study was conducted with the approval by the Central Ethical Committee of the Ministry of Health, Poland, in accordance with the tenets of the Declaration of Helsinki.
Results and Discussion
The screening of the ATM gene in 24 AT families revealed 38 changes in the DNA sequence. The rate of DNA alterations in this series of AT patients is approximately 80% (38 detected variants/48 expected mutations). The mutation types are diverse, including 21 nonsense (55.3%), 12 splicing (31.6%), 3 large genomic deletions (7.9%) and 2 missense alterations (5.3%). All ATM changes and further details are shown in Table2. The majority of AT patients were compound heterozygotes. Only two patients out of 24 were found to be homozygous (AT24 [c.4007_4008insA; c.4007_4008insA], AT37 [c.9021_9022insA; c.9021_9022insA]). As published previously, few recurring mutations were detected in Polish AT patients (c.5932G>T, c.6095G>A, c.7630-2A>C, c.7010_7011delGT) (Telatar et al. 1998; Mitui et al. 2005; Demuth et al. 2011). The c.6095G>A and c.7630-2A>C are splicing mutations causing exon skipping, 43 and 53, respectively. The most frequent mutations among our AT patients are: c.6095G>A (5 times), c.7630-2A>C (6), c.5932G>T (6), c.7010_7011delGT (2). Recurrent mutations cover 76.3% (29/38) of all detected mutations. Several families in the Polish population had newly diagnosed DNA alterations (10/38; 21.05%). Ten changes in the ATM gene were novel: c.8441delC, c.6145T>G, c.434T>G, c.6754_6754delA, c.4007_4008insA, c.7606G>A, c.3402+30_3402+32delATC, deletion of exons 19-20, deletion of exon 63, deletion of exons 62 and 63 of ATM gene). Seven newly discovered ATM alterations resulted in the exchange of the amino acid into a stop codon or are products of a frameshift error generating the stop codon and truncating the protein product. The remaining new alterations are substitutions and one intronic variant. We identified the replacement of non-polar, hydrophobic leucine by basic amino arginine at position 145 and the substitution of aromatic tyrosine for acidic amino aspartic acid at position 2049 in the protein sequence. To predict the biological effect of two missense changes, in silico analysis was performed using the Protein Variation Effect Analyzer (PROVEAN, J. Craig Venter Institute). The algorithm of analysis predicted c.434T>G (score −3.403) and c.6145T>G (score −6.956) to be pathogenic. A multiple sequence alignment was made with the Align Grantham Variation Grantham Deviation program (Align GVGD, International Agency for Research on Cancer, Lyon, France), and is presented in Table3.
Table 2.
ATM mutations of 24 families with AT
| Patients | DNA level | Protein level | Consequence | Status | Genotype |
|---|---|---|---|---|---|
| AT01 | c.8441delG | p.Glu2814LysfsTer43 | Truncation | Novel | Compound heterozygote |
| c.6095G>A | Exon 43 skipped | Aberrant splicing | Li and Swift (2000); Mitui et al. (2005); Sandoval et al. (1999); Telatar et al. (1998) | ||
| AT02 | c.3402+30_3402+32delATC | ? | ? | Novel | Compound heterozygote |
| AT02.1 | c.3402+30_3402+32delATC | ? | ? | Novel | Carrier |
| AT02.2 | Excluded c.3402+30_3402+32delATC | ||||
| AT06 | c.6145T>G | p.Tyr2049Asp | Missense | Novel | Compound heterozygote |
| c.434T>G, | p.Leu145Arg | Missense | Novel | ||
| AT07 | c.6754_6754delA | p.Thr2252ProfsTer5 | Truncation | Novel | Compound heterozygote |
| c.6095G>A | Exon 43 skipped | Aberrant splicing | Li and Swift (2000); Mitui et al. (2005); Sandoval et al. (1999); Telatar et al. (1998) | ||
| AT7.1 | c.6754_6754delA | p.Thr2252ProfsTer5 | Truncation | Novel | Compound heterozygote |
| c.6095G>A, | Exon 43 skipped | Aberrant splicing | Li and Swift (2000); Mitui et al. (2005); Sandoval et al. (1999); Telatar et al. (1998) | ||
| AT08 | c.7630-2A>C | Exon 54 skipped | Aberrant splicing | Li and Swift (2000); Mitui et al. (2005); Sandoval et al. (1999); Telatar et al. (1998) | Compound heterozygote |
| AT08.1 | c.7630-2A>C | Exon 54 skipped | Aberrant splicing | Li and Swift (2000); Mitui et al. (2005); Sandoval et al. (1999); Telatar et al. (1998) | Carrier |
| AT10 | c.6095G>A, | Exon 43 skipped | Aberrant splicing | Li and Swift (2000); Mitui et al. (2005); Sandoval et al. (1999); Telatar et al. (1998) | Compound heterozygote |
| AT12 | c. 7630-2A>C | Exon 54 skipped | Aberrant splicing | Li and Swift (2000); Mitui et al. (2005); Sandoval et al. (1999); Telatar et al. (1998) | Compound heterozygote |
| AT13 | Deletion of 62 and 63 exons | Truncation | Novel | Compound heterozygote | |
| c.5932G>T | p.Glu1978Ter | Truncation | Birrell et al. (2005); Li and Swift (2000); Mitui et al. (2005) | ||
| AT15 | c.1179_1180delGG | p.Trp393Ter | Truncation | Buzin et al. (2003) | Compound heterozygote |
| AT19 | c.6095G>A | Exon 43 skipped | Aberrant splicing | Li and Swift (2000); Mitui et al. (2005); Sandoval et al. (1999); Telatar et al. (1998) | Compound heterozygote |
| AT21 | c.7010_7011delGT | Mitui et al. (2005); Telatar et al. (1996) | Compound heterozygote | ||
| c.5932G>T | p.Glu1978Ter | Truncation | Birrell et al. (2005); Li and Swift (2000); Mitui et al. (2005) | ||
| AT23 | c.381_381delA | p.Thr127ThrfsTer2 | Truncation | Babaei et al. (2005); Castellvi-Bel et al. (1999); Mitui et al. (2005) | Compound heterozygote |
| c.3402+30_3402+32delATC | ? | ? | Novel | ||
| AT23.1 | Excluded c.381_381delA | Carrier | |||
| c.3402+30_3402+32delATC | ? | ? | Novel | ||
| AT23.2 | c.381_381delA | p. Thr127Thr fsTer2 | Truncation | Babaei et al. (2005); Castellvi-Bel et al. (1999) Mitui et al. (2005) | Carrier |
| Excluded c.3402+30_3402+32delATC | |||||
| AT24 | c.4007_4008insA | p.Phe1336PhefsTer3 | Truncation | Novel | Homozygote |
| c.4007_4008insA | p.Phe1336PhefsTer3 | Truncation | Novel | ||
| AT24.1 | c.4007_4008insA | p.Phe1336PhefsTer3 | Truncation | Novel | |
| AT26 | c.7606G>A | p.Gly2536Ter | Truncation | Novel | Compound heterozygote |
| AT27 | c.3402+30_3402+32delATC | ? | ? | Novel | Compound heterozygote |
| c.5932G>T | p.Glu1978Ter | Truncation | Birrell et al. (2005); Mitui et al. (2005) | ||
| AT27.1 | c.5932G>T | p.Glu1978Ter | Truncation | Birrell et al. (2005); Li and Swift (2000); Mitui et al. (2005) | Carrier |
| AT27.2 | c.3402+30_3402+32delATC | ? | ? | Novel | Carrier |
| AT28 | c.5932G>T | p.Glu1978Ter | Truncation | Birrell et al. (2005); Li and Swift (2000); Mitui et al. (2005) | Compound heterozygote |
| c.7630-2A>C | Exon 54 skipped | Aberrant splicing | Li and Swift (2000); Mitui et al. (2005); Sandoval et al. (1999); Telatar et al. (1998) | ||
| AT28.1 | c.5932G>T | p.Glu1978Ter | Truncation | Birrell et al. (2005); Li and Swift (2000); Mitui et al. (2005) | Carrier |
| Excluded c.7630-2A>C | |||||
| AT30 | c.6095G>A | Exon 43 skipped | Aberrant splicing | Li and Swift (2000); Mitui et al. (2005); Sandoval et al. (1999); Telatar et al. (1998) | Compound Heterozygote |
| AT31 | c.2250G>A | Aberrant splicing | Byrd et al. (1996); Mitui et al. (2003); Sandoval et al. (1999) | Compound heterozygote | |
| c.7630-2A>C | Exon 54 skipped | Aberrant splicing | Li and Swift (2000); Mitui et al. (2005); Sandoval et al. (1999); Telatar et al. (1998) | ||
| AT33 | c.7630-2A>C | Exon 54 skipped | Aberrant splicing | Li and Swift (2000); Mitui et al. (2005); Sandoval et al. (1999); Telatar et al. (1998) | Compound heterozygote |
| c.5932G>T | p.Glu1978Ter | Truncation | Birrell et al. (2005); Li and Swift (2000); Mitui et al. (2005) | ||
| AT33.1 | c.7630-2A>C | Exon 54 skipped | Aberrant splicing | Li and Swift (2000); Mitui et al. (2005); Sandoval et al. (1999); Telatar et al. (1998) | Compound heterozygote |
| c.5932G>T | p.Glu1978Ter | Truncation | Birrell et al. (2005); Li and Swift (2000); Mitui et al. (2005) | ||
| AT34 | Deletion of exons 19 and 20 | Truncation | Novel | Compound heterozygote | |
| Deletion of exon 63 | Truncation | Novel | |||
| AT35 | c.5932G>T | p.Glu1978Ter | Truncation | Birrell et al. (2005); Li and Swift (2000); Mitui et al. (2005) | Compound heterozygote |
| AT36 | c.3802_3802delG | p.Val1268Ter | Truncation | Mitui et al. (2003); Sandoval et al. (1999) | Compound heterozygote |
| AT37 | c.9021_9022insA | p.Arg3008ThrfsTer54 | Truncation | Mitui et al. (2005) | Homozygote |
| c.9021_9022insA | p.Arg3008ThrfsTer54 | Truncation | |||
| AT37.1 | c.9021_9022insA | p.Arg3008ThrfsTer54 | Truncation | Mitui et al. (2005) | Carrier |
| AT37.2 | c.9021_9022insA | p.Arg3008ThrfsTer54 | Truncation | Mitui et al. (2005) | Carrier |
| AT38 | c.7010_7011delGT | p.Gly2337SerfsTer35 | Truncation | Telatar et al. (1996) | Compound heterozygote |
| c.7630-2A>C | Exon 54 skipped | Aberrant splicing | Li and Swift (2000); Mitui et al. (2005); Sandoval et al. (1999); Telatar et al. (1998) | ||
| AT38.1 | Excluded c.7010_7011delGT |
On the basis of transcripts NM_000051 for ATM.
Table 3.
Multiple alignment of regions surrounding L145 (A) and Y2049 (B) of ATM across different organisms
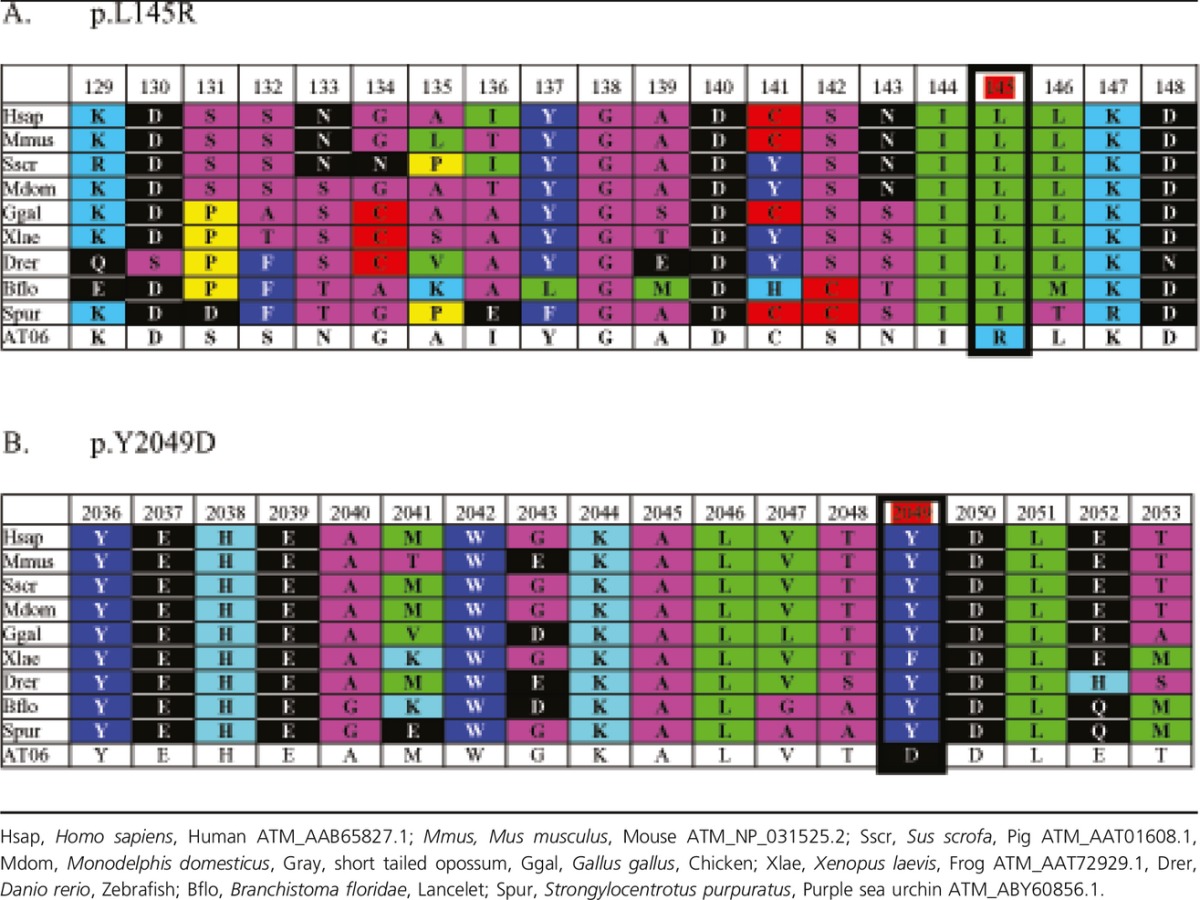
The other new change in the ATM sequence in the Polish population was c.3402+30_3402+32delATC in intron 25. Splicing defects in the ATM gene are common (Teraoka et al. 1999). Most of these involve disruption of the canonical splice sites and lead to exon skipping. Furthermore, deep intronic mutation was described previously (Sutton et al. 2004; Coutinho et al. 2005). For example, in the United Kingdom, 15% of AT families are intronic c.5762-1050A>G mutation carriers. This mutation activates a cryptic splice donor/acceptor site, resulting in the insertion of 137 nucleotides of an intronic sequence (McConville et al. 1996). The c.3402+30_3402+32delATC was identified in three of our AT families (3/24, 12.5%). This intronic variant appeared in a heterozygous state in all cases. Among these 200 controls, this intronic variant was not observed. According to data from the NHLBI Exome Sequencing Project, the ATC deletion allele has a frequency of 0.11% (14/12504) of total alleles studied and is not observed in a homozygous state. We subsequently analyzed the ATM transcripts, to investigate the possible effects of intronic deletions. No abnormal ATM transcripts were detected. Moreover, in silico analysis showed that an intronic variant may damage the transcription factor (TF) binding sites, resulting in the disruption of the Oct-1 and GATA1 binding sites and the appearance of the binding site for other TF and Pit1 sites. Up to now, there has been no report of immediate interactions between these three TF proteins and the ATM gene. Oct-1 and Pit1 belong to a large POU family of transcription factors. Oct-1 is known as a transcription factor involved in regulation of some housekeeping genes, histone H2B, snRNAs as well as in tissue-specific regulation of immunoglobulin and mediated antigen-independent B cell development. GATA1 is implicated in the reprograming of hematopoietic precursors and the regulation of G1/S cell cycle progression. Also it is known that three TFs bind to intronic regions and affect the gene expression. On the basis of these data, ATM mRNA was measured by quantitative real-time PCR with β-actin as an internal reference gene. The results showed that the mRNA level in the samples with c.3402+30_3402+32delATC is similar to the mRNA level in control cases (Fig. 1). However, the second allele without intronic variant can be up-regulated to compensate for the lack of a function of the defective allele. On the other hand, the ATM tissue-specific expression depending on Oct, Pit-1 or GATA1 is also not excluded. A total loss of the ATM protein was detected by western blotting in patients carrying this intronic variant. This observation supports the hypothesis that second allele can be up-regulated. In spite of the initial results, other functional analysis may reveal that the c.3402+30_3402+32delATC is a pathogenic mutation.
Figure 1.
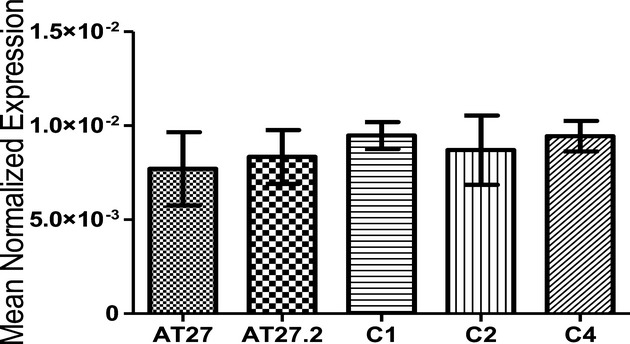
Real-time PCR results for ATM mRNA levels. ATM mRNA levels were measured by RT-PCR from controls and individuals with c.3402+30_3402+32delATC and normalized to β-actin mRNA levels. Data are expressed as mean normalized expression ± s.d. The one-way ANOVA followed by Newman-Keuls test was performed to determine the significance. There are no significant differences in expression between patients with c.3402+30_3402+32delATC and controls.
In patient AT13, the large genomic deletion of exons 62 and 63 was detected. This deletion is combined with a nonsense mutation c.5932G>T in exon 42 (Table3). Another recent interesting case is a patient with two large deletions. The first deletion encompasses two exons 19–20 and is combined with a deletion removing the last exon of ATM. Previous reports estimated that the large genomics mutations in ATM are detected in 2% to 23% of AT patients. A high percentage of large genomic mutations was described in the Japanese population. There have been a few reports showing that large genomic deletion (LGD) occurs in Brazilian, Chinese, Costa Rican, Dutch, and Japanese ataxia telangiectasia patients (Broeks et al. 1998; Coutinho et al. 2004; Nakamura et al. 2012; Huang et al. 2013). Moreover, Cavalieri et al. reported a large duplication in the ATM gene, spanning exons 4–20 (41kbp) (Cavalieri et al. 2008). LGDs were localized in a different part of the ATM gene, especially in the last two exons. Previous analyses of the genomic deletions of last two exons of the ATM gene show that mutations are caused by retro-transposable elements (long interspersed element-1, LINE1). The 3′ end and downstream sequence of the ATM gene are riddled with retrotransposons (ALU, LINE).
In summary, in this study, we confirmed the status of recurrent mutations (c.5932G>T, c.6095G>A, c.7630-2A>C) and also detected ten new ATM gene changes in Polish patients with AT. In the future, further investigations on the functional role and clinical impact of novel alterations will be performed.
Acknowledgments
Grant support: NN401098240. The study was conducted with the approval by the Central Ethical Committee of Ministry of Health, Poland, in accordance with the tenets of the Helsinki declaration.
References
- Babaei M, Mitui M, Olson ER. Gatti RA. ATM haplotypes and associated mutations in Iranian patients with ataxia-telangiectasia: recurring homozygosity without a founder haplotype. Hum. Genet. 2005;117:101–106. doi: 10.1007/s00439-005-1254-7. [DOI] [PubMed] [Google Scholar]
- Birrell GW, Kneebone K, Nefedov M, Nefedova E, Jartsev MN, Mitsui M, et al. ATM mutations, haplotype analysis, and immunological status of Russian patients with ataxia telangiectasia. Hum. Mutat. 2005;25:593. doi: 10.1002/humu.9341. [DOI] [PubMed] [Google Scholar]
- Broeks A, Deklein A, Floore AN, Muijtjens M, Kleijer WJ, Jaspers NG, et al. ATM germline mutations in classical ataxia-telangiectasia patients in the Dutch population. Hum. Mutat. 1998;12:330–337. doi: 10.1002/(SICI)1098-1004(1998)12:5<330::AID-HUMU6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Buzin CH, Gatti RA, Nguyen VQ, Wen CY, Mitui M, Sanal O, et al. Comprehensive scanning of the ATM gene with DOVAM-S. Hum. Mutat. 2003;21:123–131. doi: 10.1002/humu.10158. [DOI] [PubMed] [Google Scholar]
- Byrd PJ, Srinivasan V, Last JI, Smith A, Biggs P, Carney EF, et al. Severe reaction to radiotherapy for breast cancer as the presenting feature of ataxia telangiectasia. Br. J. Cancer. 1996;106:262–268. doi: 10.1038/bjc.2011.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlessi L, Fusar Poli E, de Filippis L. Delia D. ATM-deficient human neural stem cells as an in vitro model system to study neurodegeneration. DNA Repair (Amst) 2013;12:605–611. doi: 10.1016/j.dnarep.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellvi-Bel S, Sheikhavandi S, Telatar M, Tai LQ, Hwang M, Wang Z, et al. New mutations, polymorphisms, and rare variants in the ATM gene detected by a novel SSCP strategy. Hum. Mutat. 1999;14:156–162. doi: 10.1002/(SICI)1098-1004(1999)14:2<156::AID-HUMU7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Cavalieri S, Funaro A, Pappi P, Migone N, Gatti RA. Brusco A. Large genomic mutations within the ATM gene detected by MLPA, including a duplication of 41 kb from exon 4 to 20. Ann. Hum. Genet. 2008;72:10–18. doi: 10.1111/j.1469-1809.2007.00399.x. [DOI] [PubMed] [Google Scholar]
- Coutinho G, Mitui M, Campbell C, Costa Carvalho BT, Nahas S. Sun X. Five haplotypes account for fifty-five percent of ATM mutations in Brazilian patients with ataxia telangiectasia: seven new mutations. Am. J. Med. Genet. A. 2004;126A:33–40. doi: 10.1002/ajmg.a.20570. [DOI] [PubMed] [Google Scholar]
- Coutinho G, Xie J, Du L, Brusco A, Krainer AR. Gatti RA. Functional significance of a deep intronic mutation in the ATM gene and evidence for an alternative exon 28a. Hum. Mutat. 2005;25:118–124. doi: 10.1002/humu.20170. [DOI] [PubMed] [Google Scholar]
- Demuth I, Dutrannoy V, Marques W, Jr, Neitzel H, Schindler D, Dimova PS, et al. New mutations in the ATM gene and clinical data of 25 AT patients. Neurogenetics. 2011;12:273–282. doi: 10.1007/s10048-011-0299-0. [DOI] [PubMed] [Google Scholar]
- Derheimer FA. Kastan MB. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett. 2010;584:3675–3681. doi: 10.1016/j.febslet.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Yang L, Wang J, Yang F, Xiao Y, Xia R, et al. Twelve novel Atm mutations identified in Chinese ataxia telangiectasia patients. Neuromolecular Med. 2013;15:536–540. doi: 10.1007/s12017-013-8240-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keimling M, Volcic M, Csernok A, Wieland B, Dork T. Wiesmuller L. Functional characterization connects individual patient mutations in ataxia telangiectasia mutated (ATM) with dysfunction of specific DNA double-strand break-repair signaling pathways. Faseb J. 2011;25:3849–3860. doi: 10.1096/fj.11-185546. [DOI] [PubMed] [Google Scholar]
- Li A. Swift M. Mutations at the ataxia-telangiectasia locus and clinical phenotypes of A-T patients. Am. J. Med. Genet. 2000;92:170–177. doi: 10.1002/(sici)1096-8628(20000529)92:3<170::aid-ajmg3>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Lumsden JM, McCarty T, Petiniot LK, Shen R, Barlow C, Wynn TA, et al. Immunoglobulin class switch recombination is impaired in Atm-deficient mice. J. Exp. Med. 2004;200:1111–1121. doi: 10.1084/jem.20041074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConville CM, Stankovic T, Byrd PJ, McGuire GM, Yao QY, Lennox GG, et al. Mutations associated with variant phenotypes in ataxia-telangiectasia. Am. J. Hum. Genet. 1996;59:320–330. [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ. ATM and ataxia telangiectasia. EMBO Rep. 2004;5:772–776. doi: 10.1038/sj.embor.7400210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitui M, Campbell C, Coutinho G, Sun X, Lai CH, Thorstenson Y, et al. Independent mutational events are rare in the ATM gene: haplotype prescreening enhances mutation detection rate. Hum. Mutat. 2003;22:43–50. doi: 10.1002/humu.10232. [DOI] [PubMed] [Google Scholar]
- Mitui M, Bernatowska E, Pietrucha B, Piotrowska-Jastrzebska J, Eng L, Nahas S, et al. ATM gene founder haplotypes and associated mutations in Polish families with ataxia-telangiectasia. Ann. Hum. Genet. 2005;69:657–664. doi: 10.1111/j.1529-8817.2005.00199.x. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Du L, Tunuguntla R, Fike F, Cavalieri S, Morio T, et al. Functional characterization and targeted correction of ATM mutations identified in Japanese patients with ataxia-telangiectasia. Hum. Mutat. 2012;33:198–208. doi: 10.1002/humu.21632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman A, Srinivasan V, Barone G, Last JI, Wootton LL, Davies EG, et al. Lymphoid tumours and breast cancer in ataxia telangiectasia; substantial protective effect of residual ATM kinase activity against childhood tumours. Br. J. Cancer. 2011;105:586–591. doi: 10.1038/bjc.2011.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval N, Platzer M, Rosenthal A, Dork T, Bendix R, Skawran B, et al. Characterization of ATM gene mutations in 66 ataxia telangiectasia families. Hum. Mol. Genet. 1999;8:69–79. doi: 10.1093/hmg/8.1.69. [DOI] [PubMed] [Google Scholar]
- Staples ER, McDermott EM, Reiman A, Byrd PJ, Ritchie S, Taylor AM, et al. Immunodeficiency in ataxia telangiectasia is correlated strongly with the presence of two null mutations in the ataxia telangiectasia mutated gene. Clin. Exp. Immunol. 2008;153:214–220. doi: 10.1111/j.1365-2249.2008.03684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton IJ, Last JI, Ritchie SJ, Harrington HJ, Byrd PJ. Taylor AM. Adult-onset ataxia telangiectasia due to ATM 5762ins137 mutation homozygosity. Ann. Neurol. 2004;55:891–895. doi: 10.1002/ana.20139. [DOI] [PubMed] [Google Scholar]
- Telatar M, Wang Z, Udar N, Liang T, Bernatowska-Matuszkiewicz E, Lavin M, et al. Ataxia-telangiectasia: mutations in ATM cDNA detected by protein-truncation screening. Am. J. Hum. Genet. 1996;59:40–44. [PMC free article] [PubMed] [Google Scholar]
- Telatar M, Teraoka S, Wang Z, Chun HH, Liang T, Castellvi-Bel S, et al. Ataxia-telangiectasia: identification and detection of founder-effect mutations in the ATM gene in ethnic populations. Am. J. Hum. Genet. 1998;62:86–97. doi: 10.1086/301673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teraoka SN, Telatar M, Becker-Catania S, Liang T, Onengut S, Tolun A, et al. Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am. J. Hum. Genet. 1999;64:1617–1631. doi: 10.1086/302418. [DOI] [PMC free article] [PubMed] [Google Scholar]