

Summary
In recent years, it has become increasingly apparent that the life span of transfused platelets in circulation is regulated, at least in part, by glycan-lectin mediated mechanisms. There is clear evidence that refrigerated platelets are cleared by glycan-lectin mediated clearance mechanisms. Acute platelet cooling clusters glycoprotein (GP) Ibα receptors bearing uncovered N-acetylglucosamine (GlcNAc), and αMβ2 integrins on hepatic macrophages recognise clustered GlcNAc to rapidly clear these platelets from circulation. With prolonged refrigeration GPIbα clustering bearing uncovered galactose increases, which mediates the removal of long-term refrigerated platelets via hepatic Ashwell-Morell receptors (AMR), originally named as asialoglycoprotein receptors. In contrast, little is known about the molecular mechanisms of transfused room temperature platelet clearance. This review examines the role of glycan-lectin mediated clearance of exogenous, i.e. transfused chilled platelet clearance and briefly addresses the current knowledge of stored platelet function, degradation and its relation to platelet clearance.
To be clinically effective, transfused platelets must circulate and function in clotting (i.e. prevent or stop bleeding). Currently, the gold standard test to evaluate platelet products is in vivo survival and count increment of transfused radiolabelled platelets [1, 2]. It is assumed that if a platelet product circulates normally, it should function appropriately. However, both parameters fail to assess the functional quality of transfused platelets. Furthermore, efficacy of a transfused platelet product is clearly patient-dependent, complicating the assessment of platelet functionality [3]. Undeniably, clinical experience shows that transfused platelets work. Improvements in treatment have rendered fatal haemorrhage less common today, although thrombocytopenic patients receiving prophylactic transfusions in randomised platelet “trigger” trials have had clinically significant bleeding complications (WHO Grades 2–4), at rates of 17–21.5%, irrespective of study arm [4, 5]. Therefore, it seems reasonable to question whether current bleeding-rates could be reduced by improved platelet products. Platelet refrigeration could represent one way to improve transfused platelet quality and extend platelet shelf life by reducing the risk of bacterial growth.
Platelet function and clearance: are they related?
For more than thirty years the “dogma” held that cold-stored platelets do not circulate because they do not function. One key concept that has emerged from our refrigerated platelet studies is that a platelet’s ability to survive in the circulation may be entirely separate from its ability to function in haemostasis [6, 7], thus challenging the dogma. This notion is supported by several human studies performed in the early 1970s, which suggest that platelets stored at 4°C have better in vivo function than those stored at room temperature despite having poor survival in the circulation. Becker, Aster and colleagues [8] tested the effects of platelets refrigerated for ≤72 hours in thrombocytopenic patients. The patients, with pre-transfusion bleeding times of >15 minutes, were given fresh platelet concentrates, room temperature platelet concentrates that had been stored for 24, 48 or 72 hours, or 4°C platelet concentrates that had been stored for the same periods. Bleeding times were measured one hour after transfusion. Platelets refrigerated for 48 or 72 hours corrected the bleeding time in 63% of cases, while room temperature stored platelets corrected the bleeding time in only 24% of patients. In similar studies performed in aspirin-treated volunteers, bleeding times were corrected almost to baseline within four hours after transfusion with 4°C platelets. In contrast, almost no effect was seen at this early time-point in subjects receiving platelets stored at room temperature for 72 hours. At 24 hours post-transfusion, substantial correction of the bleeding time was seen in the recipients of room temperature platelets [8, 9]. Another study showed that refrigerated (≤72hours) platelets transfused into 41 leukaemia patients effectively stopped bleeding and were considered as “safe” in over 100 transfused patients [10]. Similar observations were made by Handin and Valeri [11], who found that room temperature platelets corrected the bleeding time of aspirin treated volunteers 24 hours post-transfusion, but had no effect immediately post-transfusion, suggesting that loss of in vitro platelet function may be reversible upon transfusion [11]. Following these clinical studies, the suggestion was made that platelets should be stored at 4°C if intended to treat acute/active bleeding (i.e. trauma or surgical patients), while platelets used for bleeding prophylaxis should be stored at room temperature [12, 13]. However, this approach never gained acceptance, as studies performed subsequently failed to confirm that transfused room temperature platelets showed a significant delay in haemostatic function. A study by Slichter and Harker showed that room temperature platelets consistently corrected the bleeding times of aplastic thrombocytopenic patients within two and half hours. In contrast refrigerated platelets (<72 hours) corrected the bleeding time in 6/8 cases shortly post-transfusion, but the effect was not sustained beyond two and half hours [14, 15]. Aster and colleagues repeated the bleeding time experiments done previously in their laboratory [15], administering either room temperature or refrigerated platelets to 41 thrombocytopenic patients and reported that platelets stored at room temperature for 24 hours produced the greatest bleeding time correction one hour post-transfusion, while platelets refrigerated for 72 hours yielded the least bleeding time correction, although the difference was not statistically significant. The investigators attributed the relative ineffectiveness of room temperature platelets in their earlier study to inadequate low storage volume [14].
Based on these in vivo data it seems safe to conclude that the sole reason platelets are currently stored at room temperature is that chilled platelets do not circulate for an acceptable period of time in the recipient. It appears that the mechanisms dictating platelet removal from the circulation, including refrigerated platelets, are independent of platelet function.
Platelet shape and clearance: are they related?
Another prevailing dogma has been for years that chilled platelets do not circulate because they change shape. Undoubtedly, platelet activation is accompanied by shape change [16]. Platelets become activated during room temperature storage [17–19]. Key mediators of thrombosis stored in the α-granules of resting platelets, such as β-thromboglobulin (β-TG) and platelet factor 4, accumulate in the platelet storage medium over time [20]. Moreover, platelet surface P-selectin expression increases during room temperature storage [21], indicating that platelets continuously release their granule content, i.e. become activated. It is therefore likely that room temperature stored platelets change shape, however, room temperature platelets circulate once transfused.
Whether cooling itself causes platelet activation is controversial. In many ways, platelets stored at 4°C seem to be less activated than platelets stored at room temperature. For example, 4°C platelet storage does not lead to β-TG release [20]. On the other hand, platelet chilling induces rapid irreversible disc-to-sphere shape changes, i.e. changes in shape [22] and P-selectin surface expression [23–25]. Despite initial enthusiasm for the use of P-selectin as a marker of platelet quality in transfusion settings, P-selectin levels alone do not predict platelet survival after transfusion [26–29]. Studies using platelets lacking P-selectin [28] or platelets activated with thrombin [30, 31] showed that P-selectin exposure or loss of platelet discoid shape had no effect on platelet clearance. Conversely, spherical platelets characteristic for mice lacking β1-tubulin circulate normally [6, 32, 33]. In support of this notion, we and others have shown that preserving the discoid shape of refrigerated platelets using actin-remodelling inhibitors did not diminished their clearance [6, 32, 33].
Conflicting results using in vitro tests comparing refrigerated and room temperature platelet metabolic efficacy add to the controversy about the equality of cold-platelets. It is assumed that both the retention of platelet discoid shape, as measured photometrically by the extent of shape change (ESC), and hypotonic shock response (HSR) indicating metabolic efficacy, report on platelet viability [12, 34, 35]. Not surprisingly, platelets stored at room temperature perform better in both tests because platelets rapidly lose their “discoid shape” when chilled, implying that refrigerated platelets are not metabolically efficient and viable [8, 12, 22]. Most investigators, however, agree that 4°C stored platelet products show better pH stability [36], a reduced rate of glycolysis, and a better response when stimulated by ADP, epinephrine or collagen than room temperature stored platelets [8], implying that refrigerated non-discoid platelets function better. Taken together, these studies contradict the dogma that the discoid shape of a platelet per se predict its function or survival.
Apoptosis, degradation and metabolic processes in transfused platelet clearance
Without a doubt the intrinsic apoptotic machinery regulates, at least in part, endogenous platelet survival (for review see [37]). Does phosphatidylserine (PS) exposure per se trigger the engulfment of platelets? PS is the archetypal ‘eat me’ signal displayed by cells undergoing apoptosis. However, platelets also expose PS when activated by a range of physiological agonists [38, 39]. In this context, it facilitates assembly of the prothrombinase complex, which plays an essential role in generating thrombin [40]. Whether the apoptotic machinery is required for this type of PS exposure is unclear. Storage of platelets induces PS exposure independent of the storage temperature [23, 25, 41]. However, as in “normal” platelet activation processes, the role of the apoptotic machinery in PS exposure during platelet storage needs to be established, especially if surface PS reliably predicts transfused platelet survival.
Low pH levels <6.0 have been consistently associated with severely diminished platelet viability [42, 43]. Platelets stored at room temperature are continuously metabolically active. Metabolic products, such as lactate, accumulate during room temperature storage, leading to a drop in pH [44]. In contrast, at 4°C there is minimal lactate accumulation, and the pH does not decrease [36] posing the question; what do changes in pH during storage mean for platelet survival?
Glycan composition and its effects on platelet survival
Loss of sialic acid is also thought to influence platelet life span by exposing penultimate galactose (Gal) residues, effecting the recognition and phagocytosis by asialoglycoprotein receptors (ASGPRs) [45–48] [7, 49, 50]. Similar to erythrocytes, in vitro desialylated platelets are cleared rapidly from circulation [51, 52]. Loss of sialic acid during storage of platelets has been reported, independent of storage temperature [53]. However no consistent studies have been conducted in humans to determine the extent of sialic acid loss and its effects on platelet recovery and survival. How do platelets lose sialic acid? An intriguing thought is that low pH potentially promotes platelet-derived sialidase activity during platelet storage. Bacterially derived sialidases have been shown to affect platelet survival in mammals [50, 54]. It is therefore tempting to speculate that bacteria-derived sialidases deplete sialic acid from platelets during storage thereby severely affecting platelet viability.
It appears that platelet clearance is regulated in a complex fashion. For example, chilled platelet clearance is not mediated by loss of platelet function or shape change. Release of granule contents per se does not seem to influence platelet survival. Our lab has demonstrated that loss of sialic acid and galactose from platelet glycoproteins during cold storage leads to removal by lectin-mediated mechanisms. Whether change in platelet glycan composition predicts platelet survival per se still needs to be determined.
Lectins in platelet clearance
The clearance of fresh room temperature platelets is equally divided between spleen and liver [6]. However, little is known about the molecular mechanisms mediating their clearance in both organs. For example, it is assumed, but has not been proven that macrophages mediate the clearance of room temperature platelets. In contrast, cooled transfused platelets are predominately cleared in the liver [6]. We have defined two previously unsuspected, carbohydrate-dependent platelet clearance mechanisms, which will be detailed in the next sections.
The macrophage αM lectin-domain: an overview
αMβ2 (or CR3, CD11b/CD18, MAC-1) has two main functions. First, it mediates adhesion and migration of leukocytes into inflammatory sites in tissues via binding to the intercellular adhesion molecule (ICAM)-1 expressed on stimulated endothelium [55]. Second, αMβ2 serves as a phagocytic receptor for the iC3b fragment of complement [56, 57]. The αMβ2 receptor shares functional characteristics with other integrins including the bidirectional signalling via conformational changes in the extracellular region that are produced by inside-out signalling [58, 59]. αMβ2, like all integrins, consists of two chains: the αM- and the β2 -chain. αM contains the ligand binding I-domain, a cation-binding region, and a lectin-site. Protein ligands bind to partially overlapping sites contained within the I-domain [60] and include ICAM-(1–2), fibrinogen, iC3b, [61], and GPIbα [62, 63]. Interestingly, the αM domain also contains a cation-independent lectin-site [64], which binds to glycans (specifically GlcNAc) on microbial surface polysaccharides and β-glucan [65]. C3 opsonised microorganisms display iC3b in combination with cell wall polysaccharides, such that both the I-domain and lectin-site of αMβ2 become attached to microbial pathogens, stimulating phagocytosis and cytotoxic degranulation [66]. Target cells bearing only iC3b, but not αM binding glycans, do not trigger phagocytosis and/or degranulation, despite avid attachment of the target cells to the I-domain. In contrast, glycan structures that are large enough to cross-link the lectin domains of multiple membrane surface αMβ2 molecules, stimulate degranulation and the release of inflammatory mediators in the absence of the iC3b-opsonin (for review see [67]).
Role of the αM-lectin in transfused cold-platelet clearance
The clearance of fresh room temperature platelets is equally divided between spleen and liver. However, little is known about the molecular mechanisms mediating their clearance in these organs. For example, it is assumed, but has not been proven, that macrophages mediate the clearance of room temperature platelets. In contrast, cooled transfused platelets are predominately cleared in the liver. We defined a previously unsuspected, carbohydrate-dependent platelet clearance mechanism. The hepatic macrophage carbohydrate-binding integrin αMβ2 selectively recognises glycans on clustered GPIbα subunits of the von Willebrand factor receptor (VWF-R) following short-term (hours) platelet cooling, resulting in the phagocytosis and clearance of platelets in vivo in mice and in vitro by human THP-1 macrophages [6, 62, 68, 69]. Experiments using αM-deficient, but not VWF-, complement- or P-selectin-deficient mice showed marked improvement in the survival of short-term cooled platelets. [6]. Subsequent work narrowed carbohydrate recognition by integrin αMβ2 to uncovered βGlcNAc on glycans within GPIbα’s N-terminal ligand binding-domain [62, 68]. Removal of sialic acid (desialylation) exposes galactose and de-galactosylation reveals βGlcNAc. Exposure on of βGlcNAc-residues on mammalian circulating cells is unexpected and is either the result of incomplete glycan processing or the result of a degrading process that occurs in blood. βGlcNAc-exposure would be expected to be kept at a minimum, as loss of covering glycan residues is normally considered as a clearance signal. Therefore, lectins recognising βGlcNAc bind only minimally to circulating room temperature platelets. Conversely, refrigerated platelets have markedly increased lectin-binding, suggesting that altered epitope presentation and/or clustering of exposed βGlcNAc on GPIbα can facilitate lectin binding to refrigerated platelets and chilled platelet clearance. What causes these alterations of specific carbohydrate epitopes on platelet glycoproteins during refrigeration? Actin rearrangement during refrigeration is likely to initiate surface VWF-R redistribution from linear arrays into aggregates [6]. Glycan and VWF-R clustering is detected early after refrigeration [6, 68], but increases with long-term platelet storage and refrigeration in plasma, as discussed later.
A potential method for preventing the rapid clearance of refrigerated platelets for transfusion represented the enzymatic galactosylation of surface βGlcNAc residues on platelet glycoproteins using a β1,4-galactosyltransferase (β4GalT). Surprisingly, both human and murine platelets have functional platelet galactosyltransferase(s) on their surface which simply transfer galactose onto uncovered βGlcNAc of human or mouse GPIbα after addition of UDP-galactose [68]. Although galactosylation markedly improved the survival of mouse platelets chilled for hours [68], this manoeuvre theoretically provided a new ligand for ASGPRs. Hence, it was surprising that refrigerated mouse platelet circulation could be improved by galactosylation. We postulated that the number of exposed βGlcNAc residues on GPIbα was small, such that even after clustering and galactosylation, the galactose density was insufficient to engage galactose-recognising lectins [68]. A phase I clinical trial administering autologous, radiolabelled galactosylated apheresis platelets refrigerated for 48 hours in plasma into human volunteers clearly showed that the galactosylation procedure did not extend their circulation time [41]. Evidently, this trial showed that additional mechanisms were involved in the clearance of platelets refrigerated for days in plasma.
The Ashwell-Morell receptor (AMR): an overview
It is “common knowledge” that the liver controls the removal of exogenously administered desialylated glycoproteins (termed asialoglycoproteins) from circulation [70–72]. The hepatic lectin (HL) or AMR was identified as a galactose-binding receptor using preparations of asialo-ceruloplasmin, free of sialic acid with exposed terminal galactose (for review see [73]). Today, the AMR is one of multiple C-type lectins that bind asialoglycoproteins. Other lectins that bind asialoglycoproteins include the Kupffer cell receptor, the macrophage galactose receptor and galectins. The AMR consists of transmembrane glycoproteins termed hepatic lectin-1 or asialoglycoprotein-1 (HL-1 or asgr-1) and hepatic lectin-2 or asialoglycoprotein-2 (HL-2 or asgr-2) (for review see [73]). Both glycoproteins are type-2 transmembrane proteins with a ~40-amino acid N-terminal cytoplasmic domain, a ~80-amino acid extracellular stalk, and an ~130-amino acid C-terminal carbohydrate recognition domain. HL-1 and HL-2 are predominantly expressed in liver hepatocytes [74, 75] and their expression is induced rapidly upon birth implying that the foetus lacks this particular mechanism of removing desialylated circulating glycoproteins [76]. Both HL-1 and HL-2 are highly conserved among mammalian species and may have originated from a single ancestral gene (for review see [73]). Particularly, the human and mouse genes share >85% amino acid sequence identity between the two subunits. Homo- and hetero-oligomerisation of the mammalian HL-1 and HL-2 glycoproteins to form functional variants of the AMR has been observed in various cellular contexts (for review see [73]). This variability in subunit combinations to form functional complexes facilitates a two-state pathway of receptor-mediated endocytosis (for review see [73]).
Mammalian ASGPRs generally mediate the capture and endocytosis of a wide range of exogenously administered glycoproteins that carry Gal or N-acetylgalactosamine (GalNAc) residues at the termini of their glycan chains (for review see [73]). More recent findings have indicated that some α2,6-sialylated glycans are also ligands for the ASGPR-mediated clearance [77]. Specificities of ASGPR and their relative binding affinities are dependent upon glycan structures, ligand size and the precise spatial arrangement and clustering of the glycan chains [78, 79], although binding specificities may alter by cooperation and competition for specific glycans [80, 81].
The AMR is localised at the vascular side of the hepatocyte cell surface and is therefore ideally positioned to remove and degrade potentially detrimental glycoproteins. Its role is specifically recognised in the design of therapeutic glycosylated proteins, i.e. adding sialic acid to the repertoire of the glycoprotein, to keep its therapeutic levels in circulation [82].
The role of the AMR in transfused platelet clearance
Using a murine transfusion model, we have found that as with short-term cooling, platelets refrigerated for 48 hours in plasma (designated throughout the following text as long-term refrigerated platelets) are removed from the recipients’ circulation by the liver [7, 83] but, unexpectedly by hepatocytes [7] and not macrophages. Macrophages, including Kupffer cells play only a minor role in removing long-term refrigerated platelets, in contrast to our previous findings for short-term cooled platelet clearance [6] as demonstrated in experiments using mice that were depleted of macrophages by injecting toxic clodronate-encapsulated liposomes. Removal of macrophages greatly improved recoveries and survivals of short-term cooled platelets, confirming our earlier results. In contrast, the recoveries and survivals of long-term refrigerated platelets were not affected by macrophage depletion, showing that removal of long-term refrigerated platelets is macrophage-independent and mediated by hepatocytes [7]. Not surprisingly, long-term refrigerated and galactosylated platelets are cleared slightly faster independent of macrophage depletion [7]. Interestingly, these studies also revealed that macrophages immediately remove a large fraction of transfused platelets independent of storage and temperature (~40%) [7]. Our findings in mice, irresistibly agree with loss of ~30–40% platelet recovery consistently reported (and accepted by the transfusion community) following transfusion of fresh platelets into healthy volunteers [41]. The mechanism of “macrophage-mediated” fresh platelet removal remains unclear but once characterised may prove as useful to develop methods to improve “fresh-platelet” recovery following transfusion.
How does the AMR recognize long-term refrigerated platelets? We reported that short-term cooled platelets have exposed/clustered βGlcNAc residues that are recognized and phagocytosed by the αMβ2 hepatic macrophage receptors [6, 68]. In contrast, platelets refrigerated for long periods have severely increased galactose exposure, as evidenced by the galactose-binding lectins. Galactose-exposure presents a ligand for ASGPRs. Consistent with this hypothesis, co-injection of asialofetuin, a competitive inhibitor of the ASGPR, restored the recovery and circulation of long-term refrigerated platelets, but not of short-term refrigerated platelets. Conversely, asialofetuin inhibited phagocytosis of human platelets refrigerated for up to 10 days by the hepatic cell line HepG2 in vitro [7]. Experiments using mice lacking Asgr-1 or Asgr-2 subunits of the AMR showed a significantly improved recovery and circulation of long-term refrigerated platelets, providing clear evidence for the removal of long-term refrigerated platelets by hepatic AMR (Fig. 1). It is noteworthy that fresh isolated platelets have better recoveries and survivals in AMR-lacking mice (Fig. 1). Our studies and studies by other investigators point to the importance of a hepatic-based platelet removal system that uses its AMR to recognise and remove platelets expressing desialylated glycans on their surface [7, 49, 50].
Fig. 1. The Ashwell-Morell receptor mediates hepatic recognition and clearance of long-term-refrigerated platelets.
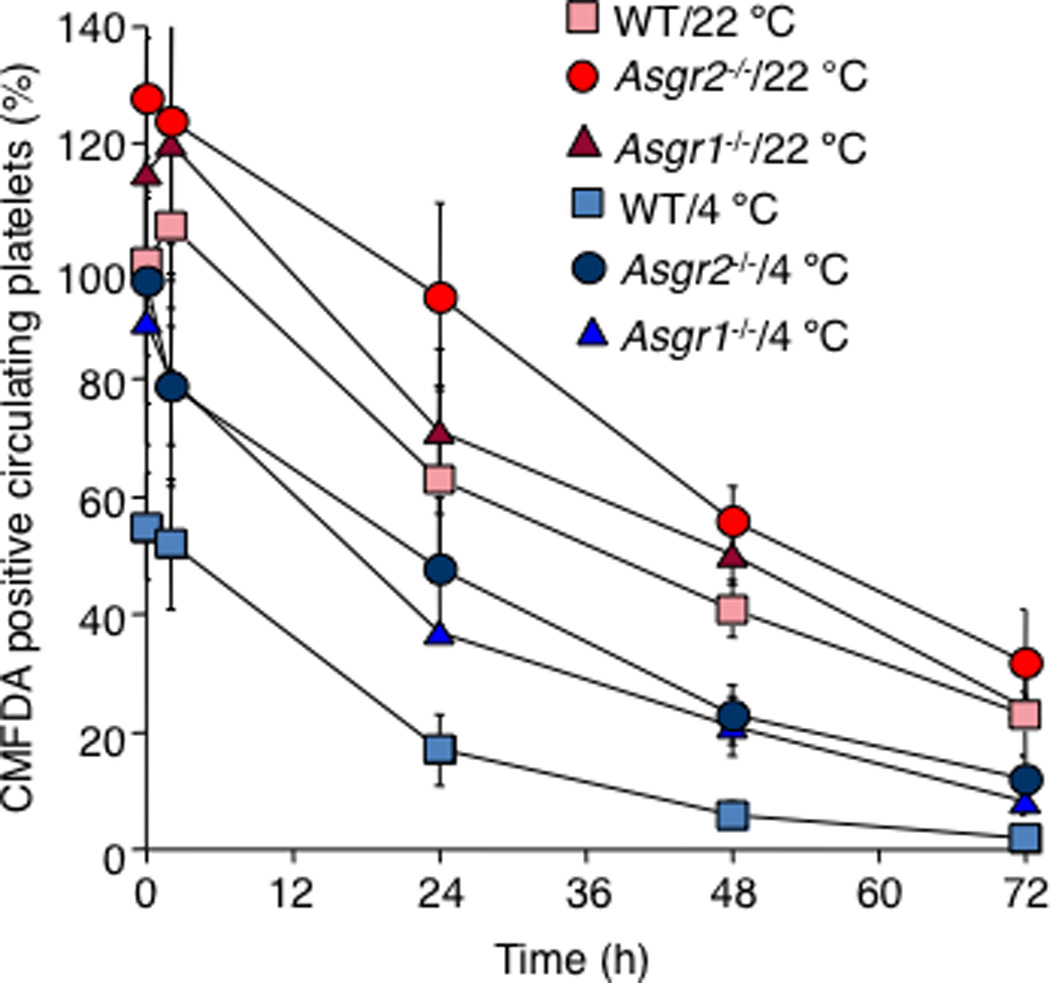
Survival of transfused long-term–refrigerated (4°C) or fresh platelets (22°C) in wild type (WT) mice and in mice lacking asialoglycoprotein receptor-1 or asialoglycoprotein receptor -2 (Asgpr1−/− and Asgpr2−/−, respectively). Note that the recoveries and survival of transfused refrigerated and room temperature platelets are improved in Asgpr1−/− and Asgpr2−/− mice (figure adapted from [7]).
Desialylated glycans reside on GPIbα following long-term refrigeration
GPIbα contains O- and N-linked glycans [84]. Removal of the N-terminal 282 residues of GPIbα from human platelets using the snake venom protease mocarhagin [85] or O-sialoglycoprotein endopeptidase eliminates at least two putative N-glycan residues, as well as the VWF-binding region of GPIbα [85]. Removal of GPIbα’s 45 kDa domain from mouse or human platelets significantly improved the circulation of long-term refrigerated mouse platelets and prevented human platelet ingestion by HepG2 cells in vitro, pointing to the fact that most uncovered galactose residues reside within the external domain of GPIbα initiating clearance by AMR [7]. However, the exact GPIbα glycan structure(s) that engages the interaction with the AMR need(s) to be determined. A scheme of both chilled-platelet clearance mechanisms is shown in Fig. 2.
Fig. 2. Lectin receptors mediating cooled platelet clearance.
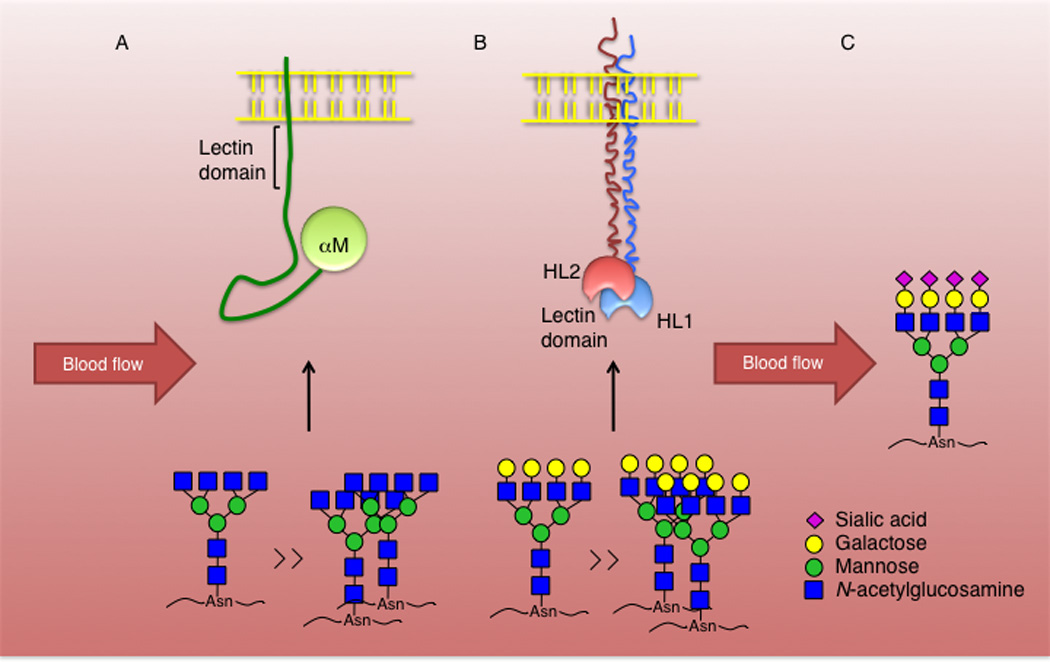
(A) Acutely chilled platelets have clustered GPIbα subunits bearing glycans with N-acetylglucosamine terminal structures, which engage with the αM lectin domain of the β2 integrin to initiate phagocytosis by liver macrophages. Only the αM domain is demonstrated. Clustering or increase of N-acetylglucosamine terminal glycan structures can reflect an increase in binding affinity to the αM carbohydrate-recognizing domain. N-linked glycan ligands are shown. (B) Prolonged refrigeration increases GPIbα clustering and exposure of galactose terminal glycan structures, which bind to the hepatic AMR initiating removal of long-term refrigerated platelets from circulation. Both HL-1 and HL-2 subunits form a functional AMR with multiantennary ligand specificities. Increased binding avidity can reflect the presence of clustered multiantennary galactose-terminal structures. (C) Platelets with sialylated glycan structures continue to circulate.
Is the presentation of GPIbα important for the engagement of refrigerated platelets with the AMR? Platelets isolated from ST3GalIV−/− mice bear desialylated surface glycans as they lack α2–3-sialyltransferase activity [50, 86] and are removed by AMR from the circulation. The extent of galactose exposure on refrigerated platelets is less than that present on the surface of ST3GalIV+/− platelets, but ST3GalIV+/− platelets retain their ability to circulate with normal lifetimes [49, 50, 86]. Clustering of GPIbα substantially increases with prolonged refrigeration [7]. Hence, clustering of platelet GPIbα subunits with cooling might amplify the galactose signal enhancing binding to AMR [68]. However, the functional relationship between GPIbα clustering and platelet clearance by AMR remains to be established.
The mechanism of galactose exposure on long-term refrigerated platelets remains to be determined. Our recent results indicate that surface sialidase activity significantly increases during cold-platelet storage when compared to the sialidase activity measured during room temperature storage (manuscript in preparation). Increase in sialidase activity could substantially contribute to desialylation of platelet surface glycans during storage, revealing underlying galactose.
Are other glycoproteins than GPIbα glycans involved in the clearance of cooled platelets? VWF binding to platelets increases with storage [7]. It appears that preferentially desialylated VWF binds to long-term refrigerated platelets, indicating that desialylation of VWF molecules and/or of platelet glycoproteins may promote binding to platelets. In support of this notion we found the desialylated VWF isolated from ST3Gal-IV−/− mice binds significantly better to platelets than wild type VWF [49] and desialylated VWF is removed by AMR [50]. Proteolytic removal of the GPIbα N-terminal region deprives GPIbα of its VWF-binding domain and bound VWF. It is tempting to speculate that VWF-glycans contribute to recognition of platelets by AMR.
Endogenous platelet clearance by αMβ2 and AMR
The liver is a major anatomic site of normal platelet clearance. Scavenger receptors on Kupffer cells, which represent ~15% of liver cells, contribute to ~15% of normal platelet clearance in humans [87]. The localisation of Kupffer cells within the liver sinusoids ensures that they are positioned to efficiently remove “unwanted” material from the circulatory system. We have shown that mice lacking the αMβ2 macrophage receptor have increased platelet counts, indicating that circulating platelets have exposed βGlcNAc while they circulate and are removed by this receptor. Liver sinusoidal endothelial cells mediate clearance via hyaluronan-receptor-, or mannose-receptor-mediated endocytosis [88–90]. However, if any of these receptors plays a role in endogenous platelet removal needs to be determined. Hepatocytes comprise the majority of cells within the liver (~60%) and are physically separated from the sinusoids by an endothelial cell barrier and the space of Disse [91]. However, fenestrations in the sinusoidal endothelial cells allow for interaction via hepatocyte microvilli protrusions into the lumen [92]. Hepatocytes are not generally considered as phagocytotic though they are capable of internalising particles ranging in sizes from a few nanometers up to 1.5 µm [93]. In addition, cooperation between liver cells allows hepatocytes to become more phagocytic when the amount of exogenous material exceeds the capacity of Kupffer cells [94] [95]. Furthermore, platelets have been observed to translocate to hepatocytes in response to cytokine signals from Kupffer cells [96].
Since the discovery of the AMR over 35 years ago, few endogenous ligands have been identified. Mice lacking either one of the two AMR subunits appear normal and surprisingly do not accumulate asialoglyco-proteins or -lipids in their circulation [97–99]. α2,3-linked sialic acid can mask underlying ASGPR ligands on glycoproteins. Recent studies revealed that endogenous ASGPR rapidly clear platelets and VWF with reduced α2,3-linked sialic acid during pathologic conditions of rapid desialylation, such as bloodstream infection with Streptococcus pneumoniae expressing sialidase (neuraminidase) activity. Removal of desialylated platelets by AMR retarded the onset of severe haematologic changes that are indicative of acute disseminated intravascular coagulation [50]. Bacterial contamination of platelet units stored for transfusion with Streptococcus pneumoniae and transfusion associated sepsis have been reported [100]. Platelet transfusion-associated sepsis is a major side effect of current transfusion practices. It is likely that, as in mice, bacteria-derived sialidases desialylate platelets during storage, a process that may initiate removal of stored platelets by AMR. Studies on the AMR and its ligands may provide new opportunities to explore the biology of coagulation in sepsis-induced disseminated intravascular coagulation. These studies may also prove useful to develop better platelet storage methods. Our studies of platelet binding and internalisation report the ability of hepatocytes to ingest platelets with glycoproteins bearing terminal galactose structures [7, 49, 50], an AMR-dependent mechanism in which both HL-1 and HL-2 subunits are necessary. Does the AMR play a role in removal of endogenous platelets in non-pathological states? Our recent novel studies evaluate whether platelets become desialylated while circulating and if the clearance mechanisms of endogenous platelets is mediated through the action of the AMR. It appears that the AMR plays a role in endogenous desialylated (senile) platelet removal. Studies on the AMR and its endogenous platelets ligands may provide new opportunities to explore novel regulatory mechanisms of platelet production and removal.
Conclusions
Transfused platelets lacking sialic acid or galactose on their membranes are recognised as “foreign” and are removed by lectins (β2-integrin and AMR) in the liver. Endogenous platelet homeostasis under pathologic and non-pathologic conditions is regulated at least in part by both lectin-mediated actions. The exact impact on endogenous platelet clearance is currently under investigation.
Acknowledgements
This work was supported by US National Institutes of Health grant PO1 HL056949 and grant HL089224.
References
- 1.Guidance for industry for platelet testing and evaluation of platelet substitute products [Internet] Rockville, MD: Center for Biologics Evaluation and Research (CBER); Food and Drug Administration; 1999. pp. 1–7. Available from: http//www.fda.gov/cber/gdlns/platelet.htm. [Google Scholar]
- 2.Murphy S, Rebulla P, Bertolini F, Holme S, Moroff G, Snyder E, Stromberg R. In vitro assessment of the quality of stored platelet concentrates. The BEST (Biomedical Excellence for Safer Transfusion) Task Force of the International Society of Blood Transfusion. Transfus Med Rev. 1994;8:29–36. doi: 10.1016/s0887-7963(94)70095-x. [DOI] [PubMed] [Google Scholar]
- 3.Ishida A, Handa M, Wakui M, Okamoto S, Kamakura M, Ikeda Y. Clinical factors influencing posttransfusion platelet increment in patients undergoing hematopoietic progenitor cell transplantation--a prospective analysis. Transfusion. 1998;38:839–847. doi: 10.1046/j.1537-2995.1998.38998409004.x. [DOI] [PubMed] [Google Scholar]
- 4.Rebulla P, Finazzi G, Marangoni F, Avvisati G, Gugliotta L, Tognoni G, Barbui T, Mandelli F, Sirchia G. The threshold for prophylactic platelet transfusions in adults with acute myeloid leukemia. Gruppo Italiano Malattie Ematologiche Maligne dell'Adulto. N Engl J Med. 1997;337:1870–1875. doi: 10.1056/NEJM199712253372602. [DOI] [PubMed] [Google Scholar]
- 5.Wandt H, Frank M, Ehninger G, Schneider C, Brack N, Daoud A, Fackler-Schwalbe I, Fischer J, Gackle R, Geer T, Harms P, Loffler B, Ohl S, Otremba B, Raab M, Schonrock-Nabulsi P, Strobel G, Winter R, Link H. Safety and cost effectiveness of a 10 x 10(9)/L trigger for prophylactic platelet transfusions compared with the traditional 20 x 10(9)/L trigger: a prospective comparative trial in 105 patients with acute myeloid leukemia. Blood. 1998;91:3601–3606. [PubMed] [Google Scholar]
- 6.Hoffmeister K, Felbinger T, Falet H, Denis C, Bergmeier W, Mayadas T, von Andrian U, Wagner D, Stossel T, Hartwig J. The clearance mechanism of chilled blood platelets. Cell. 2003;10:87–97. doi: 10.1016/s0092-8674(02)01253-9. [DOI] [PubMed] [Google Scholar]
- 7.Rumjantseva V, Grewal PK, Wandall HH, Josefsson EC, Sorensen AL, Larson G, Marth JD, Hartwig JH, Hoffmeister KM. Dual roles for hepatic lectin receptors in the clearance of chilled platelets. Nat Med. 2009;15:1273–1280. doi: 10.1038/nm.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becker G, Tuccelli M, Kunicki T, Chalos M, Aster R. Studies of platelet concentrates stored at 22°C and 4°C. Transfusion. 1973;13:61–68. doi: 10.1111/j.1537-2995.1973.tb05442.x. [DOI] [PubMed] [Google Scholar]
- 9.Valeri CR. Hemostatic effectiveness of liquid-preserved and previously frozen human platelets. N Engl J Med. 1974;290:353–358. doi: 10.1056/NEJM197402142900702. [DOI] [PubMed] [Google Scholar]
- 10.Silva VA, Miller WV. Platelet transfusion survey in a regional blood program. Transfusion. 1977;17:255–260. doi: 10.1046/j.1537-2995.1977.17377196361.x. [DOI] [PubMed] [Google Scholar]
- 11.Handin R, Valeri C. Hemostatic effectiveness of platelets stored at 22oC. N Engl J Med. 1971;285:538–543. doi: 10.1056/NEJM197109022851003. [DOI] [PubMed] [Google Scholar]
- 12.Kattlove HE. Platelet preservation--what temperature? A rationale for strategy. Transfusion. 1974;14:328–330. doi: 10.1111/j.1537-2995.1974.tb04540.x. [DOI] [PubMed] [Google Scholar]
- 13.Valeri CR. Circulation and hemostatic effectiveness of platelets stored at 4 C or 22 C: studies in aspirin-treated normal volunteers. Transfusion. 1976;16:20–23. doi: 10.1046/j.1537-2995.1976.16176130832.x. [DOI] [PubMed] [Google Scholar]
- 14.Slichter S, Harker L. Preparation and storage of platelet concentrates. II. Storage variables influencing platelet viability and function. Brit J Haemat. 1976;34:403–419. doi: 10.1111/j.1365-2141.1976.tb03587.x. [DOI] [PubMed] [Google Scholar]
- 15.Filip D, Aster R. Relative hemostatic effectiveness of human platelets stored at 4oC and 22oC. J Lab Clin Med. 1978;91:618–624. [PubMed] [Google Scholar]
- 16.Hartwig JH. The platelet: form and function. Semin Hematol. 2006;43:S94–S100. doi: 10.1053/j.seminhematol.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Rinder HM, Snyder EL. Activation of platelet concentrate during preparation and storage. Blood Cells. 1992;18:445–456. discussion 57–60. [PubMed] [Google Scholar]
- 18.Rinder HM, Murphy M, Mitchell JG, Stocks J, Ault KA, Hillman RS. Progressive platelet activation with storage: evidence for shortened survival of activated platelets after transfusion. Transfusion. 1991;31:409–414. doi: 10.1046/j.1537-2995.1991.31591263195.x. [DOI] [PubMed] [Google Scholar]
- 19.Rinder HM, Ault KA. Platelet activation and its detection during the preparation of platelets for transfusion. Transfus Med Rev. 1998;12:271–287. doi: 10.1016/s0887-7963(98)80003-5. [DOI] [PubMed] [Google Scholar]
- 20.Snyder EL. Release of beta-thromboglobulin during storage of platelet concentrates. Vox Sang. 1981;40(Suppl 1):115–116. [PubMed] [Google Scholar]
- 21.Perrotta PL, Perrotta CL, Snyder EL. Apoptotic activity in stored human platelets. Transfusion. 2003;43:526–535. doi: 10.1046/j.1537-2995.2003.00349.x. [DOI] [PubMed] [Google Scholar]
- 22.Zucker M, Borrelli J. Reversible alteration in platelet morphology produced by anticoagulants and by cold. Blood. 1954;28:524–534. [PubMed] [Google Scholar]
- 23.Babic AM, Josefsson EC, Bergmeier W, Wagner DD, Kaufman RM, Silberstein LE, Stossel TP, Hartwig JH, Hoffmeister KM. In vitro function and phagocytosis of galactosylated platelet concentrates after long-term refrigeration. Transfusion. 2007;47:442–451. doi: 10.1111/j.1537-2995.2007.01134.x. [DOI] [PubMed] [Google Scholar]
- 24.Sandgren P, Hansson M, Gulliksson H, Shanwell A. Storage of buffy-coat-derived platelets in additive solutions at 4 degrees C and 22 degrees C: flow cytometry analysis of platelet glycoprotein expression. Vox Sang. 2007;93:27–36. doi: 10.1111/j.1423-0410.2007.00912.x. [DOI] [PubMed] [Google Scholar]
- 25.Leytin V, Allen DJ, Gwozdz A, Garvey B, Freedman J. Role of platelet surface glycoprotein Ibalpha and P-selectin in the clearance of transfused platelet concentrates. Transfusion. 2004;44:1487–1495. doi: 10.1111/j.1537-2995.2004.04042.x. [DOI] [PubMed] [Google Scholar]
- 26.Rinder H, Murphy M, Mitchell J, Stocks J, Ault K, Hillman R. Progressive platelet activation with storage: evidence for shortened survival of activated platelets after transfusion. Transfusion. 1991;31:409–414. doi: 10.1046/j.1537-2995.1991.31591263195.x. [DOI] [PubMed] [Google Scholar]
- 27.Triulzi D, Kickler T, Braine H. Detection and significance of alpha granule membrane protein 140 expression on platelets collected by apheresis. Transfusion. 1992;32:529–533. doi: 10.1046/j.1537-2995.1992.32692367196.x. [DOI] [PubMed] [Google Scholar]
- 28.Berger G, Hartwell D, Wagner D. P-selectin and platelet clearance. Blood. 1998;92:4446–4452. [PubMed] [Google Scholar]
- 29.Michelson A, Barnard M, Hechtman H, MacGregor H, Connolly R, Loscalzo J, Valeri C. In vivo tracking of platelets: circulating degranulated platelets rapidly lose suface P-selectin but continue to circulate and function. Proc Natl Acad Sci, USA. 1996;93:11877–11882. doi: 10.1073/pnas.93.21.11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adelman B, Michelson A, Loscalzo J, Greenberg J, Handin R. Plasmin Effect on Platelet Glycoprotein Ib-von Willebrand Factor Interactions. Blood. 1985;65:32–40. [PubMed] [Google Scholar]
- 31.Reimers HJ, Kinlough-Rathbone RL, Cazenave JP, Senyi AF, Hirsh J, Packham MA, Mustard JF. In vitro and in vivo functions of thrombin-treated platelets. Thromb Haemost. 1976;35:151–166. [PubMed] [Google Scholar]
- 32.Italiano JJ, Bergmeier W, Tiwari S, Falet H, Hartwig J, Hoffmeister K, Andre P, Wagner D, Shivdasani R. Mechanisms and implications of platelet discoid shape. Blood. 2003;101:4789–4796. doi: 10.1182/blood-2002-11-3491. [DOI] [PubMed] [Google Scholar]
- 33.Valeri C, Ragno G, Marks P, Kuter D, Rosenberg R, Stossel T. Effect of thrombopoietin alone and a combination of cytochalasin B and ethylene glycol bis(beta-aminoethyl ether) N,N'-tetraacetic acid-AM on the survival and function of autologous baboon platelets stored at 4 degrees C for as long as 5 days. Transfusion. 2004;44:865–870. doi: 10.1111/j.1537-2995.2004.03326.x. [DOI] [PubMed] [Google Scholar]
- 34.Kim BK, Baldini MG. The platelet response to hypotonic shock. Its value as an indicator of platelet viability after storage. Transfusion. 1974;14:130–138. doi: 10.1111/j.1537-2995.1974.tb04504.x. [DOI] [PubMed] [Google Scholar]
- 35.Murphy S, Gardner FH. Room temperature storage of platelets. Transfusion. 1976;16:2–3. doi: 10.1046/j.1537-2995.1976.16176130831.x. [DOI] [PubMed] [Google Scholar]
- 36.Slichter SJ. In vitro measurements of platelet concentrates stored at 4 and 22 degree C: correlation with posttransfusion platelet viability and function. Vox Sang. 1981;40(Suppl 1):72–86. doi: 10.1111/j.1423-0410.1981.tb00741.x. [DOI] [PubMed] [Google Scholar]
- 37.Kile BT. The role of the intrinsic apoptosis pathway in platelet life and death. J Thromb Haemost. 2009;7(Suppl 1):214–217. doi: 10.1111/j.1538-7836.2009.03366.x. [DOI] [PubMed] [Google Scholar]
- 38.Bevers EM, Tilly RH, Senden JM, Comfurius P, Zwaal RF. Exposure of endogenous phosphatidylserine at the outer surface of stimulated platelets is reversed by restoration of aminophospholipid translocase activity. Biochemistry. 1989;28:2382–2387. doi: 10.1021/bi00432a007. [DOI] [PubMed] [Google Scholar]
- 39.Bevers EM, Comfurius P, van Rijn JL, Hemker HC, Zwaal RF. Generation of prothrombin-converting activity and the exposure of phosphatidylserine at the outer surface of platelets. Eur J Biochem. 1982;122:429–436. doi: 10.1111/j.1432-1033.1982.tb05898.x. [DOI] [PubMed] [Google Scholar]
- 40.Rosing J, Bevers EM, Comfurius P, Hemker HC, van Dieijen G, Weiss HJ, Zwaal RF. Impaired factor X and prothrombin activation associated with decreased phospholipid exposure in platelets from a patient with a bleeding disorder. Blood. 1985;65:1557–1561. [PubMed] [Google Scholar]
- 41.Wandall HH, Hoffmeister KM, Sorensen AL, Rumjantseva V, Clausen H, Hartwig JH, Slichter SJ. Galactosylation does not prevent the rapid clearance of long-term, 4 degrees C-stored platelets. Blood. 2008;111:3249–3256. doi: 10.1182/blood-2007-06-097295. [DOI] [PubMed] [Google Scholar]
- 42.Murphy S. Metabolic patterns of platelets--impact on storage for transfusion. Vox Sang. 1994;67(Suppl 3):271–273. doi: 10.1111/j.1423-0410.1994.tb04592.x. [DOI] [PubMed] [Google Scholar]
- 43.Slichter SJ, Harker LA. Preparation and storage of platelet concentrates. II. Storage variables influencing platelet viability and function. Br J Haematol. 1976;34:403–419. doi: 10.1111/j.1365-2141.1976.tb03587.x. [DOI] [PubMed] [Google Scholar]
- 44.Murphy S, Gardner FH. Platelet storage at 22 degrees C; metabolic, morphologic, and functional studies. J Clin Invest. 1971;50:370–377. doi: 10.1172/JCI106504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crook M. Platelet sialic Acid and its significance to platelet life-spans. Platelets. 1990;1:167. doi: 10.3109/09537109009005484. [DOI] [PubMed] [Google Scholar]
- 46.Crook M. Sialic Acid: its importance to platelet function in health and disease. Platelets. 1991;2:1–10. doi: 10.3109/09537109109005496. [DOI] [PubMed] [Google Scholar]
- 47.Steiner M, Vancura S. Asymmetrical loss of sialic acid from membrane glycoproteins during platelet aging. Thromb Res. 1985;40:465–471. doi: 10.1016/0049-3848(85)90283-x. [DOI] [PubMed] [Google Scholar]
- 48.Reimers HJ, Greenberg J, Cazenave JP, Packham MA, Mustard JF. Experimental modification of platelet survival. Adv Exp Med Biol. 1977;82:231–233. doi: 10.1007/978-1-4613-4220-5_48. [DOI] [PubMed] [Google Scholar]
- 49.Sorensen AL, Rumjantseva V, Nayeb-Hashemi S, Clausen H, Hartwig JH, Wandall HH, Hoffmeister KM. Role of sialic acid for platelet life span: exposure of beta-galactose results in the rapid clearance of platelets from the circulation by asialoglycoprotein receptor-expressing liver macrophages and hepatocytes. Blood. 2009;114:1645–1654. doi: 10.1182/blood-2009-01-199414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grewal PK, Uchiyama S, Ditto D, Varki N, Le DT, Nizet V, Marth JD. The Ashwell receptor mitigates the lethal coagulopathy of sepsis. Nat Med. 2008;14(6):648–655. doi: 10.1038/nm1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kotze H, van Wyk V, Badenhorst P, Heyns A, Roodt J, Lotter M. Influence of platelet membrane sialic acid and platelet-associated IgG on ageing and sequestration of blood platelets in baboons. Thromb Haemost. 1993;18:676–680. [PubMed] [Google Scholar]
- 52.Greenberg J, Packham M, Cazenave J, Reimers H, Mustard J. Effects on platelet function of removal of platelet sialic acid by neuraminidase. Lab Invest. 1975;32:476–484. [PubMed] [Google Scholar]
- 53.Soslau G, Giles J. The loss of sialic acid and its prevention in stored human platelets. Thromb Res. 1982;26:443–455. doi: 10.1016/0049-3848(82)90316-4. [DOI] [PubMed] [Google Scholar]
- 54.Tribulatti MV, Mucci J, Van Rooijen N, Leguizamon MS, Campetella O. The trans-sialidase from Trypanosoma cruzi induces thrombocytopenia during acute Chagas' disease by reducing the platelet sialic acid contents. Infec. Immun. 2005;73:201–207. doi: 10.1128/IAI.73.1.201-207.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Springer T. Traffic signals for lymphocyte recirculation and leucocyte emigration: The multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 56.Petty HR, Todd RF., III Receptor-receptor interactions of complement receptor type 3 in neutrophil membranes. J Leukoc Biol. 1993;54:492–494. doi: 10.1002/jlb.54.5.492. [DOI] [PubMed] [Google Scholar]
- 57.Xia Y, Borland G, Huang J, Mizukami I, Petty H, Todd Rr, Ross G. Function of the lectin domain of Mac-1/complement receptor type 3 (CD11b/CD18) in regulating neutrophil adhesion. J Immunol. 2002;169:6417–6426. doi: 10.4049/jimmunol.169.11.6417. [DOI] [PubMed] [Google Scholar]
- 58.Brown E, Hogg N. Where the outside meets the inside: integrins as activators and targets. Immunol Lett. 1996;54:189–193. doi: 10.1016/s0165-2478(96)02671-5. [DOI] [PubMed] [Google Scholar]
- 59.Newton R, Thiel M, Hogg N. Signaling mechanisms and the activation of leukocyte integrins. J Leukoc Biol. 1997;61:422–426. doi: 10.1002/jlb.61.4.422. [DOI] [PubMed] [Google Scholar]
- 60.Diamond M, Garcia-Aguilar J, Bickford J, Corbi A, Springer T. The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct adhesion ligands. J Cell Biol. 1993;120:1031–1043. doi: 10.1083/jcb.120.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Santoso S, Sachs U, Kroll H, Linder M, Ruf A, Preissner K, Chavakis T. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J Exp Med. 2002;196:679–691. doi: 10.1084/jem.20020267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Josefsson E, Gebhard H, Stossel T, Hartwig J, Hoffmeister K. The macrophage alphaMbeta2 integrin alphaM lectin domain mediates the phagocytosis of chilled platelets. J Biol Chem. 2005;280:18025–18032. doi: 10.1074/jbc.M501178200. [DOI] [PubMed] [Google Scholar]
- 63.Simon D, Chen Z, Xu H, Li C, Dong J-f, McIntire L, Ballantyne C, Zhang L, Furman M, Berndt M, Lopez J. Platelet glycoprotein Iba is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18) J Exp Med. 2000;192:193–204. doi: 10.1084/jem.192.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thornton B, Vetvicka V, Pitman M, Glodman R, Ross G. Analysis of the sugar specifty and molecular location of the b-glucan-binding lectin site of compement receptor type 3 (CD11b/D18) J Immonol. 1996;156:1235–1246. [PubMed] [Google Scholar]
- 65.Ross GD. Role of the lectin domain of Mac-1/CR3 (CD11b/CD18) in regulating intercellular adhesion. Immunol Res. 2002;25:219–227. doi: 10.1385/IR:25:3:219. [DOI] [PubMed] [Google Scholar]
- 66.Cain J, Newman S, Ross G. Role of complement receptor type three and serum opsonins in the neutrophil response to yeast. Complement. 1987;4:75–86. doi: 10.1159/000463011. [DOI] [PubMed] [Google Scholar]
- 67.Ross GD. Regulation of the adhesion versus cytotoxic functions of the Mac-1/CR3/alphaMbeta2-integrin glycoprotein. Crit Rev Immunol. 2000;20:197–222. [PubMed] [Google Scholar]
- 68.Hoffmeister K, Josefsson E, Isaac N, Clausen H, Hartwig J, Stossel T. Glycosylation restores survival of chilled blood platelets. Science. 2003;301:1531–1534. doi: 10.1126/science.1085322. [DOI] [PubMed] [Google Scholar]
- 69.Badlou B, Spierenburg G, Ulrichts H, Deckmy H, Smid W, Akkerman J. Role of glycoprotein Ibalpha in phagocytosis of platelets by macrophages. Transfusion. 2006;46:2090–2099. doi: 10.1111/j.1537-2995.2006.01034.x. [DOI] [PubMed] [Google Scholar]
- 70.Pricer WE, Jr, Hudgin RL, Ashwell G, Stockert RJ, Morell AG. A membrane receptor protein for asialoglycoproteins. Methods Enzymol. 1974;34:688–691. doi: 10.1016/s0076-6879(74)34090-6. [DOI] [PubMed] [Google Scholar]
- 71.Morell AG, Gregoriadis G, Scheinberg IH, Hickman J, Ashwell G. The role of sialic acid in determining the survival of glycoproteins in the circulation. J Biol Chem. 1971;246:1461–1467. [PubMed] [Google Scholar]
- 72.Hudgin RL, Pricer WE, Jr, Ashwell G, Stockert RJ, Morell AG. The isolation and properties of a rabbit liver binding protein specific for asialoglycoproteins. J Biol Chem. 1974;249:5536–5543. [PubMed] [Google Scholar]
- 73.Grewal PK. The Ashwell-Morell receptor. Methods Enzymol. 479:223–241. doi: 10.1016/S0076-6879(10)79013-3. [DOI] [PubMed] [Google Scholar]
- 74.Monroe RS, Huber BE. The major form of the murine asialoglycoprotein receptor: cDNA sequence and expression in liver, testis and epididymis. Gene. 1994;148:237–244. doi: 10.1016/0378-1119(94)90694-7. [DOI] [PubMed] [Google Scholar]
- 75.Park JH, Kim KL, Cho EW. Detection of surface asialoglycoprotein receptor expression in hepatic and extra-hepatic cells using a novel monoclonal antibody. Biotechnol Lett. 2006;28:1061–1069. doi: 10.1007/s10529-006-9064-0. [DOI] [PubMed] [Google Scholar]
- 76.Zalik SE, Thomson LW, Ledsham IM. Expression of an endogenous galactose-binding lectin in the early chick embryo. J Cell Sci. 1987;88(Pt 4):483–493. doi: 10.1242/jcs.88.4.483. [DOI] [PubMed] [Google Scholar]
- 77.Park EI, Mi Y, Unverzagt C, Gabius HJ, Baenziger JU. The asialoglycoprotein receptor clears glycoconjugates terminating with sialic acid alpha 2,6GalNAc. Proc Natl Acad Sci U S A. 2005;102:17125–17129. doi: 10.1073/pnas.0508537102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rensen P, Sliedregt L, Ferns M, Kieviet E, Rossenberg Sv, Leeuwen Sv, Berkel Tv, Biessen E. Detemination of the upper size limit for uptake and processing of ligands by the asialoglycoprotein receptor on hepacytes in vitro and in vivo. J Biol Chem. 2001;276:37577–37584. doi: 10.1074/jbc.M101786200. [DOI] [PubMed] [Google Scholar]
- 79.Van der Smissen P, Vael T, Courtoy PJ, Baudhuin P. Ligand-induced clustering of asialoglycoprotein receptors on rat hepatocytes at 4 degrees C. Eur J Cell Biol. 1993;60:122–130. [PubMed] [Google Scholar]
- 80.Wahrenbrock M, Varki A. Multiple hepatic receptors cooperate to eliminate secretory mucins aberrantly entering the bloodstream: are circulating carcer mucins the "tip of the iceberg"? Cancer Res. 2006;66:2433–2441. doi: 10.1158/0008-5472.CAN-05-3851. [DOI] [PubMed] [Google Scholar]
- 81.Lee YC, Townsend RR, Hardy MR, Lonngren J, Arnarp J, Haraldsson M, Lonn H. Binding of synthetic oligosaccharides to the hepatic Gal/GalNAc lectin. Dependence on fine structural features. J Biol Chem. 1983;258:199–202. [PubMed] [Google Scholar]
- 82.Drickamer K. C-type lectin-like domains. Curr Opin Struct Biol. 1999;9:585–590. doi: 10.1016/s0959-440x(99)00009-3. [DOI] [PubMed] [Google Scholar]
- 83.Valeri C, Giorgio A, Macgregor H, Ragno G. Circulation and distribution of autotransfused fresh, liquid-preserved and cryopreserved baboon platelets. Vox Sang. 2002;83:347–351. doi: 10.1046/j.1423-0410.2002.00229.x. [DOI] [PubMed] [Google Scholar]
- 84.Tsuji T, Osawa T. The carbohydrate moiety of human platelet glycocalicin: the structures of the major Asn-linked sugar chains. J Biochem. 1987;101:241–249. doi: 10.1093/oxfordjournals.jbchem.a121897. [DOI] [PubMed] [Google Scholar]
- 85.Berndt M, Gregory C, Kabral A, Zola H, Fournier D, Castaldi P. Purification and preliminary characterization of the glycoprotein Ib complex in the human platelet membrane. Eur J Biochem. 1985;151:637–649. doi: 10.1111/j.1432-1033.1985.tb09152.x. [DOI] [PubMed] [Google Scholar]
- 86.Ellies L, Ditto D, Levy G, Wahrenbrock M, Ginsburg D, Varki A, Le D, Marth J. Sialyltransferase ST3Gal-IV operates as a dominant modifier of hemostasis by concealing asialoglycoprotein receptor ligands. Proc Natl Acad Sci U S A. 2002;99:10042–10047. doi: 10.1073/pnas.142005099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stratton JR, Ballem PJ, Gernsheimer T, Cerqueira M, Slichter SJ. Platelet destruction in autoimmune thrombocytopenic purpura: kinetics and clearance of indium-111-labeled autologous platelets. J Nucl Med. 1989;30:629–637. [PubMed] [Google Scholar]
- 88.Zhou B, Weigel JA, Fauss L, Weigel PH. Identification of the hyaluronan receptor for endocytosis (HARE) J Biol Chem. 2000;275:37733–37741. doi: 10.1074/jbc.M003030200. [DOI] [PubMed] [Google Scholar]
- 89.Malovic I, Sorensen KK, Elvevold KH, Nedredal GI, Paulsen S, Erofeev AV, Smedsrod BH, McCourt PA. The mannose receptor on murine liver sinusoidal endothelial cells is the main denatured collagen clearance receptor. Hepatology. 2007;45:1454–1461. doi: 10.1002/hep.21639. [DOI] [PubMed] [Google Scholar]
- 90.Elvevold K, Simon-Santamaria J, Hasvold H, McCourt P, Smedsrod B, Sorensen KK. Liver sinusoidal endothelial cells depend on mannose receptor-mediated recruitment of lysosomal enzymes for normal degradation capacity. Hepatology. 2008;48:2007–2015. doi: 10.1002/hep.22527. [DOI] [PubMed] [Google Scholar]
- 91.Wisse E, De Zanger RB, Charels K, Van Der Smissen P, McCuskey RS. The liver sieve: considerations concerning the structure and function of endothelial fenestrae, the sinusoidal wall and the space of Disse. Hepatology. 1985;5:683–692. doi: 10.1002/hep.1840050427. [DOI] [PubMed] [Google Scholar]
- 92.Warren A, Le Couteur DG, Fraser R, Bowen DG, McCaughan GW, Bertolino P. T lymphocytes interact with hepatocytes through fenestrations in murine liver sinusoidal endothelial cells. Hepatology. 2006;44:1182–1190. doi: 10.1002/hep.21378. [DOI] [PubMed] [Google Scholar]
- 93.Soji T, Murata Y, Ohira A, Nishizono H, Tanaka M, Herbert DC. Evidence that hepatocytes can phagocytize exogenous substances. Anat Rec. 1992;233:543–546. doi: 10.1002/ar.1092330408. [DOI] [PubMed] [Google Scholar]
- 94.Kmiec Z. Cooperation of liver cells in health and disease. Adv Anat Embryol Cell Biol. 2001;161:III–XIII. 1–151. doi: 10.1007/978-3-642-56553-3. [DOI] [PubMed] [Google Scholar]
- 95.Malarkey DE, Johnson K, Ryan L, Boorman G, Maronpot RR. New insights into functional aspects of liver morphology. Toxicol Pathol. 2005;33:27–34. doi: 10.1080/01926230590881826. [DOI] [PubMed] [Google Scholar]
- 96.Nakamura M, Shibazaki M, Nitta Y, Endo Y. Translocation of platelets into Disse spaces and their entry into hepatocytes in response to lipopolysaccharides, interleukin-1 and tumour necrosis factor: the role of Kupffer cells. J Hepatol. 1998;28:991–999. doi: 10.1016/s0168-8278(98)80348-6. [DOI] [PubMed] [Google Scholar]
- 97.Ishibashi S, Hammer RE, Herz J. Asialoglycoprotein receptor deficiency in mice lacking the minor receptor subunit. J Biol Chem. 1994;269:27803–27806. [PubMed] [Google Scholar]
- 98.Braun JR, Willnow TE, Ishibashi S, Ashwell G, Herz J. The major subunit of the asialoglycoprotein receptor is expressed on the hepatocellular surface in mice lacking the minor receptor subunit. J Biol Chem. 1996;271:21160–21166. doi: 10.1074/jbc.271.35.21160. [DOI] [PubMed] [Google Scholar]
- 99.Tozawa R, Ishibashi S, Osuga J, Yamamoto K, Yagyu H, Ohashi K, Tamura Y, Yahagi N, Iizuka Y, Okazaki H, Harada K, Gotoda T, Shimano H, Kimura S, Nagai R, Yamada N. Asialoglycoprotein receptor deficiency in mice lacking the major receptor subunit. Its obligate requirement for the stable expression of oligomeric receptor. J Biol Chem. 2001;276:12624–12628. doi: 10.1074/jbc.M011063200. [DOI] [PubMed] [Google Scholar]
- 100.Katayama T, Kamiya M, Hoshina S, Masuoka H, Nishiwaki K, Sano K, Hagino T, Kobayashi M. [Fatal septic shock and rhabdomyolysis following transfusion of platelet concentrates contaminated with Streptococcus pneumoniae] Rinsho Ketsueki. 2003;44:381–385. [PubMed] [Google Scholar]