

Abstract
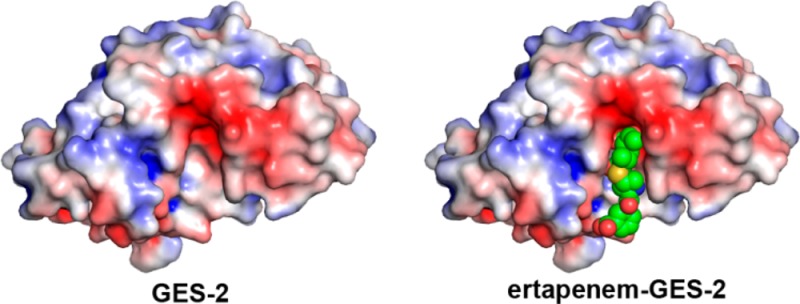
Carbapenems are the last resort antibiotics for treatment of life-threatening infections. The GES β-lactamases are important contributors to carbapenem resistance in clinical bacterial pathogens. A single amino acid difference at position 170 of the GES-1, GES-2, and GES-5 enzymes is responsible for the expansion of their substrate profile to include carbapenem antibiotics. This highlights the increasing need to understand the mechanisms by which the GES β-lactamases function to aid in development of novel therapeutics. We demonstrate that the catalytic efficiency of the enzymes with carbapenems meropenem, ertapenem, and doripenem progressively increases (100-fold) from GES-1 to -5, mainly due to an increase in the rate of acylation. The data reveal that while acylation is rate limiting for GES-1 and GES-2 for all three carbapenems, acylation and deacylation are indistinguishable for GES-5. The ertapenem–GES-2 crystal structure shows that only the core structure of the antibiotic interacts with the active site of the GES-2 β-lactamase. The identical core structures of ertapenem, doripenem, and meropenem are likely responsible for the observed similarities in the kinetics with these carbapenems. The lack of a methyl group in the core structure of imipenem may provide a structural rationale for the increase in turnover of this carbapenem by the GES β-lactamases. Our data also show that in GES-2 an extensive hydrogen-bonding network between the acyl-enzyme complex and the active site water attenuates activation of this water molecule, which results in poor deacylation by this enzyme.
Production of β-lactamases constitutes the major mechanism of resistance to β-lactam antibiotics in Gram-negative bacteria. These enzymes attack and cleave the β-lactam ring of the antibiotics before the drugs can reach their intended targets, the penicillin binding proteins.1 The first β-lactamases discovered had relatively narrow-spectrum activity and were capable of hydrolyzing penicillins and some narrow-spectrum cephalosporins.2 Introduction of expanded-spectrum cephalosporins into clinical use resulted in the selection of extended-spectrum β-lactamase (ESBLs), enzymes capable of hydrolyzing these antibiotics.2 To combat bacterial infections caused by the ESBL-producing pathogens, the carbapenem subclass of β-lactam antibiotics was developed. To withstand this new challenge, bacteria have acquired (from mostly unidentified sources) carbapenemases, novel β-lactamases capable of destroying carbapenems.3 Production of carbapenemases by bacterial pathogens in conjunction with other resistance mechanisms, such as porin deficiency or upregulation of efflux pumps, is especially worrisome as carbapenems are used as the last resort drugs for treatment of life-threatening infections caused by antibiotic-resistant pathogens.4
β-Lactamases are divided into four classes; those of classes A, C, and D are active site serine enzymes, while those of class B are zinc-dependent.5 Class A is the largest of the four and includes a variety of enzyme families such as TEM, SHV, GES, and KPC, among others.5 The active site of class A β-lactamases is composed of several highly conserved residues, including Ser-70, Lys-73, Ser-130, Asn-132, and Glu-166 (ABL numbering).5,6 The catalytic Ser-70 attacks the β-lactam ring of the antibiotic to yield an acyl-enzyme intermediate.5,7 Subsequently, Glu-166 acts as a general base to activate a water molecule for deacylation of the enzyme.4 In addition, all known class A carbapenemases contain the invariant Cys-69 and Cys-238 amino acid residues that form a disulfide bond thought to be important for the stability and carbapenemase activity of these enzymes.5,8
GES-1, the first representative of the GES family of enzymes, was identified in a clinical strain of Klebsiella pneumoniae in 1998.9 GES-1 was found to confer resistance to penicillins and broad-spectrum cephalosporins, but not to monobactams or carbapenems.9 A few years later GES-5, a single amino acid derivative of GES-1, was shown to confer resistance to carbapenem antibiotics in clinical bacterial isolates.10−12 In following years, the number of GES enzymes identified has risen to 24 variants, increasing more than 89% since 2005 (http://lahey.org/studies/other.asp).3 Many of these GES variants exhibit significant carbapenemase activity and are increasingly being identified in a variety of Gram-negative bacteria, including Pseudomonas aeruginosa, Enterobacter cloacae, K. pneumoniae, Kluyvera intermedia, and Acinetobacter baumannii.13−16
The GES-1, -2, and -5 enzymes differ by only one amino acid at position 170. This residue is usually a highly conserved Asn in class A β-lactamases. GES-2 indeed does contain an Asn at position 170; however, in GES-1 and -5, this residue is a Gly and Ser, respectively. Whereas GES-1 and -2 are considered ESBLs, GES-5 is a carbapenemase due to its increased catalytic activity toward imipenem and ability to confer imipenem resistance in clinically important bacterial pathogens.17 To gain insights into the differences in the carbapenemase activity of the GES β-lactamases, we examined the steady-state and presteady-state kinetics for GES-1, -2, and -5 with imipenem.17 Subsequent to the introduction of this first carbapenem into clinical practice, novel carbapenems have been designed to expand the arsenal of these last resort antibiotics. To determine whether the structural differences between various carbapenems impact their interaction with the GES family of β-lactamases, we performed a comprehensive kinetic analysis of the GES-1, -2, and -5 enzymes with three carbapenem antibiotics, meropenem, doripenem, and ertapenem, and determined the X-ray structure of GES-2 in complex with ertapenem.
Experimental Procedures
Protein Expression and Purification
The vectors for expression of the GES enzymes were described previously.17 GES-1, -2, and -5 were expressed in Escherichia coli BL21(DE3) cells using the T7 expression system.18 Briefly, bacteria were grown in 500 mL of LB broth supplemented with kanamycin (60 μg/mL) at 37 °C, at 180 rpm to OD600 = 0.4. Protein overexpression was induced by addition of isopropyl-β-d-1-thigalactopyranoside (1 mM). After an additional 24 h growth at 25 °C, the media were collected and concentrated in Centricon-70 tubes with a 10 kDa molecular weight cut off (Millipore). Next, the media were dialyzed against 20 mM Tris, pH 7.5 at 4 °C and applied to a DEAE (Bio-Rad) column. The protein was eluted with a linear NaCl gradient (0–1 M) in 20 mM Tris, pH 7.5. Subsequently, after buffer exchange into 20 mM MES, pH 5.5, the protein was purified further using a High S (Bio-Rad) column. Elution of the protein was achieved using a linear NaCl gradient (0–1 M) in 20 mM MES, pH 5.5. Finally, fractions containing GES were combined and dialyzed against 50 mM NaPi, 100 mM NaCl, pH 7, and stored at 4 °C. The purity of the proteins was verified by SDS-PAGE. The protein concentration was determined by using the absorbance at 280 nm and ε = 24 075 M–1 cm–1.
Data Collection and Analysis
Kinetic data were monitored using either a Cary 60 spectrophotometer (Agilent) or an SFM-300 stopped-flow instrument (Bio-Logic) at 22 °C. Nonlinear regression was performed with Prism 6 (GraphPad Software, Inc.) using data from at least three different measurements.
Determination of Steady-State Kinetic Parameters
Reactions containing increasing concentrations of carbapenem (5–50 μM) in 50 mM NaPi, 100 mM NaCl, pH 7.0 were initiated by mixing with GES-1, -2, or -5 under multiple concentrations (0.001–5 μM). The change in absorbance associated with the opening of the β-lactam ring was monitored at the following wavelengths: ertapenem (λ = 295 nm and Δε = −10 940 cm–1 M–1), doripenem (λ = 299 nm and Δε = −11 540 cm–1 M–1), and meropenem (λ = 298 nm and Δε = −7200 cm–1 M–1). The linear portion of each reaction’s time course was used to determine the steady-state velocities (v). The steady-state parameters, kcat and Km, were calculated by plotting the observed rate constants (kobs = v/[E]) as a function of substrate concentration and fitting the data nonlinearly using the Michaelis–Menten equation.
Determination of Dissociation Constants
To determine the dissociation constant, Ks, the carbapenems were treated as inhibitors, while the chromogenic cephalosporin nitrocefin (λ = 500 nm and Δε = +15 900 cm–1 M–1) was used as a competing substrate. The data were analyzed using the Morrison equation (eq 1),
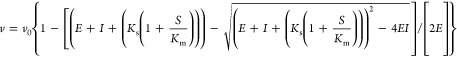 |
1 |
where v is defined as the steady-state velocity in the presence of inhibitor, v0 is the steady-state velocity in the absence of inhibitor, E is the concentration of enzyme, I is the concentration of inhibitor, Ks is the dissociation constant for the inhibitor, S is the concentration of nitrocefin, and Km is the Michaelis constant for nitrocefin. Reactions containing 50 mM NaPi, 100 mM NaCl, pH 7.0, varying amounts of inhibitor and 100 μM nitrocefin were started by mixing with GES-1, -2, or -5 (2, 100, or 1 nM, respectively). The change in absorbance was monitored for nitrocefin hydrolysis. The linear portion of each reaction was used to determine the steady-state velocities.
Determination of the Microscopic Rate Constants Describing Acylation and Deacylation
Single turnover conditions were utilized to determine the acylation rate constant (k2) for GES-1, -2, and -5 with meropenem, ertapenem, doripenem, and imipenem (λ = 297 nm and Δε = −10 930 cm–1 M–1). Reactions containing 50 mM NaPi, 100 mM NaCl, pH 7.0 and 5–10 μM carbapenem were initiated by the addition of increasing concentrations of enzyme (25–250 μM). In all cases, acylation of the GES enzymes was biphasic and data were fit with eq 2,
| 2 |
where At is defined as the absorbance at time t, A0 is the starting absorbance, A∞ is the final absorbance, Ffast is the fraction of the fast phase of the reaction, kfast is the fast phase first-order rate constant and kslow is the slow phase first-order rate constant.
The deacylation rate constant (k3) was determined using the enzyme reactivation method.19 Each GES enzyme was first incubated with an excess of carbapenem (10 × Km) in 50 mM NaPi, 100 mM NaCl, pH 7.0 for 0.5–20 min at 22 °C. Subsequently, the mixtures were diluted 50–1000-fold into buffer containing either nitrocefin (800 μM for GES-1 and 50 μM for GES-2) or cephalothin (300 μM for GES-5) to yield a final concentration of these reporter substrates 5–10-fold above their Km values. The final concentrations of the GES enzymes in the reaction mixture were 5 nM for GES-1 and GES-2 and 20 nM for GES-5. Incubation periods and dilution ratios were varied to ensure full recovery of enzyme activity. Control reactions were performed using the same conditions as described above but in the absence of carbapenem. The rate of enzyme reactivation was measured at either 500 nm (nitrocefin) or 284 nm (cephalothin). The k3 values were determined by fitting the data with eq 3 using the absorbance versus time traces,
| 3 |
where At, A0, and t are as defined in eq 2, vs is the steady-state velocity, and k3 is the rate constant for deacylation.
Calculation of the Microscopic Rate Constants Describing Carbapenem Binding and Release
The following relationships are derived from Scheme 1.
Scheme 1. Minimal Enzymatic Reaction for Class A β-Lactamases.
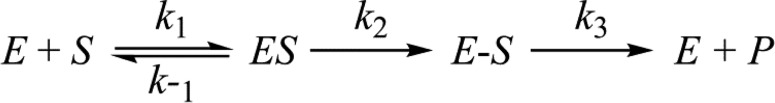
| 4 |
| 5 |
| 6 |
| 7 |
The rate constants (k1 and k–1) were calculated using the empirically determined parameters for kcat, Km, Ks, k2,slow, and k3 using the relationships described in eqs 4–7.
Protein Crystallization and Preparation of the Ertapenem-GES-2 Complex
GES-2 was crystallized as previously described.20 Soaking experiments were carried out at 4 °C. Ertapenem (Figure 1) was dissolved in crystallization buffer to a final concentration of 50 mM, and the GES-2 crystals were soaked in this solution for varying times (30 s, 1 min, 2 and 5 min) prior to passing them through a cryoprotectant solution comprised of crystallization buffer plus 30% ethylene glycol and flash cooling in liquid nitrogen. The ertapenem-soaked GES-2 crystals were subsequently screened for diffraction quality using the high-throughput Stanford Automated Mounter robotic system21 on beamline BL7-1 at the Stanford Synchrotron Radiation Lightsource.
Figure 1.

Chemical structures of the carbapenems ertapenem, meropenem, imipenem, and doripenem. The core and tail regions are indicted, as well as functional groups referred to in the text.
Diffraction Data Collection and Processing
The GES-2 crystals grown under these conditions belonged to space group P21 with cell dimensions a = 42.94 Å, b = 81.47 Å, c = 71.99 Å, β = 101.9°, and diffracted to 1.4 Å resolution. The Matthews coefficient22 assuming two molecules in the asymmetric unit, is 2.1 Å3/Da (42% solvent content). A complete data set comprising 360 images with an oscillation angle of 0.5° was collected from a single ertapenem-GES-2 crystal on SSRL beamline BL9-2, using a Rayonix MX325 detector with X-rays at 12658 eV (0.97945 Å). The images were processed with XDS23 and scaled and merged with SCALA.24 Additional data collection statistics are given in Table 1.
Table 1. Data Collection and Refinement Statistics for Ertapenem-GES-2.
| Data collection | |
| resolution (Å) | 37.7–1.40 |
| reflections, observed/unique | 350293/94348 |
| Rmergea (%) | 6.0 (67.9) |
| I/σI | 13.4 (2.3) |
| completeness (%) | 99.6 (98.1) |
| CC1/2b | 99.8 (64.4) |
| multiplicity | 3.7 |
| Wilson B (Å2) | 13.5 |
| Refinement | |
| Rwork/Rfree/Rallc | 0.158/0.197/0.160 |
| protein atoms/solvent atoms | 4167/486 |
| average B – protein (A/B) (Å2) | 15.7/15.2 |
| – solvent (Å2) | 28.5 |
| – ertapenem (A/B) (Å2) | 33.3/34.9 |
| rmsd bonds (Å) | 0.006 |
| rmsd angles (deg) | 1.22 |
| Ramachandran plot, residues in allowed regions (%) | 98.4 |
Numbers in Parentheses Relate to the Highest Resolution Shell, 1.45–1.40 Å
Rmerge = Σ(Ii – <I>)/ΣIi where Ii = the observed intensity of a given reflection and <I> is the mean intensity for all observations of that reflection.
The correlation between two random half-sets of data.
R = Σ||Fo| – |Fc||/|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes. Rall is calculated using all unique reflections.
Structure Solution and Refinement
The ertapenem–GES-2 structure was solved by molecular replacement (MR) with the program MOLREP25 using the apo-GES-2 structure previously reported20 as the starting model. Prior to the MR calculation, the side chain of residue 170 was truncated to glycine, all water molecules were removed from the apo-GES-2 model, and the atomic displacement parameters were set to a uniform value of 20.0 Å2. The MR solutions for both structures were initially refined using REFMAC,26 and subsequent rounds of refinement used PHENIX27 and manual model building with COOT.28 The final ertapenem–GES-2 structure, refined at 1.4 Å resolution to a Rwork and Rfree of 0.158 and 0.197, respectively, contains 533 residues (A, 268 residues, and B, 265 residues), 486 water molecules, 6 iodide ions, and 2 ertapenem molecules. The atomic coordinates for the crystal structure of the ertapenem-GES-2 complex have been deposited in the Protein Data Bank (PDB accession code 4QU3), along with the observed structure factors. Final refinement statistics are given in Table 1.
Results and Discussion
Steady-State Kinetics
We determined the steady-state kinetic parameters for GES-1, -2, and -5 for meropenem, ertapenem, and doripenem. Examining the measured Km values reveals that saturation of GES-1, -2, and -5 for turnover of these carbapenems is achieved at low micromolar concentrations (Table 2), similar to those reported for turnover of imipenem by these enzymes.17 When the three β-lactamases are compared, the Km values for GES-2 are 2–5-fold higher than those for GES-1 and -5. Overall, Km values are similar for the three GES enzymes, an indication that the apparent affinity does not contribute significantly to the observed 10–100 fold differences in the catalytic efficiencies (kcat/Km) of the GES β-lactamases.
Table 2. Steady-State Kinetic Parameters for the Hydrolysis of Carbapenems by GES Enzymes.
| enzyme | parameter | meropenem | ertapenem | doripenem |
|---|---|---|---|---|
| GES-1 | kcat (s–1) | 0.00094 ± 0.00004 | 0.00076 ± 0.00002 | 0.00063 ± 0.00003 |
| Km (μM) | 0.9 ± 0.5 | 2.2 ± 0.4 | 1.0 ± 0.7 | |
| kcat/Km (M–1 s–1) | (1.0 ± 0.6) × 103 | (3.5 ± 0.6) × 102 | (6.3 ± 4.4) × 102 | |
| Ks (μM) | 0.34 ± 0.04 | 0.25 ± 0.03 | 0.18 ± 0.02 | |
| GES-2 | kcat (s–1) | 0.0033 ± 0.0002 | 0.0062 ± 0.0002 | 0.0024 ± 0.0002 |
| Km (μM) | 4.1 ± 0.8 | 4.0 ± 0.6 | 5 ± 1 | |
| kcat/Km (M–1 s–1) | (8.1 ± 1.6) × 102 | (1.6 ± 0.2) × 103 | (4.4 ± 1.2) × 102 | |
| Ks (μM) | 1.2 ± 0.1 | 1.7 ± 0.2 | 1.0 ± 0.1 | |
| GES-5 | kcat (s–1) | 0.076 ± 0.004 | 0.053 ± 0.002 | 0.046 ± 0.001 |
| Km (μM) | 1.0 ± 0.7 | 2.0 ± 0.6 | 0.9 ± 0.2 | |
| kcat/Km (M–1 s–1) | (7.6 ± 5.3) × 104 | (2.7 ± 0.8) × 104 | (5 ± 1) × 104 | |
| Ks (μM) | 0.32 ± 0.08 | 0.25 ± 0.03 | 0.25 ± 0.03 |
Indeed, in all cases, the dissociation constants (Ks) for the noncovalent enzyme–carbapenem complex were 2–10-fold lower than the Km values and not significantly different from one another (Table 2), indicating that acylation (k2) proceeds faster than dissociation of the substrates from the ES complex (k–1). This was also observed for turnover of imipenem by GES-1, -2, and -5.17 GES-2 has 4–8-fold higher Ks values, indicating weaker binding of the carbapenems than observed with GES-1 and -5. These results indicate that substitution of the conserved Asn-170 in GES-2 with Gly or Ser as in GES-1 and GES-5, respectively, increases the affinity of the enzyme for meropenem, ertapenem, and doripenem. In contrast, this trend was not observed for imipenem, where GES-5 exhibited a weaker affinity for this carbapenem than GES-2.17
Despite the high affinity of the GES β-lactamases for the carbapenems, the enzymes have relatively poor catalytic efficiency for these substrates due to very inefficient turnover rates (kcat). The kcat values for GES-1 were the lowest, ranging from 0.00063 to 0.00094 s–1 (Table 2). In general, kcat progressively increases 4–8-fold when comparing GES-1 to GES-2 and 9–20-fold between GES-2 and GES-5, reaching values of 0.046 to 0.076 s–1 for the latter (Table 2). The differences in kcat values for meropenem, ertapenem, and doripenem for each individual enzyme are small, indicating that structural differences in the tail regions of these substrates (Figure 1) do not influence their turnover rates significantly. Imipenem, the only carbapenem whose core region lacks a methyl group on the C1 carbon, is turned over by the GES enzymes with roughly 10-fold higher efficiencies.17
When the catalytic efficiencies (kcat/Km) of each enzyme were compared, GES-1 and -2 for meropenem, ertapenem, and doripenem were the least proficient and had kcat/Km values ranging from 102 to 103 M–1 s–1 (Table 2). These values were generally comparable for the two enzymes except in the case of GES-2 for ertapenem, where the catalytic efficiency is approximately 4-fold higher than observed for GES-1. Overall, GES-2 has higher kcat and Km values when compared to GES-1. GES-5 was the most proficient enzyme with efficiencies on the order of 104 M–1 s–1, approximately 10–100-fold higher than GES-1 and -2. The observed catalytic efficiencies of GES-1, -2, and -5 correlate well with the minimal inhibitory concentration (MIC) values for these carbapenems.17
Pre-Steady-State Kinetics
We next examined the pre-steady-state kinetics of the GES enzymes. We measured the microscopic rate constant k2 which describes the rate constant for acylation. The carbapenems were reacted with an excess of each GES enzyme to force single turnover conditions. All three enzymes displayed biphasic reaction time courses for each of the substrates in this study. However, it must be noted that we were unable to achieve saturation for the faster acylation phase with up to a 50-fold excess of enzyme (the solubility limit of the enzymes), indicating that these rates must be faster. Representative data for each enzyme with meropenem can be seen in Figure 2. The initial faster phase of acylation (k2,fast) was very similar for GES-1 and -2 with meropenem, ertapenem, and doripenem, ranging from 31 to 61 s–1 depending on the substrate, while for GES-5, k2,fast ranged from 52 to 180 s–1 (Table 3). For all three enzymes, the faster phase of acylation comprised approximately one-third of the total reaction. Prominent differences were observed for the second, slower phase of acylation (k2,slow), where single turnover conditions only required a 5-fold excess of each of the enzymes (Table 3). The GES-1 and -2 k2,slow acylation rate constants with meropenem, ertapenem, and doripenem ranged between 0.0012 and 0.0073 s–1. In general, GES-5 experienced 30–70-fold faster acylation with these three carbapenems when comparing to GES-1 and 10–40-fold faster than GES-2, with k2,slow ranging from 0.063 to 0.087 s–1. Biphasic kinetics were also observed for all three β-lactamases with imipenem. Overall the k2 rate constants were faster for imipenem with all three enzymes, with a 2–9-fold increase for k2,fast and 4–19-fold increase for k2,slow, depending on the substrate (Table 3).
Figure 2.

Time courses for acylation of GES-1, -2, and -5 by meropenem. The GES enzymes were reacted in at least a 5-fold excess with meropenem. Representative time courses for acylation are shown for GES-1 (A and B), GES-2 (C and D), and GES-5 (E and F). The faster phase is shown in the panels on the left, while the slower phase is shown in the panels on the right. The line of best fit is shown in black.
Table 3. Microscopic Rate Constants for the Hydrolysis of Carbapenems by GES Enzymes.
| enzyme | parameter | meropenem | ertapenem | doripenem | imipenem |
|---|---|---|---|---|---|
| GES-1 | k1 (M–1 s–1)b | 1.6 × 103 | 9.4 × 102 | 1.4 × 103 | 5 × 103 |
| k–1 (s–1)b | 5.4 × 10–4 | 2.3 × 10–4 | 2.5 × 10–4 | 5 × 10–5 | |
| k2,fast (s–1) | >50 ± 2 | >40 ± 1 | >61 ± 1 | >360 ± 10 | |
| k2,slow (s–1) | 0.0012 ± 0.0001 | 0.0023 ± 0.0001 | 0.0015 ± 0.0001 | 0.014 ± 0.001 | |
| k3 (s–1) | 0.0056 ± 0.0001 | 0.010 ± 0.001 | 0.0055 ± 0.0001 | 0.03 ± 0.01a | |
| GES-2 | k1 (M–1 s–1)b | 6.9 × 102 | 1.9 × 103 | 5.0 × 102 | 2.8 × 104 |
| k–1 (s–1)b | 8.2 × 10–4 | 3.3 × 10–3 | 5.0 × 10–4 | 6.1 × 10–3 | |
| k2,fast (s–1) | >51 ± 1 | >31 ± 1 | >51 ± 1 | >100 ± 10 | |
| k2,slow (s–1) | 0.0023 ± 0.0001 | 0.0073 ± 0.0001 | 0.0025 ± 0.0001 | 0.043 ± 0.001 | |
| k3 (s–1) | 0.021 ± 0.001 | 0.020 ± 0.001 | 0.024 ± 0.001 | 0.028 ± 0.001a | |
| GES-5 | k1 (M–1 s–1)b | 5.5 × 104 | 2.3 × 104 | 3.6 × 104 | 1.6 × 105 |
| k–1 (s–1)b | 1.7 × 10–2 | 4.6 × 10–3 | 8.5 × 10–3 | 7.9 × 10–2 | |
| k2,fast (s–1) | >97 ± 2 | >52 ± 1 | >180 ± 10 | >450 ± 10 | |
| k2,slow (s–1) | 0.087 ± 0.001 | 0.076 ± 0.001 | 0.063 ± 0.001 | 0.35 ± 0.01 | |
| k3 (s–1) | 0.10 ± 0.01 | 0.10 ± 0.01 | 0.051 ± 0.001 | 0.45a |
Data from ref (17).
Not measured experimentally.
Biphasic kinetics have previously been observed for acylation of GES-5 by imipenem.17 In this case, the phenomenon was attributed to imipenem’s Z and E side chain rotational isomers. This is likely not the reason for the biphasic kinetics observed here since meropenem, ertapenem, and doripenem do not have side chain rotational isomers (Figure 1). However, these antibiotics’ side chains are rotationally flexible, and thus it cannot be ruled out that the GES enzymes recognize different conformers of the carbapenems, giving rise to the observed biphasic kinetics. Another possible explanation is the existence of two different enzyme populations that are acylated by the substrates at different rates during steady-state turnover. It has to be noted that such conformational changes have not yet been observed in the reported crystal structures of this family of enzymes.17,20,29−32 However, it is quite plausible that not all available enzyme conformations are amenable for crystallization and the crystal structures represent only the most stable of them.
Next, k3, the microscopic rate constant for deacylation, was measured. Figure 3 shows sample data for the three enzymes with meropenem. The deacylation rate constants for GES-5 with meropenem, ertapenem, and doripenem were 2–5-fold higher than for GES-2 and another 2–4-fold higher than for GES-1 (Table 3). The deacylation rate constants for the three newer carbapenems were 3–5-fold slower for GES-1 and 4–9-fold slower for GES-5 than those for imipenem. For GES-2 these values were identical for all four carbapenem substrates (Table 3).17
Figure 3.

Reactivation of GES-1, -2, and -5 after incubation with meropenem. The GES enzymes were incubated with meropenem (10–20 min for GES-1, 2–5 min for GES-2 and 30 s for GES-5) at 10-fold above Km, then diluted 1:1000 (panel A, GES-1), 1:200 (panel B, GES-2), or 1:50 (panel C, GES-5) in either nitrocefin (GES-1 and -2) or cephalothin (GES-5) (shown in gray). The line of best fit is shown in black. Control reactions (black) were run in the absence of meropenem.
As both k2 and k3 can contribute to kcat (see eq 4), we calculated the theoretical kcat values using k3 and both k2,fast and k2,slow and compared the result with the experimentally determined kcat values. These calculations demonstrate that for GES-1 and GES-2 only the k2,slow values result in calculated kcat values that match those empirically determined for all four carbapenems, an indication that this slow phase of acylation is relevant for the steady state, while k2,fast is not. For GES-5 both k2,fast and k2,slow result in kcat values similar to those experimentally determined, an indication that both phases of acylation could be relevant for the steady state.
Both k2 and k3 can contribute to the rate-limiting step of the reaction pathway for carbapenem turnover. For both the GES-1 and -2 β-lactamases, the k2,slow values for meropenem, ertapenem, and doripenem are 3–10-fold slower than k3, an indication that acylation is likely the rate-limiting step in the turnover of these substrates. However, for GES-5, the k2,slow and k3 values for meropenem, ertapenem, and doripenem were very similar, suggesting either acylation or deacylation could be the rate-limiting step. The acylation and deacylation rate constants for each individual enzyme are very similar for meropenem, ertapenem, and doripenem, indicating that structural differences between these carbapenems do not play a significant role in their turnover. Somewhat different results were observed for turnover of imipenem by these GES enzymes. Since the k2,slow and k3 values for this antibiotic differed by 2-fold or less, acylation or deacylation could be rate limiting for turnover of imipenem by all three enzymes (Table 3).
Pre-steady-state kinetic rate constants k1 and k–1 describe substrate binding and release. These rate constants were calculated for GES-1, -2, and -5 using our experimental data for meropenem, doripenem, and ertapenem (Table 3) and eqs 4 through 7. Resulting values for k1 and k–1 were very similar (less than 2-fold difference) when either the k2,fast or k2,slow values were used for calculations. Further increases in k2,fast did not change the calculated k1 and k–1 values. As only k2,slow is likely relevant in the steady state (see above), only data obtained by using the rate constant k2,slow are shown in Table 3. In all cases, the second-order rate constant k1 for carbapenem binding is highest for GES-5, indicating that this enzyme binds carbapenems more efficiently. The same trend is also seen for carbapenem release. Similar to the trend observed for the k2,slow and k3 values, the k1 and k–1 values for each GES enzyme are independent of the three newer carbapenems. These data demonstrate that the structural differences between meropenem, doripenem, and ertapenem do not influence their binding or release.
Structure of the Ertapenem–GES-2 Complex
The results of this study show that meropenem, ertapenem, and doripenem behave kinetically very similar for each individual GES enzyme, yet differ from the reported kinetic data for imipenem (Table 3).17 To gain insight into the structural determinants responsible for differences in the kinetics for imipenem versus the other three carbapenems, we have determined the high resolution structure of the acyl-enzyme complex of GES-2 with ertapenem.
Electron density maps calculated following MR clearly indicated the presence of the asparagine side chain at residue 170, and an acyl-enzyme intermediate in the active site of GES-2 (Figure 4A). Density corresponding to the pyrroline ring, the 6α-hydroxyethyl, and the C3 carboxylate (Figure 1) were clearly evident, and an ertapenem molecule was added and refined with partial occupancy. The pyrroline ring adopts the Δ2 tautomeric form, with the C2 atom sp2 hybridized and the S21 sulfur coplanar with the pyrroline ring. The final ertapenem occupancy values were 0.85 and 0.7 Å2 for molecules A and B respectively. The 2Fo – Fc density for ertapenem is continuous as far as the sulfur atom, with additional pieces of density visible for the pyrollidine and carboxyphenyl rings. The core of ertapenem, comprising the pyrroline, carboxylate, and hydroxyethyl moieties, is anchored by a total of eight hydrogen bonds to the surrounding protein molecule (Figure 4B). In contrast, the tail of the ertapenem molecule, comprising the pyrrolidine and carboxyphenyl rings, makes no direct contacts with the protein and projects into the external milieu, although there is a water-mediated hydrogen bond between the terminal carboxylate group and the side chain of Asp-276 (Figure 4B). Meropenem, ertapenem, and doripenem have an identical core structure and differ only in their tail groups (Figure 1). This provides a structural rationale for the observed similarities in the kinetic parameters (k2,slow, k3, k1, and k–1) for each individual GES enzyme with these three carbapenems, as only the core of the antibiotics appears to interact with the enzymes.
Figure 4.

GES-2-ertapenem structure. (A) Stereoview of the final 2Fo– Fc electron density (blue mesh) contoured at 1 σ for some of the GES-2 residues (green) in the active site. The ertapenem molecule (magenta sticks) is shown in the initial Fo– Fc difference density (pink mesh) calculated following molecular replacement, contoured at 2.5 σ. The side chain of Asn170 is also shown in the same Fo– Fc electron density map since this residue was truncated to a glycine in the MR model used. The deacylating water molecule (W2) is also indicated as a red sphere. (B) Stereoview of the hydrogen bonding interactions between ertapenem (magenta sticks) and the GES-2 protein (green). (C) Superposition of the imipenem complexes of GES-1 and GES-5 onto the ertapenem-GES-2 complex (green). Ertapenem is shown as thick magenta sticks and imipenem as thin yellow and cyan sticks for GES-1 and GES-5, respectively. The GES-1 and GES-5 proteins are not shown for clarity.
Superpositions of the ertapenem–GES-2 complex with the imipenem complexes of GES-1 and -529 show that the cores of ertapenem and imipenem are bound in a very similar way in the three enzymes, with the exception of the 6α-hydroxyethyl group attached to the C6 atom, which adopts a different conformation in GES-2 compared to GES-1 and -5. In the GES-2 complex, the hydroxyl group and the terminal carbon C62 (Figure 1) point away from the Asn-170 side chain, and the hydroxyl group makes a hydrogen bonding interaction with the side chain of Asn-132 (Figure 4B). Conversely, in the absence of the asparagine side chain in both GES-1 and -5, the hydroxyethyl group is rotated 120° such that the C62 carbon points toward residue 170 (Figure 4C), with the hydroxyl group still able to donate a hydrogen bonding interaction to Asn-132.29 Were the hydroxyethyl group to adopt this conformation in GES-2, a severe steric clash with the Asn-170 side chain would ensue. Thus, the presence of the Asn-170 side chain in GES-2 is solely responsible for the conformational rearrangement of the 6α-hydroxyethyl group in the GES-2-ertapenem complex. Indeed, although the core parts of ertapenem and imipenem differ by the presence in ertapenem of a methyl group attached to the C1 carbon atom (Figure 1), this group is unlikely to influence the spatial orientation of the 6α-hydroxyethyl of carbapenems. The spatial disposition of the methyl group relative to the hydroxyethyl group is such that the presence of the methyl does not constrain the hydroxyethyl, and this group is free to rotate about the C6–C61 bond. The superpositions also show that the tail of the imipenem molecule in GES-1 and -5 projects in a different direction compared to the ertapenem tail in GES-2 (Figure 4C). In this regard, the presence of the methyl group in ertapenem does have a steric effect, forcing the pyrrolidine ring to point away from the pyrroline ring such that the C1A atom of the pyrrolidine is in a transoid orientation relative to the C1 methyl (Figure 4C). In contrast, the first carbon atom (C22) of the imipenem (iminomethyl)aminoethyl tail is almost coplanar with the pyrroline ring, an orientation facilitated by the absence of the methyl (Figure 4C). Although there is no direct evidence that the difference in the orientation of the tail groups explains the variation in the kinetics between imipenem and the other carbapenems, the structural difference is intriguing.
Superposition of the ertapenem–GES-2 complex onto apo-GES-2 gives a root-mean-square deviation (rmsd) in atomic positions of 0.14 Å for all 533 Cα atoms. Inspection of the active site of both structures shows that upon ertapenem binding, there are only minor movements of the active site residues. The most striking difference between the two structures is the rearrangement of water in the active site. In apo-GES-2, three water molecules are observed, and the binding of ertapenem displaces two of these, one from the oxyanion hole (W1, Figure 5) and the other near the 6α-hydroxyethyl group (W3, Figure 5). The deacylating water (W2, Figures 4A and 5) is also present in the apo-GES-2 structure, and upon acylation this water molecule moves approximately 0.4 Å in a direction away from the incoming 6α-hydroxyethyl moiety, yet remains tightly bound in the active site, and at full occupancy. This is in distinct contrast to both GES-1 and -5, where in the former, the deacylating water molecule is completely displaced from the binding site following acylation by imipenem, and in GES-5 the active site is partially occupied by either imipenem (at about 70%) or a water molecule (at about 30%).29 The W2 water molecule in GES-2 sits in a pocket between the conserved Glu-166 and the catalytic Ser-70 and is involved in at least three interactions, accepting a hydrogen bond from the 6α-hydroxyethyl side group of the ertapenem, and donating two hydrogen bonds to the Oδ1 atom of Asn-170 and the Glu-166 side chain. The asparagine side chain, in turn, donates hydrogen bonds to Glu-166 and the carbonyl oxygen of Pro-167 (Figure 5). Moreover, there appears to be an additional transient interaction with the Sδ atom of Met-169 (Figure 5). This latter residue adopts two alternate conformations with an occupancy ratio of about 70:30. The transient hydrogen bond with W2 is made with the lower occupancy conformation.
Figure 5.
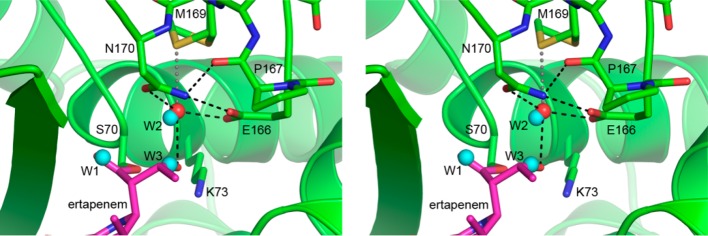
Superposition of ertapenem–GES-2 with apo-GES-2. The active site of GES-2 is shown (green) centered on the deacylating water (W2, red sphere). The locations of three water molecules in the apo-GES-2 structure are represented as cyan spheres. The W1 water represents the acylating water molecule which is ejected from the oxyanion hole upon ertapenem binding.
The structure of the GES-2–ertapenem complex provides insight into why W2 is retained in this enzyme upon acylation, whereas it is displaced in both GES-1 and -5. In GES-2, not only does the Asn-170 side chain provide a hydrogen bond to W2, it also forces the 6α-hydroxyethyl group of the ertapenem to rotate and present the hydroxyl group to the W2 atom for an additional hydrogen bond. This creates a significantly more stable water binding site compared to the equivalent binding site in either GES-1 or -5. Although the presence of a fully occupied tightly bound deacylating water molecule in GES-2 might suggest that this enzyme would have a more efficient deacylation step than GES-5, our results show that it has a deacylation rate intermediate between that of GES-1 and GES-5. Clearly, the mechanism by which deacylation is initiated, presumably via activation of the water by deprotonation, plays a critical role. The Glu-166 residue should be the major proton acceptor in the active site and would, under normal circumstances, readily deprotonate a water molecule bound in the active site. However, the presence of the transient hydrogen bond between the water and the partially occupied Met-169 side chain might presumably make the water molecule less susceptible to deprotonation, since three acceptors (Asn-170, Glu-166, and Met-169) are, at least for a portion of the time, all vying for the two protons on the water molecule. This would conceivably delocalize the protons to a certain extent and make deprotonation less favored.
Conclusions
GES-type β-lactamases are clinically important enzymes producing resistance to a variety of β-lactam antibiotics. Single amino acid substitutions in GES β-lactamases can result in the enhancement of their catalytic activity against carbapenems, last resort antibiotics used for treatment of life-threatening infections. Among the three GES enzymes in this study, GES-1 is the weakest and GES-5 is the strongest carbapenemase, with GES-2 intermediate between these two. Structural and molecular dynamics simulation analyses of GES-1 and GES-5 have demonstrated that acylation of either of these enzymes by a carbapenem antibiotic results in the displacement of the deacylating water from their active sites.29 As a result, both GES-1 and GES-5 require a water molecule from the external milieu to enter the active site to facilitate deacylation, and the structural features of the active sites of these two enzymes make this process very inefficient in GES-1 but significantly more efficient in GES-5 which is a bona fide carbapenemase.29 Contrary to what has been observed in GES-1 and -5, the deacylating water molecule is retained in the active site of GES-2 due primarily to the presence of the canonical asparagine residue at position 170. The structural differences in the GES-2 active site give rise to a tightly bound deacylating water molecule, and it is the abundance and nature of the hydrogen bonding interactions between the acyl-enzyme complex and this water molecule that attenuate its activation for deacylation and subsequently make this step less efficient in GES-2 when compared to GES-5.
Moreover, our studies have demonstrated that while there is a significant difference in the catalytic efficiency of GES-1, -2, and -5 for all four carbapenem antibiotics studied, three of them, meropenem, ertapenem and doripenem, behave kinetically very similar for each individual GES enzyme, while differing from imipenem, which is a better substrate. Superpositions of the crystal structures of the acyl-enzyme intermediates of GES-2 with ertapenem and GES-1 and -5 with imipenem show that while the cores of ertapenem and imipenem are bound in a similar way, their tails project in different directions. The presence of the extra methyl group in ertapenem forces the tail to occupy an orientation different from that of the tail of imipenem (Figure 4C). As the core structures of meropenem and doripenem are identical to that of ertapenem their tails likely would be similarly oriented. Differences in the size and orientation of the tail group of imipenem and the other carbapenems may affect protein dynamics associated with catalysis and thus result in the observed variation in the kinetics. In summary, results of our studies provide novel information on how structural differences between various GES enzymes and structural differences between carbapenem substrates affect the efficiency of catalysis. This information provides valuable insights regarding the mechanism of carbapenemase activity of the GES family of β-lactamases and is essential for the design of new antibiotics and inhibitors of these clinically important enzymes.
Glossary
Abbreviations
- ESBL
extended-spectrum β-lactamase
- MIC
minimal inhibitory concentration
- MR
molecular replacement
- rmsd
root-mean-square deviation
This work was supported by National Institutes of Health (NIH) Grant AI089726 (S.B.V.). Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy (BES, BER) and by the National Institutes of Health (NCRR, BTP, NIGMS). The project described was also supported by Grant Number 5 P41 RR001209 from the NCRR, a component of the National Institutes of Health.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Bush K.; Jacoby G. A. (2010) Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 54, 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K. (2013) Proliferation and significance of clinically relevant β-lactamases. Ann. N. Y. Acad. Sci. 1277, 84–90. [DOI] [PubMed] [Google Scholar]
- Bush K.; Fisher J. F. (2011) Epidemiological expansion, structural studies, and clinical challenges of new β-lactamases from gram-negative bacteria. Annu. Rev. Microbiol. 65, 455–478. [DOI] [PubMed] [Google Scholar]
- Fisher J. F.; Meroueh S. O.; Mobashery S. (2005) Bacterial resistance to β-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 105, 395–424. [DOI] [PubMed] [Google Scholar]
- Walther-Rasmussen J.; Hoiby N. (2007) Class A carbapenemases. J. Antimicrob. Chemother. 60, 470–482. [DOI] [PubMed] [Google Scholar]
- Ambler R. P.; Coulson A. F.; Frere J. M.; Ghuysen J. M.; Joris B.; Forsman M.; Levesque R. C.; Tiraby G.; Waley S. G. (1991) A standard numbering scheme for the class A β-lactamases. Biochem. J. 276, 269–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher J. F.; Mobashery S. (2009) Three decades of the class A beta-lactamase acyl-enzyme. Curr. Protein Pept. Sci. 10, 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majiduddin F. K.; Palzkill T. (2003) Amino acid sequence requirements at residues 69 and 238 for the SME-1 β-lactamase to confer resistance to β-lactam antibiotics. Antimicrob. Agents Chemother. 47, 1062–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirel L.; Le Thomas I.; Naas T.; Karim A.; Nordmann P. (2000) Biochemical sequence analyses of GES-1, a novel class A extended-spectrum β-lactamase, and the class 1 integron In52 from Klebsiella pneumoniae. Antimicrob. Agents Chemother. 44, 622–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vourli S.; Giakkoupi P.; Miriagou V.; Tzelepi E.; Vatopoulos A. C.; Tzouvelekis L. S. (2004) Novel GES/IBC extended-spectrum β-lactamase variants with carbapenemase activity in clinical enterobacteria. FEMS Microbiol. Lett. 234, 209–213. [DOI] [PubMed] [Google Scholar]
- Jeong S. H.; Bae I. K.; Kim D.; Hong S. G.; Song J. S.; Lee J. H.; Lee S. H. (2005) First outbreak of Klebsiella pneumoniae clinical isolates producing GES-5 and SHV-12 extended-spectrum β-lactamases in Korea. Antimicrob. Agents Chemother. 49, 4809–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae I. K.; Lee Y. N.; Jeong S. H.; Hong S. G.; Lee J. H.; Lee S. H.; Kim H. J.; Youn H. (2007) Genetic and biochemical characterization of GES-5, an extended-spectrum class A β-lactamase from Klebsiella pneumoniae. Diagn. Microbiol. Infect. Dis. 58, 465–468. [DOI] [PubMed] [Google Scholar]
- Manageiro V.; Ferreira E.; Canica M.; Manaia C. M. (2013) GES-5 among the β-lactamases detected in ubiquitous bacteria isolated from aquatic environment samples. FEMS Microbiol. Lett. 351, 64–69. [DOI] [PubMed] [Google Scholar]
- Poirel L.; Bonnin R. A.; Nordmann P. (2012) Genetic support and diversity of acquired extended-spectrum β-lactamases in Gram-negative rods. Infect. Genet. Evol. 12, 883–893. [DOI] [PubMed] [Google Scholar]
- Bonnin R. A.; Rotimi V. O.; Al Hubail M.; Gasiorowski E.; Al Sweih N.; Nordmann P.; Poirel L. (2013) Wide dissemination of GES-type carbapenemases in Acinetobacter baumannii isolates in Kuwait. Antimicrob. Agents Chemother. 57, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro V. B.; Zavascki A. P.; Rozales F. P.; Pagano M.; Magagnin C. M.; Nodari C. S.; da Silva R. C.; Dalarosa M. G.; Falci D. R.; Barth A. L. (2014) Detection of bla(GES-5) in carbapenem-resistant Kluyvera intermedia isolates recovered from the hospital environment. Antimicrob. Agents Chemother. 58, 622–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frase H.; Shi Q.; Testero S. A.; Mobashery S.; Vakulenko S. B. (2009) Mechanistic basis for the emergence of catalytic competence against carbapenem antibiotics by the GES family of β-lactamases. J. Biol. Chem. 284, 29509–29513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F. W.; Rosenberg A. H.; Dunn J. J.; Dubendorff J. W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185, 60–89. [DOI] [PubMed] [Google Scholar]
- Koerber S. C.; Fink A. L. (1987) The analysis of enzyme progress curves by numerical differentiation, including competitive product inhibition and enzyme reactivation. Anal. Biochem. 165, 75–87. [DOI] [PubMed] [Google Scholar]
- Frase H.; Smith C. A.; Toth M.; Champion M. M.; Mobashery S.; Vakulenko S. B. (2011) Identification of products of inhibition of GES-2 β-lactamase by tazobactam by X-ray crystallography and spectrometry. J. Biol. Chem. 286, 14396–14409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A. E.; Ellis P. J.; Miller M. D.; Deacon A. M.; Phizackerley R. P. (2002) An automated system to mount cryo-cooled protein crystals on a synchrotron beam line, using compact sample cassettes and a small-scale robot. J. Appl. Crystallogr. 35, 720–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews B. W. (1968) Solvent contents of protein crystals. J. Mol. Biol. 33, 491–497. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82. [DOI] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025. [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D Biol. Crystallogr. 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D Biol. Crystallogr. 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Smith C. A.; Frase H.; Toth M.; Kumarasiri M.; Wiafe K.; Munoz J.; Mobashery S.; Vakulenko S. B. (2012) Structural basis for progression toward the carbapenemase activity in the GES family of β-lactamases. J. Am. Chem. Soc. 134, 19512–19515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. A.; Caccamo M.; Kantardjieff K. A.; Vakulenko S. (2007) Structure of GES-1 at atomic resolution: insights into the evolution of carbapenamase activity in the class A extended-spectrum β-lactamases. Acta Crystallogr., Sect. D Biol. Crystallogr. 63, 982–992. [DOI] [PubMed] [Google Scholar]
- Bebrone C.; Bogaerts P.; Delbruck H.; Bennink S.; Kupper M. B.; Rezende de Castro R.; Glupczynski Y.; Hoffmann K. M. (2013) GES-18, a new carbapenem-hydrolyzing GES-Type β-lactamase from Pseudomonas aeruginosa that contains Ile80 and Ser170 residues. Antimicrob. Agents Chemother. 57, 396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbruck H.; Bogaerts P.; Kupper M. B.; Rezende de Castro R.; Bennink S.; Glupczynski Y.; Galleni M.; Hoffmann K. M.; Bebrone C. (2012) Kinetic and crystallographic studies of extended-spectrum GES-11, GES-12, and GES-14 β-lactamases. Antimicrob. Agents Chemother. 56, 5618–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]