

Abstract
Extended-spectrum β-lactamases (ESBLs) pose a threat to public health because of their ability to confer resistance to extended-spectrum cephalosporins such as cefotaxime. The CTX-M β-lactamases are the most widespread ESBL enzymes among antibiotic resistant bacteria. Many of the active site residues are conserved between the CTX-M family and non-ESBL β-lactamases such as TEM-1, but the residues Ser237 and Arg276 are specific to the CTX-M family, suggesting that they may help to define the increased specificity for cefotaxime hydrolysis. To test this hypothesis, site-directed mutagenesis of these positions was performed in the CTX-M-14 β-lactamase. Substitutions of Ser237 and Arg276 with their TEM-1 counterparts, Ala237 and Asn276, had a modest effect on cefotaxime hydrolysis, as did removal of the Arg276 side chain in an R276A mutant. The S237A:R276N and S237A:R276A double mutants, however, exhibited 29- and 14-fold losses in catalytic efficiency for cefotaxime hydrolysis, respectively, while the catalytic efficiency for benzylpenicillin hydrolysis was unchanged. Therefore, together, the Ser237 and Arg276 residues are important contributors to the cefotaximase substrate profile of the enzyme. High-resolution crystal structures of the CTX-M-14 S70G, S70G:S237A, and S70G:S237A:R276A variants alone and in complex with cefotaxime show that residues Ser237 and Arg276 in the wild-type enzyme promote the expansion of the active site to accommodate cefotaxime and favor a conformation of cefotaxime that allows optimal contacts between the enzyme and substrate. The conservation of these residues, linked to their effects on structure and catalysis, imply that their coevolution is an important specificity determinant in the CTX-M family.
A wide range of bacterial infectious diseases are treated with β-lactam antibiotics. Unfortunately, resistant bacteria have evolved mechanisms to escape the lethal action of these drugs. The majority of resistance to β-lactam antibiotics results from the expression of β-lactamases, which are enzymes that hydrolyze the amide bond of the β-lactam ring of the drugs to inactivate these antimicrobials.1
In the early 1980s, extended-spectrum oxyimino-cephalosporins, including cefotaxime, were developed that are poor substrates for class A serine β-lactamases such as TEM-1 and SHV-1.2−4 These antibiotics are characterized by bulky side chains containing an oxyimino group and, for many, an aminothiazole ring (Figure 1).5 The extensive clinical use of these antibiotics has resulted in the emergence of extended-spectrum β-lactamases (ESBLs), which are class A enzymes capable of hydrolyzing these drugs.
Figure 1.

Structures of β-lactam antibiotics. Cefotaxime, cephalothin, and benzylpenicillin with atom positions numbered.
The CTX-M family of class A ESBLs first appeared in the late 1980s and has since become the most widespread plasmid-mediated source of resistance to oxyimino-cephalosporins in Gram-negative bacteria.4,6 The CTX-M-14 β-lactamase is a common CTX-M-type enzyme among antibiotic resistant bacteria.3 It was initially found in isolates of Klebsiella pneumoniae, Shigella sonnei, and Escherichia coli and has been isolated from a large range of clinical bacteria, mainly from the Enterobacteriaceae family.3 A defining feature of the CTX-M enzymes is that they hydrolyze the oxyimino-cephalosporin, cefotaxime, with approximately 1000-fold higher catalytic efficiency (kcat/Km) than other class A β-lactamases such as TEM-1 and SHV-1.4 Although the overall folds are structurally similar, the CTX-M enzymes share less than 40% amino acid sequence identity with TEM-1 and SHV-1.4 The nature of the catalytic and structural features of the CTX-M enzymes responsible for their high activity toward cefotaxime is an important question. Structural analysis of variants of the TEM and SHV enzymes capable of hydrolyzing oxyimino-cephalosporins showed that the substitutions act on the omega loop or the β3 strand that outline the active site to increase the active site volume to accommodate the larger cefotaxime and ceftazidime substrates.7,8 X-ray structures of CTX-M enzymes with and without substrate revealed a narrow active site that expands significantly due to a conformational change upon cefotaxime binding.5,9,10,11 This expansion is accompanied by the rupture of a hydrogen bond between main chain groups of Asn170 and Asp240 that connects the omega loop to the β3 strand.11
Despite the lack of overall high sequence identity, the majority of the active site residues that make contact with substrate in TEM-1 and CTX-M-14 are identical or represent conservative changes.12−14 Of the remaining positions, two stand out as differences that may contribute to the enhancement of the cefotaximase activity in the CTX-M family. In the TEM/SHV-type enzymes, a conserved arginine residue at position 244 interacts with the C-3/4 carboxyl group common to β-lactam antibiotics to stabilize the substrate and transition state complexes of benzylpenicillin15,16,17 (Figure 2). In the CTX-M family, this arginine is replaced by Thr244, but a different positive charge is present in the region due to an arginine at position 276. In the TEM-1/SHV-1 enzymes, position 276 is occupied by an asparagine5,9 (Table 1 and Figure 2). However, substitution of the analogous Arg276 with a number of amino acids in the CTX-M-4 or CTX-M-1 enzymes has little effect on penicillin G and cephalothin hydrolysis and only a modest (2–3-fold) effect on the hydrolysis of cefotaxime.13,18 These results suggest a relatively minor contribution of Arg276 toward determining the substrate specificity, despite the fact that it is conserved throughout the CTX-M family.
Figure 2.
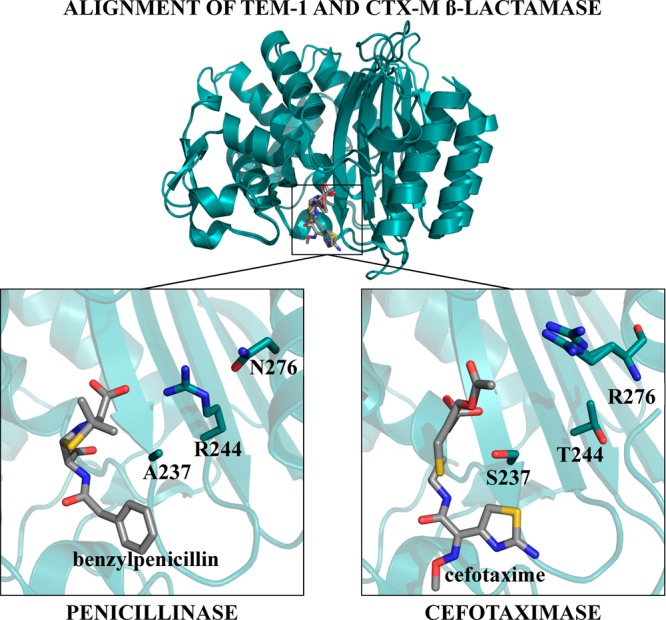
View of residue positions 237, 244, and 276 in the TEM-1 and CTX-M β-lactamases. A structural alignment between TEM-1 β-lactamase (PDB ID: 1FQG) and CTX-M-9 β-lactamase (PDB ID: 3HLW) is shown in teal cartoon with ligands shown as gray sticks. The left panel shows residues Ala237 and Arg244 and Asn276 in TEM-1 β-lactamase (teal ribbon) with benzylpenicillin (gray sticks) in the active site. The TEM-1 β-lactamase is representative of class A enzymes that efficiently hydrolyze penicillins and early generation cephalosporins. In TEM-1 β-lactamase, Arg244 is directed toward the carboxyl group of benzylpenicillin. The right panel shows residues Ser237, Thr244, and Arg276 in CTX-M-9 β-lactamase (teal ribbon) with cefotaxime (gray sticks) in the active site. The hydroxyl group of Ser237 forms a hydrogen bond with the C4 carboxylate of cefotaxime. Arg276 is directed toward the C3 methyl acetate group and the C4 carboxylate of cefotaxime, although it does not make direct interactions with these groups. Enzymes are shown in teal, with their respective ligands shown as gray sticks. Nitrogen atoms are shown in blue, oxygen atoms are shown in red, and sulfur atoms are shown in yellow.
Table 1. Amino Acid Residues at Positions 237, 244, and 276 in the Representative Penicillinase and CTX-M Family β-Lactamases.
| 237 | 244 | 276 | |
|---|---|---|---|
| TEM-1 (1M40a) | Ala | Arg | Asn |
| SHV-2 (1N9B) | Ala | Arg | Asn |
| CTX-M-1b | Ser | Thr | Arg |
| CTX-M-2 | Ser | Thr | Arg |
| CTX-M-8 | Ser | Thr | Arg |
| CTX-M-25 | Ser | Thr | Arg |
| CTX-M-9 (1YLW) | Ser | Thr | Arg |
| CTX-M-14 (1YLT) | Ser | Thr | Arg |
PDB codes are provided in parentheses for available structures.
CTX-M-1, 2, 8, 9, and 25 are representative enzymes from each subgroup of CTX-M-β-lactamases. CTX-M-14 is a member of the CTX-M-9 subgroup.
The CTX-M enzymes contain a serine at position 237, whereas this position is an alanine in TEM/SHV enzymes (Table 1 and Figure 2). X-ray structures of a CTX-M-9 S70G mutant in complex with benzylpenicillin or cefotaxime show that Ser237 makes contact with cefotaxime, including a hydrogen bond with the C-4 carboxyl group. However, Ser237 does not contact bound benzylpenicillin.11 A S237A substitution in CTX-M-4 resulted in little effect on benzylpenicillin hydrolysis but, surprisingly, also showed only a modest decrease in activity for cefotaxime hydrolysis.19
The sequence conservation among the CTX-M family and the prior structural observations suggest that Arg276 and Ser237 would contribute significantly to the enhanced specificity of this family toward cefotaxime. Thus, the rather modest effect of the individual mutations on cefotaxime hydrolysis is unexpected. Because of their conservation in the CTX-M family and the structural differences in this region compared to the TEM-type enzymes, we hypothesized that the two residues might interact cooperatively so that mutating both residues in the same molecule would have a larger effect on catalysis than expected from the individual mutations.14 In this study, we tested this hypothesis by constructing single and double mutant combinations of residues 237 and 276 in CTX-M-14 β-lactamase and assessed kinetic and structural effects of the mutations with first-generation and extended-spectrum antibiotics. The kinetic and structural results argue that these two conserved residues in the CTX-M family work together to position the substrate and significantly enhance cefotaxime hydrolysis.
Materials and Methods
Bacterial Strains and Plasmids
E. coli K12 XL1-Blue strain (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′ proAB lacIqZΔM15 Tn10 (Tetr)] was obtained from Stratagene (La Jolla, CA) and used in site-directed mutagenesis experiments. The E. coli RB791 strain was used for protein expression and purification. The blaCTX-M-14 gene was inserted into the pTP123 plasmid, and the resulting plasmid was used as template for site-directed mutagenesis and subsequent expression of mutant enzymes in E. coli RB791.14
Site-Directed Mutagenesis
All CTX-M-14 mutants were created using the QuikChange kit (Stratagene, La Jolla, CA). Primers were obtained from Integrated DNA Technologies (Coralville, IA). The following primers introduced the underlined mutations into pTP123 CTX-M-14 (complementary primer not shown):
S237A (5′-gtgataagaccggcgccggcgactacggca-3′);
R276A (5′-cgcagagagccgcgccgatgtgctggct-3′);
R276N (5′-cgcagagagccgcaatgatgtgctggct-3′).
All CTX-M-14 mutant constructs were verified by DNA sequencing (Lone Star Laboratories, Houston, TX).
Determination of Minimum Inhibitory Concentrations
In vivo antibiotic resistance levels were determined by E-test (Ab Biodisk, Sweden) in accordance with the manufacturer’s recommendations to evaluate the function of CTX-M-14 mutants in E. coli RB791 cells. Cultures were inoculated with a single colony and grown overnight at 37 °C. Overnight cultures were diluted 1:100 and plated on LB agar. An E-test strip was then added to a single plate, which was placed at 37 °C overnight before the resistance level was recorded.
Protein Purification
E. coli RB791 cells transformed with the relevant pTP123 CTX-M-14 construct were selected on LB agar containing 12.5 μg/mL chloramphenicol. Single colonies were used to inoculate 20 mL of LB supplemented with 12.5 μg/mL chloramphenicol, and cultures were grown overnight at 37 °C. Fifteen milliliters of the overnight culture was used to inoculate 1.5 L of LB containing 12.5 μg/mL chloramphenicol, and the resulting cultures were allowed to grow to mid log (OD600 ∼ 0.7). The cultures were grown at 23 °C overnight after addition of 300 μL of 1 M IPTG to the 1.5 L cultures, resulting in a final concentration of 0.2 mM. Cells were harvested by centrifugation and frozen for at least an hour at −80 °C. To release the periplasmic contents, the cells were resuspended in 30 mL of 10 mM Tris, pH 8.0, 20% sucrose, and 1 mM EDTA and incubated on ice. After 20 min, 40 mL of cold, sterile water was added and mixed. The insoluble material was pelleted by centrifugation. The supernatant was filtered and passed through a HiTrap SP column (Amersham, GE Healthcare, Piscataway, NJ). A linear NaCl gradient was used to elute the CTX-M-14 enzyme. The purity of the β-lactamase containing fractions was determined by SDS-PAGE, and the pooled fractions were dialyzed overnight into 50 mM sodium phosphate buffer, pH 7.0. The concentration of active β-lactamase was determined by titrating the CTX-M-14 enzyme with β-lactamase inhibitory protein-II (BLIP-II), which is a potent inhibitor (0.5 pM Ki) of the CTX-M-14 enzyme.20 Varying concentrations (μM) of BLIP-II were incubated with CTX-M-14 enzyme for 30 min at room temperature in a 96-well plate in a total volume of 200 μL. Following incubation, the chromogenic substrate nitrocefin was added to a final concentration of 30 μM, and the initial velocity of nitrocefin hydrolysis was followed by absorbance at 482 nm using a plate reader to determine the percent BLIP-II occupancy of β-lactamase (Tecan Infinite 200 PRO, Männedorf, Switzerland). The percent inhibition versus inhibitor concentration was fit with a linear equation and extrapolated to 100% inhibition to obtain the active enzyme concentration.21
Enzyme Kinetics
Steady-state Michaelis–Menten kinetic parameters were determined on a Beckman-Coulter spectrophotometer model DU 800 (Fullerton, CA). Substrate hydrolysis was monitored at the following wavelengths: benzylpenicillin, 233 nm; cefotaxime, 264 nm; cephalothin, 262 nm; nitrocefin, 482 nm. Reactions were performed in 50 mM sodium phosphate buffer, pH 7.0, with 1 mg/mL BSA at 30 °C. BSA was added to buffer in order to stabilize β-lactamase, which is present at low concentrations in enzyme assays. BSA was excluded for the benzylpenicillin reactions due to an overlap in protein absorbance with the wavelength used to monitor the reaction. To determine Km and kcat values, initial velocity rates were fitted over a range of substrate concentrations to a Michaelis–Menten curve using GraphPad Prism 6, and standard errors to these fits are reported. The errors reported for kcat/Km are calculated by summing the percent errors of both parameters.
Determination of Statistical Significance for kcat/Km values
Enzymatic efficiency (kcat/Km) was determined as previously described, and significance was determined using GraphPad Prism 6 unpaired t-test for the CTX-M-14 as compared to the various mutants. ** denotes a p-value less than 0.005, and *** denotes a p-value less than 0.0005.
ΔΔG Calculations
ΔΔG was calculated using the following equation
| 1 |
Using this equation, an increase in catalytic efficiency (kcat/Km) upon mutation would result in a negative ΔΔG value, whereas a decrease in catalytic efficiency would be reported as a positive change in free energy. Error for ΔΔG values was calculated using the following equation
| 2 |
where SEM represents standard error of the mean, WT represents wild-type CTX-M-14, and mut represents the mutant enzyme.
Fluorescence Scan of CTX-M-14 and Mutants
A fluorescence scan was performed on CTX-M-14 as well as CTX-M-14 S237A and CTX-M-14 S237A:R276A to ensure no secondary structural changes occurred in the mutant structures. This was also done to ensure that the crystal structures accurately depicted the structures seen in bulk solution. The protein was diluted to 2 μM in 50 mM sodium phosphate buffer, pH 7.0. The protein was excited at 280, and fluorescence intensity was measured from 295 to 545 nm using a Cary Eclipse Fluorescent Spectrophotometer from Agilent Technologies (Santa Clara, CA). The fluorescence intensity was normalized for all measurements, and each protein was measured in at least duplicate.
Crystallization and Structure Determination
The CTX-M-14 S70G, S70G:S237A, and S70G:S237A:R276A proteins were dialyzed into 5 mM Tris, pH 7.0, and 50 mM NaCl and were concentrated to 10 mg/mL for crystallization. Diffraction-quality crystals were obtained in 30% PEG4K containing 0.2 M ammonium sulfate. The protein and mother liquor were mixed at a 1:1 ratio in a 2 μL drop and grown by hanging drop vapor diffusion. The crystals were harvested and cryoprotected with 25% glycerol added to the mother liquor. For the structures in complex with cefotaxime, crystals were soaked in the mother liquor at pH 8.0 (adjusted with 1 M Tris) containing 50 mM cefotaxime overnight and then cryoprotected in 25% glycerol, 50 mM cefotaxime, 30% PEG4K, 0.2 M ammonium sulfate, pH 8.0. X-ray diffraction data sets were collected on beamline 5.0.2 (Advanced Light Source, Berkeley, CA) for all structures except CTX-M-14 S70G, which was collected at Baylor College of Medicine. The data sets for S70G and S70G:S237A were processed using iMosfilm, and the MolRep program from CCP4 was used for molecular replacement using CTX-M-14 (1YLT) as the search model.22−24 The diffraction data sets for S70G:S237A:R276A were processed with HKL2000 software, and the Phaser program from the CCP4 package was used for molecular replacement using the wild-type CTX-M-14 (1YLT) as the search molecule.25,26 One molecule was found in the asymmetric unit for all crystals. All models were fitted to the density in Coot.27 The structures were continually refined through the use of Coot and Phenix using default anisotropic corrections. Ordered solvent was added in Coot. Structures were analyzed using the LigPlot+ program.28 Hydrogen bond and hydrophobic interactions were analyzed using Ligplus with distance criterion of 2.6 and 3.3 Å for hydrogen bonds and carbon–carbon distance of 3.6 and 4.5 Å for hydrophobic interactions. Molecular graphics images for Figures 3 and 4 were generated using PyMOL v1.3.29 Molecular graphics images for Figure 5 were produced using the UCSF Chimera package from the Computer Graphics Laboratory, University of California, San Francisco (supported by NIH P41 RR-01081).30
Figure 3.
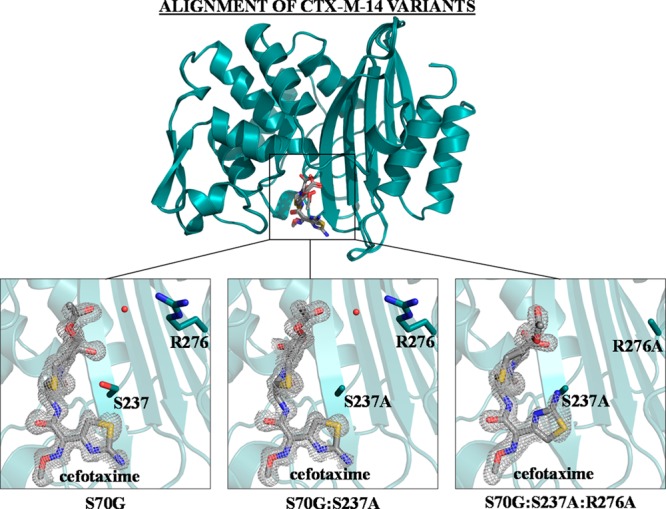
Electron density of cefotaxime in complex with the CTX-M-14 mutant enzymes. An alignment of CTX-M-14 mutants S70G, S70G:S237A, and S70G:S237A:R276A is shown on top, with the enzyme shown as teal ribbon and the substrate depicted as gray sticks. Below, the Fo – Fc difference map is shown in gray at 3σ. CTX-M-14 positions 237 and 276 are shown as teal sticks, with the enzyme shown in ribbon and cefotaxime shown in gray. The water molecule that forms hydrogen bonds with both Arg276 and cefotaxime in the S70G and S70G:S237A structures is displayed as a red sphere. Nitrogen atoms are shown in blue, oxygen atoms are shown in red, and sulfur atoms are shown in yellow. Images generated in PyMOL.29
Figure 4.

Hydrogen bond and hydrophobic interactions in CTX-M-14 mutant enzymes with cefotaxime. Hydrogen bonds are shown as dotted black lines and labeled with their distance (Å) in green. The CTX-M-14 mutant enzymes are shown as dark gray sticks, and the ligand is shown as gray sticks. Carbons atoms are shown in black, nitrogen, in blue, sulfur, in yellow, and oxygen, in red. Water molecules are shown in blue. Enzyme residues that form hydrophobic interactions with the ligand are shown with red shell. Images generated using the LigPlot+ program. (A) CTX-M-14 S70G structure with cefotaxime bound. (B) CTX-M-14 S70G:S237A structure with cefotaxime bound. (C) CTX-M-14 S70G:S237A:R276A with cefotaxime bound.
Figure 5.

Hydrogen-bonding network between cefotaxime and the CTX-M-14 mutants. The enzyme in the S70G–cefotaxime structure is shown as blue sticks, S70G:S237A–cefotaxime structure is shown as green sticks, and S70G:S237A:R276A–cefotaxime structure is shown as orange sticks. Cefotaxime is shown as gray sticks throughout. Only residues that form hydrogen bonds with the C4 carboxylate or aminothiazole ring on cefotaxime are shown. Water molecules that form hydrogen bonds with both the substrate and enzyme are shown in red. All hydrogen bonds are depicted as black dotted lines and are labeled with their distance (Å). (A) C4 carboxylate group is shown with hydrogen bonds to CTX-M-14 residues indicated. (B) The hydrogen-bonding network between the cefotaxime aminothiazole ring and the mutant CTX-M-14 enzymes. (C) The hydrogen-bonding network between cefotaxime methyl acetate group and the mutant CTX-M-14 enzymes. Images generated in Chimera.30
Results
Contributions of Ser237 and Arg276 to β-Lactam Hydrolysis
Although most of the active site residues are conserved between the TEM-1/SHV-1 penicillinases and CTX-M β-lactamases, residues Ser237 and Arg276 differ from their counterparts in TEM-1/SHV-1 (Table 1 and Figure 2). On the basis of this, it was hypothesized that Ser237 and Arg276 contribute to the specificity of CTX-M enzymes for cefotaxime hydrolysis. To test this idea, the mutations S237A, R276N, and R276A were introduced singly and in combination into the CTX-M-14 enzyme. The S237A and R276N substitutions convert these positions to the equivalent TEM-1 residue, whereas R276A was introduced to test the impact of removing both positive charge and all hydrogen-bonding capacity from the side chain.
To test the in vivo activity of the CTX-M-14 mutant enzymes, minimum inhibitory concentrations (MICs) were evaluated in E. coli with plasmids encoding the various mutants (Supporting Information Table S1). MICs were determined for benzylpenicillin, cephalothin, and cefotaxime substrates. The S237A mutant caused a decrease in resistance levels for cefotaxime, but it showed no change in resistance levels for benzylpenicillin or cephalothin. Similar results were seen for the R276A and R276N single mutants and the S237A:R276A double mutant. The S237A:R276N double mutant caused no change in resistance levels for benzylpenicillin, but it showed decreased resistance levels for both cephalothin and cefotaxime. Overall, these results suggest that positions 237 and 276 play an important role in the activity of CTX-M-14 enzyme for cefotaxime and less pronounced roles in the activity toward benzylpenicillin and cephalothin (Supporting Information Table S1).
After expression in E. coli and purification, the steady-state kinetic parameters kcat, Km, and kcat/Km were determined for the three substrates benzylpenicillin, cephalothin, and cefotaxime (Table 2). The kinetic parameter kcat/Km reflects the hydrolysis of substrate under conditions where the substrate concentration is low compared to the Km, a situation that is likely to reflect the specificity of the enzyme in vivo. In contrast, kcat reflects the conversion of bound substrate to product under saturating conditions, an indicator of how a mutation affects the energetics of the enzyme–substrate complex compared to the enzyme–transition state complex.
Table 2. Kinetic Parameters of CTX-M-14 β-Lactamase and Mutant Enzymes.
| CTX-M-14 | S237A | R276A | R276N | S237A:R276A | S237A:R276N | |
|---|---|---|---|---|---|---|
| Benzylpenicillin | ||||||
| Km (μM)a | 36 ± 4 | 38 ± 6 | 46 ± 11 | 45 ± 5 | 136 ± 6 | 118 ± 8 |
| kcat (s–1)a | 179 ± 6 | 373 ± 16 | 176 ± 13 | 204 ± 6 | 838 ± 13 | 850 ± 21 |
| kcat/Km (μM–1 s–1)b | 5.0 ± 0.7 | 9.8 ± 2 | 3.8 ± 1 | 4.5 ± 0.6 | 6.2 ± 0.4 | 7.2 ± 0.7 |
| Cephalothin | ||||||
| Km (μM)a | 88 ± 21 | 75 ± 12 | 264 ± 30 | 196 ± 17 | 140 ± 12 | 175 ± 11 |
| kcat (s–1)a | 1429 ± 96 | 997 ± 43 | 2493 ± 110 | 1580 ± 52 | 532 ± 16 | 522 ± 12 |
| kcat/Km (μM–1 s–1)b | 16 ± 5 | 13 ± 3 | 9 ± 1 | 8 ± 1 | 3.8 ± 0.4 | 3.0 ± 0.3 |
| Cefotaxime | ||||||
| Km (μM)a | 60 ± 9 | 151 ± 11 | 88 ± 10 | 119 ± 8 | 737 ± 87 | 770 ± 68 |
| kcat (s–1)a | 175 ± 8 | 173 ± 5 | 86 ± 2 | 56 ± 2 | 122 ± 9 | 94 ± 5 |
| kcat/Km (μM–1 s–1)b | 2.9 ± 0.2 | 1.1 ± 0.1 | 1.0 ± 0.1 | 0.50 ± 0.05 | 0.20 ± 0.04 | 0.10 ± 0.01 |
Errors reported for this parameter are the standard error of the mean calculated from multiple experiments.
Errors reported for this parameter are calculated by summing the percent errors of Km and kcat.
The S237A substitution in CTX-M-14 shifted the substrate specificity to improve catalytic efficiency for benzylpenicillin hydrolysis and decrease cefotaxime hydrolysis (p-value of 0.001 when compared to wild-type), consistent with an alanine at position 237 in the TEM-1/SHV-type enzymes that hydrolyze cefotaxime slowly (Table 2 and Supporting Information Figure S1). The substitution of R276A or R276N had little impact on the kinetic parameters for benzylpenicillin hydrolysis and also decreased catalytic efficiency for cefotaxime hydrolysis by 3–6-fold (p-values less than 0.001 when compared to wild-type) (Table 2 and Supporting Information Figure S1). The R276A and R276N results suggest the loss of positive charge, rather than a loss of a hydrogen bond or other side chain contacts, accounts for the decreased hydrolysis of cefotaxime for the Arg276 mutants (Table 2).
Effects of Double Mutants
Since Ser237 and Arg276 are both conserved in the CTX-M family, mutations of both side chains (S237A:R276A or S237A:R276N) were introduced at the same time. The effects of the double mutations on the kinetic parameters for benzylpenicillin, cephalothin, and cefotaxime hydrolysis are shown in Table 2. For benzylpenicillin hydrolysis, neither of the double mutants had an effect on kcat/Km, but both increased kcat by about 4-fold. The increases in kcat suggest that the Ser237 and Arg276 side chains together inhibit turnover of benzylpenicillin once substrate is bound to CTX-M-14.
In contrast, the S237A:R276A double mutant exhibits a 4-fold reduction in kcat/Km for cephalothin hydrolysis and a 14-fold decrease in kcat/Km for cefotaxime (p-value of 0.0002 when compared to wild-type) (Table 2 and Supporting Information Figure S1). The S237A:R276N mutations caused a 4-fold decrease in kcat/Km for cephalothin and a larger, 29-fold, decrease in kcat/Km for cefotaxime (p-value of 0.0002 when compared to wild-type) (Supporting Information Figure S1). Therefore, the 237 and 276 side chains together enhance the hydrolysis of cephalosporin antibiotics but have only small effects on the hydrolysis of benzylpenicillin. Furthermore, the effects of the mutations on kcat/Km are significantly larger for the oxyimino-cephalosporin cefotaxime versus cephalothin hydrolysis (Supporting Information Figure S1). In the double mutant, replacing Arg276 with either asparagine or alanine has a similar effect on catalysis for all substrates, implying a role for positive charge at position 276 on the observed effects.
An interesting finding to emerge from the kinetic analysis of the S237A:R276A/N double mutants is that introducing both mutations into the CTX-M-14 enzyme at the same time decreases its selectivity toward cefotaxime compared to that for cephalothin and benzylpenicillin. Since Ser237 and Arg276 are conserved among the CTX-M enzymes, the results indicate these positions are important determinants of the ESBL-properties of CTX-M β-lactamases.
The fact that Ser237 and Arg276 appear together among CTX-M enzymes suggests they may work together to facilitate catalysis and, therefore, that the mutations at these positions may be coupled and nonadditive. The change in a functional property (kcat/Km, in this case) caused by a single mutation at sites X and Y can be expressed relative to the wild-type enzyme as ΔΔG(X) and ΔΔG(Y).31 Similarly, the ΔΔG(X,Y) represents the free energy differences between wild-type and the double mutant. In the absence of any cooperativity, the free energy difference of the double mutant is expected to be the sum of the free energy changes of the single mutants. However, if the there is an interaction between the two, then the observed effect of the double mutant is given by
| 3 |
where ΔGI represents the coupling free energy. The sign of the coupling free energy indicates whether the cooperativity serves to increase or decrease the observed activity relative to the wild-type.31
Analysis of the double mutant cycles for the catalytic efficiency toward the antibiotics tested reveals larger ΔGI values for the cephalosporins, cephalothin and cefotaxime, than for benzylpenicillin (Table 3). Mutations at positions 237 and 276, whether singly or in combination, have small effects (<2-fold) on the kcat/Km values for the hydrolysis of benzylpenicillin. For the cephalosporins cephalothin and cefotaxime, however, the combined mutations act cooperatively to reduce catalytic efficiency of the double mutants more than expected based on adding the effects of the single mutants (Table 3). For cefotaxime hydrolysis, the single mutation S237A decreases kcat/Km by 3-fold, and the R276N mutation decreases it by 6-fold. If the two mutations were additive, then the double mutant would be expected to decrease the kcat/Km for cefotaxime by 18-fold; however, the observed decrease was 29-fold, larger than expected based on simple additivity. Similarly, based on the single mutants, the S237A:R276A double mutant would be expected to decrease the kcat/Km for cefotaxime by 7.6-fold; however, the observed decrease was 14-fold. Thus, residues Ser237 and Arg276 work together cooperatively to facilitate cefotaxime hydrolysis.
Table 3. Free Energy Values and Additivity Relationships between Single and Double Mutants for kcat/Km.
| S237A | R276A | S237A:R276A | ΔGI | ||
|---|---|---|---|---|---|
| ΔΔG kcat/Km (kcal/mol) | benzylpenicillin | –0.40 ± 0.9 | 0.17 ± 0.05 | –0.13 ± 0.02 | 0.1 |
| cephalothin | 0.12 ± 0.04 | 0.3 ± 0.1 | 0.9 ± 0.3 | 0.4 | |
| cefotaxime | 0.58 ± 0.06 | 0.64 ± 0.07 | 1.6 ± 0.3 | 0.4 |
| S237A | R276N | S237A:R276N | ΔGI | ||
|---|---|---|---|---|---|
| ΔΔG kcat/Km (kcal/mol) | benzylpenicillin | –0.4 ± 0.9 | 0.06 ± 0.01 | –0.22 ± 0.04 | 0.1 |
| cephalothin | 0.12 ± 0.04 | 0.4 ± 0.1 | 1.0 ± 0.3 | 0.5 | |
| cefotaxime | 0.58 ± 0.06 | 1.0 ± 0.1 | 2.0 ± 0.1 | 0.4 |
X-ray Structure Determination of the CTX-M-14 Mutant Enzymes
To investigate the structural basis behind the changes in the substrate specificity for cefotaxime hydrolysis by the single and double mutant enzymes, the crystal structures of the CTX-M-14 S70G single mutant, S70G:S237A double mutant, and S70G:S237A:R276A triple mutant were determined alone and in complex with cefotaxime (Table 4 and Figure 3). The substitution of the Ser70 nucleophilic residue with glycine was included in order to visualize bound substrate in the active site in the absence of catalysis.11 The S70G substitution did not significantly alter the shape of the catalytic pocket in the apo form, as evidenced by a root-mean-square deviation (RMSD) of 0.21, 0.18, and 0.18 Å for the matching Cα atoms of the residues in the active site that form hydrogen bonds with substrate between the wild-type CTX-M-14 enzyme (PDB ID: 1YLT) with the S70G, S70G:S237A, and S70G:S237A:R276A mutants, respectively. The apoenzyme and cefotaxime-complexed structures show an RMSD of 0.12 Å for the Cα atoms of the active site for the single mutant, 0.10 Å for the double mutant, and 0.12 Å for the triple mutant, suggesting that the substrate did not induce substantial structural modifications to the enzyme. All of the CTX-M-14 structures obtained in this study crystallized in the same space group. A fluorescence scan was performed on CTX-M-14 and the S237A and S237A:R276A mutants in order to confirm that the proteins used for kinetic analysis also exhibit no large structural changes in solution that would affect intrinsic fluorescence (Supporting Information Figure S2).
Table 4. Crystallographic Data Refinement and Statistics.
| CTX-M-14 | CTX-M-14 | CTX-M-14 | CTX-M-14 | CTX-M-14 | CTX-M-14 | |
|---|---|---|---|---|---|---|
| S70G | S70G:S237A | S70G:S237A: R276A | S70G–cefotaxime | S60G:S237A–cefotaxime | S70G:S237A: R276A–cefotaxime | |
| Data Collection | ||||||
| space group | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 |
| a, b, c (Å) | 41.6, 62.3, 86.2 | 41.5, 62.2, 86.1 | 41.7, 62.4, 86.7 | 41.5, 62.7, 86.1 | 41.5, 62.6, 86.1 | 41.5, 61.7, 86.7 |
| α = β = γ (deg) | 90 | 90 | 90 | 90 | 90 | 90 |
| resolution rangea (Å) | 22.1–1.56 (1.62–1.56) | 29.3–1.17 (1.21–1.17) | 50.6–1.39 (1.42–1.39) | 29.5–1.26 (1.31–1.26) | 29.4–1.29 (1.33–1.29) | 50–1.45 (1.48–1.45) |
| Rmerge (%) | 2.2 (5.1) | 1.4 (9.6) | 7.0 (40.4) | 1.8 (14.1) | 1.9 (15.1) | 6.1 (30.0) |
| I/sigma | 39.4 (12.2) | 17.9 (4.3) | 27.5 (4.7) | 19.1 (4.9) | 17.6 (4.4) | 22.2 (5.0) |
| completeness (%) | 100 (100) | 99.9 (100) | 99.9 (100) | 99.7 (98.8) | 96.8 (93.2) | 98.9 (79.9) |
| Wilson B-factor (Å2) | 9.90 | 9.18 | 8.39 | 9.25 | 9.16 | 11.64 |
| Refinement | ||||||
| molecules per asymmetric unit | 1 | 1 | 1 | 1 | 1 | 1 |
| no. of unique reflections | 32 576 | 76 012 | 46 446 | 61 202 | 56 078 | 41 240 |
| Rwork/Rfree (%) | 14.2/16.2 | 12.6/14.3 | 15.3/18.1 | 16.2/18.2 | 16.1/18.4 | 16.1/18.0 |
| no. of protein atoms | 1962 | 1963 | 1957 | 1962 | 1961 | 1950 |
| average B-factor (Å2) | ||||||
| protein | 10.12 | 12.20 | 10.08 | 11.33 | 10.85 | 12.63 |
| solvent | 24.55 | 30.30 | 22.52 | 23.47 | 22.88 | 22.53 |
| ligand | 17.10 | 13.90 | 16.2 | |||
| RMS deviations | ||||||
| bond length (Å) | 0.006 | 0.005 | 0.006 | 0.005 | 0.006 | 0.005 |
| bond angles (deg) | 1.1 | 1.1 | 1.1 | 1.1 | 1.1 | 1.1 |
| PDB codes | 4PM6 | 4PM8 | 4PMA | 4PM5 | 4PM7 | 4PM9 |
Numbers in the parentheses indicate values for highest resolution shell.
Comparing the cefotaxime-bound CTX-M-14 mutant structures with a previously published structure of the CTX-M-9 S70G mutant complexed with cefotaxime (PDB ID: 3HLW) showed an RMSD of 0.21 Å for the Cα atoms in the active site for the single mutant, 0.21 Å for the double mutant, and 0.20 Å for the triple mutant, indicating that there are no major displacements of active site residues.11 Lastly, in an alignment of all the residues, no residue exhibited greater than a 1 Å deviation when the cefotaxime-bound double and triple mutants were compared to the cefotaxime-bound S70G mutant.
Interactions between the CTX-M-14 Mutant Enzymes and Cefotaxime
The structure of the CTX-M-14 S70G enzyme in complex with cefotaxime maintains all of the same electrostatic and hydrophobic interactions between cefotaxime in the active site as those in the previously published structure of CTX-M-9 in complex with cefotaxime.11 The previous structure of the CTX-M-9 enzyme in complex with cefotaxime, however, contains an additional cefotaxime molecule at the entrance to the active site that was proposed to indicate an entry site for the drug to the active site.11 The additional cefotaxime molecule was not observed in our CTX-M-14 S70G–cefotaxime structure, which could be the result of crystallization in a different space group.
Comparing the structures of the cefotaxime complex of CTX-M-14 S70G (wild-type), S70G:S237A, and S70G:S237A:R276A shows four major structural differences resulting from the mutations. First, upon removal of the S237 hydroxyl group (S70G:S237A), the C4 carboxylate group of cefotaxime assumes an alternative position, moving closer to Lys234 than in the wild-type enzyme. Second, the aminothiazole ring of cefotaxime is flipped 180° in the S70G:S237A:R276A–cefotaxime structure, moving away from the binding site in the wild-type structure. Third, the hydrogen bond between Asn170(O) and Asp240(N), that is characteristic of the smaller TEM-1/SHV active site, is present in the wild-type and mutant apo enzymes. This hydrogen bond is broken when cefotaxime binds to the S70G and S70G:S237A enzymes; however, it is retained in the S70G:S237A:R276A–cefotaxime structure. Finally, the overall hydrogen-bonding network between cefotaxime and the active site in the S70G:S237A:R276A structure is diminished (Figure 4).
When Ser237 is mutated to the TEM/SHV-type residue, alanine, it eliminates the hydrogen bond observed in the wild-type (S70G) cefotaxime complex between the C4 carboxylate of cefotaxime with Ser237Oγ and Ser130Oγ (Figure 5A). As a result, the C4 carboxyl group shifts into the position occupied by a water molecule in the S70G–cefotaxime structure (Figure 5A). There, it participates in the same hydrogen bonds with Lys234(N2) and Lys73(N2) as the water molecule does in the S70G–cefotaxime structure. The vast majority of the hydrophobic interactions with cefotaxime are maintained between the S70G and S70G:S237A structures (Figure 4A,B). The position of the C4 carboxyl group near where the Ser70 oxygen would normally reside is, in part, due to the S70G substitution creating space for the carboxyl in this region. Nevertheless, the result indicates the loss of interaction with Ser237Oγ leads to repositioning of the C4 carboxyl group and a lower catalytic efficiency.
A previous comparison of the structure of the CTX-M-9 S70G apoenzyme with the cefotaxime-bound structure found that the hydrogen bond between Asn170(O) and Asp240(N) is ruptured to accommodate cefotaxime in the binding pocket.11 It was hypothesized that breaking the Asn170–Asp240 hydrogen bond widens the active site to accommodate and position cefotaxime more optimally for catalysis. This hydrogen bond is also absent in the CTX-M-14 S70G–cefotaxime and S70G:S237A–cefotaxime structures reported here; however, when both Ser237 and Arg276 side chains are converted to alanine in the double mutant, the hydrogen bond between Asn170 and Asp240 is retained, even in the presence of cefotaxime. This narrows the active site in this region, making it more difficult for cefotaxime to achieve optimal positioning and decreasing kcat/Km for catalysis of cefotaxime (Figure 5B).
The removal of the side chains of Ser237 and Arg276 also allow the aminothiazole ring to flip 180° into a position that makes fewer contacts with the enzyme. The hydrogen bond between the Asp240 oxygen(Oε1) and the amino group on the aminothiazole is also lost (Figure 5B). The presence of the Ser237 hydroxyl group would be sufficient to sterically interfere with the aminothiazole ring in this flipped conformation; however, removing this steric clash is not sufficient to cause the ring to flip. The side chain of Arg276 must also be converted to alanine before the flip occurs. It is clear from this structural feature that Ser237 and Arg276 work cooperatively to stabilize the bound cefotaxime with the aminothiazole ring in a conformation that is more buried in the active site and forms more contacts with CTX-M-14 residues.
Overall, the hydrogen-bonding network between cefotaxime and the CTX-M-14 enzyme is compromised in the S70G:S237A:R276A–cefotaxime structure as compared to the others (Figure 4C). In the pseudo-wild-type S70G–cefotaxime complex, cefotaxime is anchored into the active site by 12 hydrogen bonds involving 7 residues and 3 water molecules and hydrophobic interactions with 5 residues. In the S70G:S237A:R276A–cefotaxime structure, however, cefotaxime is anchored into the active site by only 9 hydrogen bonds involving 8 enzyme residues and no water molecules as well as hydrophobic interactions with 6 residues (Figure 4C). The absence of interactions between cefotaxime and the enzyme mediated by hydrogen bonding to water molecules in the S270G:S237A:R276A structure is related to the fact that these interactions in the other structures involve bonds with the aminothiazole ring of cefotaxime, which are lost with the 180° rotation of the ring in the triple mutant (Figures 4C and 5B). It is also interesting to note that these changes in water structure and hydrogen bonding are distant from the Ser237 and Arg276 substitutions. Thus, the changes in interactions with substrate at the site of substitution propagate to change interactions far from the site of mutation.
The hydrogen bond between the C3 methyl acetate group and water is lost in the S70G:S237A:R276A structure, which could lead to increased flexibility of this group, resulting in the slightly different placement in the active site (Figure 5C). A comparison of the average B-factors of the C3 methyl acetate group in the three structures shows averages of 21.2, 13.9, and 27.9 Å2 for the single, double, and triple mutants in complex with cefotaxime, respectively. This supports the notion that loss of the hydrogen bond between the C3 methyl acetate group and water slightly increases flexibility of this region. The overall B-factors for cefotaxime in the S70G, S70G:S237A, and S70G:S237A:R276A structure are 17.1, 13.9, and 16.2 Å2, respectively.
In summary, the retention of the Asn170–Asp240 hydrogen bond, flipping of the aminothiazole ring of cefotaxime, increased flexibility of the C3 methyl acetate group, and the absence of waters that bridge interactions between the substrate and the enzyme results in reduced hydrogen bonding and hydrophobic interactions, a narrower binding pocket, and a 14–29-fold reduction in kcat/Km for cefotaxime hydrolysis. Cumulatively, these changes in the positioning and bonding network of cefotaxime in the active site support the cooperative nature of positions Ser237 and Arg276 and highlight their contributions to enhancing cefotaxime hydrolysis in the CTX-M enzymes.
Discussion
The CTX-M family of β-lactamases is a widespread source of resistance to extended-spectrum cephalosporin antibiotics among Gram-negative bacteria. A distinguishing feature of the CTX-M enzymes compared to other class A β-lactamases, such as the commonly occurring TEM-1 and SHV-1 enzymes, is their ability to efficiently hydrolyze cefotaxime. The CTX-M enzymes are also distinguished by the conservation of the active site residues Ser237 and Arg276 in that this combination does not occur in other class A β-lactamases.14 In this study, we have used kinetic and structural methods to show that Ser237 and Arg276 act cooperatively to promote cefotaxime hydrolysis through structural alterations of the active site that better accommodate the larger cefotaxime molecule. As a result, these residues contribute to the unique substrate specificity of the CTX-M family of enzymes.
The X-ray crystal structures of the CTX-M-14 S70G, S70G:S237A, and S70G:S237A:R276A mutants with and without cefotaxime bound revealed that the mutations result in flipping of the aminothiazole ring 180°, decreased hydrogen bonding with the enzyme, and, importantly, a hydrogen bond between Asn170 and Asp240 that is retained when cefotaxime is bound. Previous work by Delmas et al. indicated that binding of cefotaxime in CTX-M-9 β-lactamase results in a conformational change in the active site involving rupture of a hydrogen bond between main chain groups of Asn170 and Asp240 that connects the omega loop to the β3 strand.11 The net result of this change is to expand the active site to allow adequate positioning of the cefotaxime substrate for catalysis, which was hypothesized to contribute to the cefotaximase activity of the CTX-M enzymes.11 The results of this study support this model since the preservation of this hydrogen bond in the S70G:S237A:R276A–cefotaxime complex is associated with a significant loss of activity as a cefotaximase (14-fold).
As noted above, Ser237 and Arg276 act cooperatively to increase kcat/Km for cefotaxime hydrolysis despite the fact that they are more than 8 Å apart and not within direct contact distance. Wells et al. analyzed the additivity of approximately 25 double mutations that were beyond van der Waals contact distance in the wild-type structures of several enzymes.31 In these cases, the effects of the double mutation on binding and/or catalysis were largely additive, suggesting that the effects of mutating residues that do not directly interact with each other, as is the case for Ser237 and Arg276, are largely additive. Nonadditive effects were generally attributed to direct or indirect interactions of the two residues mediated through the protein or changes in mechanism.31 A comparison of the X-ray structures of S70G–cefotaxime and S70G:S237A–cefotaxime versus S70G:S237A:R276A–cefotaxime shows changes in substrate orientation and hydrogen bonding, as described above (Figure 5). On the basis of these results, it appears that the cooperative effect between S237A and R276A is mediated indirectly through the substrate, i.e., although the residues are not in direct contact, they are bridged through contact with cefotaxime in the enzyme–substrate complex. Thus, Ser237, Arg276, and cefotaxime together form a connected, cooperative unit.
The S237A:R276N double mutation of CTX-M-14 suggests the cooperative interactions of Ser237 and Arg276 with cefotaxime contributes approximately 30-fold to enhancing hydrolysis compared to the TEM-1/SHV1-like active site. Given that the overall sequence identity of the two enzymes is only ∼35%, the rest of the sequence changes must contribute an additional rate enhancement of 30-fold to account for the approximately 1000-fold ratio in kcat/Km for hydrolysis of cefotaxime catalyzed by CTX-M-14 compared to its hydrolysis by TEM-1. Therefore, there may be additional positions that contribute to the unique cefotaximase substrate specificity of the CTX-M enzymes.
In conclusion, CTX-M-14 residues Ser237 and Arg276 work together in the hydrolysis of β-lactam substrates to help shape the substrate specificity of the enzyme to that of an effective cefotaximase. The crystal structures of the CTX-M-14 S70G, S70G:S237A, and S70G:S237A:R276A mutants alone and in complex with cefotaxime revealed the importance of residues Ser237 and Arg276 in establishing the interactions with cefotaxime and cooperatively shaping the active site in the region of Asn170 and Asp240. Finally, the results suggest the cooperative interaction between Ser237 and Arg276 is mediated via an indirect interaction bridging through the cefotaxime substrate. Cooperative interactions among residues that contact the substrate or transition state may serve as useful points for engineering protein–small molecule interactions for binding or for catalysis.
Glossary
Abbreviations
- ESBL
extended spectrum β-lactamases
- BLIP-II
β-lactamase inhibitory protein II
- MIC
minimum inhibitory concentration
Supporting Information Available
Table S1: MICs of CTX-M-14 β-lactamase and mutant enzymes. Figure S1: Catalytic efficiency of CTX-M-14 β-lactamase and mutant enzymes. Figure S2: Fluorescence scan of CTX-M-14 β-lactamase and mutant enzymes. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The coordinates of the structures reported in this article have been deposited in the Protein Data Bank (www.rcsb.org) (PDB ID codes 4PM5, 4PM6, 4PM7, 4PM8, 4PM9, and 4PMA).
Author Contributions
⊥ C.J.A. and A.M.C. contributed equally to this work.
This work was supported by a training fellowship from the Houston Area Molecular Biophysics Training Program (NIGMS grant no. T32 GM008280) of the Gulf Coast Consortia to C.J.A., a training fellowship from the Keck Center of Biomedical Discovery Training Program of the Gulf Coast Consortia (NIH grant no. 1 R90 DA023418-03) to A.M.C., and a training fellowship from the Keck Center of Pharmacoinformatics Training Program of the Gulf Coast Consortia (NIH grant no. T90 DK070109-05) to N.G.B. This work was supported by NIH grant no. AI32956 to T.P. B.V.V.P. acknowledges support from Robert Welch Foundation (Q1279). This work was supported by The Berkeley Center for Structural Biology, supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fisher J. F.; Meroueh S. O.; Mobashery S. (2005) Bacterial resistance to beta-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 105, 395–424. [DOI] [PubMed] [Google Scholar]
- Ambler R. P.; Coulson F. W.; Frere J.-M.; Ghuysen J.-M.; Joris B.; Forsman M.; Levesque R. C.; Tiraby G.; Waley S. G. (1991) A standard numbering scheme for the class A β-lactamases. Biochem. J. 276, 269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K.; Fisher J. F. (2011) Epidemiological expansion, structural studies, and clinical challenges of new β-lactamases from Gram-negative bacteria. Annu. Rev. Microbiol. 65, 455–478. [DOI] [PubMed] [Google Scholar]
- Bonnet R. (2004) Growing group of extended-spectrum beta-lactamases: the CTX-M enzymes. Antimicrob. Agents Chemother. 48, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Delmas J.; Sirot J.; Shoichet B. K.; Bonnet R. (2005) Atomic resolution structures of CTX-M beta-lactamases: extended spectrum activities from increased mobility and decreased stability. J. Mol. Biol. 348, 349–362. [DOI] [PubMed] [Google Scholar]
- Canton R.; Gonzalez-Alba J. M.; Galan J. C. (2012) CTX-M enzymes: origin and diffusion. Front. Microbiol 3, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orencia M. C.; Yoon J. S.; Ness J. E.; Stemmer W. P.; Stevens R. C. (2001) Predicting the emergence of antibiotic resistance by directed evolution and structural analysis. Nat. Struct. Biol. 8, 238–242. [DOI] [PubMed] [Google Scholar]
- Wang X.; Minasov G.; Shoichet B. K. (2002) Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J. Mol. Biol. 320, 85–95. [DOI] [PubMed] [Google Scholar]
- Ibuka A.; Taguchi A.; Ishiguro M.; Fushinobu S.; Ishii Y.; Kamitori S.; Okuyama K.; Yamaguchi K.; Konno M.; Matsuzawa H. (1999) Crystal structure of the E166A mutant of extended-spectrum β-lactamase Toho-1 at 1.8 Å resolution. J. Mol. Biol. 285, 2079–2087. [DOI] [PubMed] [Google Scholar]
- Ibuka A. S.; Ishii Y.; Galleni M.; Ishiguro M.; Yamaguichi K.; Frere J. M.; Matsuzawa H.; Sakai H. (2003) Crystal structure of extended-spectrum beta-lactamase Toho-1: insights into the molecular mechanism for catalytic reaction and substrate specificity expansion. Biochemistry 42, 10634–10643. [DOI] [PubMed] [Google Scholar]
- Delmas J.; Leyssene D.; Dubois D.; Birck C.; Vazeille E.; Robin F.; Bonnet R. (2010) Structural insights into substrate recognition and product expulsion in CTX-M enzymes. J. Mol. Biol. 400, 108–120. [DOI] [PubMed] [Google Scholar]
- Delmas J.; Chen Y.; Prati F.; Robin F.; Shoichet B. K.; Bonnet R. (2008) Structure and dynamics of CTX-M enzymes reveal insights into substrate accommodation by extended-spectrum beta-lactamases. J. Mol. Biol. 375, 192–201. [DOI] [PubMed] [Google Scholar]
- Perez-Llarena F. J.; Carelle M.; Mallo S.; Beceiro A.; Perez A.; Villanueva R.; Romero A.; Bonnet R.; Bou G. (2008) Structure-function studies of arginine at position 276 in CTX-M β-lactamases. J. Antimicrob. Chemother. 61, 792–797. [DOI] [PubMed] [Google Scholar]
- Marciano D. C.; Brown N. G.; Palzkill T. (2009) Analysis of the plasticity of location of positive charge within the active site of the TEM-1 β-lactamase. Protein Sci. 18, 2080–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strynadka N. C. J.; Adachi H.; Jensen S. E.; Johns K.; Sielecki A.; Betzel C.; Sutoh K.; James M. N. G. (1992) Molecular structure of the acyl-enzyme intermediate in β-lactam hydrolysis at 1.7 Å resolution. Nature 359, 700–705. [DOI] [PubMed] [Google Scholar]
- Wang X.; Minasov G.; Shoichet B. K. (2002) The structural bases of antibiotic resistance in clinically derived mutant beta-lactamases TEM-30, TEM-32 and TEM-34. J. Biol. Chem. 277, 32149–32156. [DOI] [PubMed] [Google Scholar]
- Zafaralla G.; Manavathu E. K.; Lerner S. A.; Mobashery S. (1992) Elucidation of the role of arginine-244 in the turnover processes of class A beta-lactamases. Biochemistry 31, 3847–3852. [DOI] [PubMed] [Google Scholar]
- Gazouli M.; Legakis N. J.; Tzouvelekis L. S. (1998) Effect of substitution of Asn for Arg-276 in the cefotaxime-hydrolyzing class A β-lactamase CTX-M-4. FEMS Microbiol. Lett. 169, 289–293. [DOI] [PubMed] [Google Scholar]
- Gazouli M.; Tzelepi E.; Sidorenko S. V.; Tzouvelekis L. S. (1998) Sequence of the gene encoding a plasmid-mediated cefotaxime-hydrolyzing class A β-lactamase (CTX-M-4): involvement of serine 237 in cephalosporin hydrolysis. Antimicrob. Agents Chemother. 42, 1259–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N. G.; Chow D.; Ruprecht K. E.; Palzkill T. (2013) Identification of the β-lactamase inhibitor protein-II (BLIP-II) interface residues essential for binding affinity and specificity for class A β-lactamases. J. Biol. Chem. 288, 17156–17166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 2nd ed., Wiley-VCH, New York. [Google Scholar]
- Battye T. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr., Sect. D.: Biol. Crystallogr. 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025. [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode, in Methods in Enzymology: Macromolecular Crystallography, Part A (Carter C. W., and Sweet R. M., Eds.) pp 307–326, Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; Swindells M. B. (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, version 1.3, Shrödinger, LLC.
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Wells J. (1990) Additivity of mutational effects in proteins. Biochemistry 29, 8509–8517. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.