

Abstract
Protein tyrosine phosphatases (PTPs) have been the subject of considerable pharmaceutical-design efforts because of the ubiquitous connections between misregulation of PTP activity and human disease. PTP-inhibitor discovery has been hampered, however, by the difficulty in identifying cell-permeable compounds that can selectively target PTP active sites, and no PTP inhibitors have progressed to the clinic. The identification of allosteric sites on target PTPs therefore represents a potentially attractive solution to the druggability problem of PTPs. Here we report that the oncogenic PTP Shp2 contains an allosteric-inhibition site that renders the enzyme sensitive to potent and selective inhibition by cell-permeable biarsenical compounds. Because Shp2 contains no canonical tetracysteine biarsenical-binding motif, the enzyme’s inhibitor-binding site is not readily predictable from its primary or three-dimensional structure. Intriguingly, however, Shp2’s PTP domain does contain a cysteine residue (C333) at a position that is removed from the active site and is occupied by proline in other classical PTPs. We show that Shp2’s unusual cysteine residue constitutes part of a Shp2-specific allosteric-inhibition site, and that Shp2’s sensitivity to biarsenicals is dependent on the presence of the naturally occurring C333. The determinative role of this residue in conferring inhibitor sensitivity is surprising because C333’s side chain is inaccessible to solvent in Shp2 crystal structures. The discovery of this cryptic Shp2 allosteric site may provide a means for targeting Shp2 activity with high specificity and suggests that buried-yet-targetable allosteric sites could be similarly uncovered in other protein families.
The protein tyrosine phosphatases (PTPs) constitute a large family of signaling enzymes that dephosphorylate specific phosphotyrosine residues in protein substrates.1 Tight control of PTP activity is critical for maintaining appropriate levels of tyrosine-phosphorylated signaling proteins, and aberrant PTP activity contributes to a wide range of human diseases.2,3 Src-homology-2-domain-containing PTP 2 (Shp2) provides a particularly striking example of the connection between misregulation of PTP activity and human pathogenesis: germline Shp2 mutations cause Noonan and Leopard syndromes, both of which can lead to cancer predisposition.4−6 Moreover, somatic Shp2 mutations are the most common cause of sporadic juvenile myelomonocytic leukemia.7,8
Because of its associations with human disease, Shp2 has been the subject of significant pharmaceutical-discovery efforts.9−12 Although moderately selective active-site-directed inhibitors of Shp2 have been identified, Shp2-inhibitor discovery often suffers from the same limitations that have led to the general characterization of PTPs as undruggable;13 specifically, active-site-directed PTP inhibitors often suffer from a lack of target specificity (classical PTP active sites share a high degree of sequence and structural homology) and poor bioavailability (most of the known PTP-binding pharmacophores contain negatively charged phosphotyrosine mimetics that lower a putative inhibitor’s cellular permeability).
The factors that have limited success in the field of active-site-directed PTP inhibitors generally—Shp2 inhibitors specifically—point to the need for the discovery of new allosteric sites. Previous reports have shown that PTPs can be inhibited allosterically by targeting protein regions outside of their catalytic domains,14,15 and, more relevantly to the work presented here, that PTP catalytic domains can themselves contain targetable allosteric-inhibition sites.16,17 In particular, the catalytic domain of PTP1B (39% PTP-domain identity with Shp2) can be inhibited allosterically by two distinct mechanisms of action. Weismann and co-workers discovered an allosteric site on PTP1B that is approximately 20 Å from the enzyme’s active site and showed that small molecules that noncovalently bind the allosteric site are capable of inhibiting the enzyme, albeit with moderate (low micromolar) potency.17 Hansen and co-workers later demonstrated that PTP1B could be inhibited covalently via modification of a non-active-site cysteine residue (C121 in human PTP1B) by high concentrations (high micromolar to millimolar) of the electrophilic reagent 4-(aminosulfonyl)-7-fluoro-2,1,3-benzoxadiazole (ABDF).16 The selectivity of ABDF among PTPs is likely very low, however, as the compound’s amino acid target, C121, is highly conserved among mammalian classical PTPs.1
Taken together, these seminal studies on allosteric inhibition of PTP1B have been critical for establishing the idea that allosteric sites may indeed exist on PTP domains, but the compounds discovered to date that target these sites exhibit only moderate to weak potency and selectivity. No catalytic-domain allosteric sites that allow a PTP to be targeted with high selectivity have been discovered, and beyond PTP1B, allosteric-inhibition sites have not been discovered on the PTP domains of the remaining members of the classical PTP family. Here we report the discovery of a cryptic allosteric site on Shp2’s catalytic domain. The Shp2-specific allosteric-inhibition site comprises two cysteine residues, one conserved (C367, Shp2’s position analogous to C121 of PTP1B) and one at a position that is occupied by proline in most other classical PTPs (C333). Our results demonstrate that the presence of both of these cysteine residues, almost unique among classical PTPs to Shp2, renders the enzyme sensitive to selective and potent allosteric inhibition by cysteine-reactive cell-permeable biarsenical compounds.
Materials and Methods
General
All details for the cloning, mutagenesis, expression, and purification of the PTP constructs described in the text can be found in the Supporting Information. FlAsH-EDT2 (FlAsH) was synthesized as described previously.18−20 ReAsH-EDT2 was purchased from Life Technologies as a 2 mM stock solution in dimethyl sulfoxide (DMSO). Fluorescein was purchased from Acros. Arsenic trioxide (As2O3) was purchased from Sigma-Aldrich. Stock solutions and dilutions of biarsenical compounds and fluorescein were prepared in DMSO, which was added to the negative controls for all assays that investigated these compounds. Stock solutions and dilutions of As2O3 were prepared in aqueous sodium hydroxide (250 μM), which was added to the negative controls for all assays that investigated this compound. All PTP assays were performed in triplicate; error bars and “±” values represent the standard deviations of at least three independent experiments unless noted otherwise.
Phosphatase Activity and Inhibition Assays with Small-Molecule Substrates (pNPP and DiFMUP)
PTP assays using p-nitrophenyl phosphate (pNPP) as a substrate were conducted in a total volume of 200 μL, containing PTP buffer [50 mM 3,3-dimethyl glutarate (pH 7.0), 1 mM EDTA, and 50 mM NaCl], enzyme (50 nM), and pNPP (0.625–10 mM). PTP reactions were quenched by the addition of 40 μL of 5 M NaOH, and the absorbances (405 nm) of 200 μL of the resulting solutions were measured on a VersaMax plate reader (Molecular Devices). Kinetic constants were determined by fitting the data to the Michaelis–Menten equation using SigmaPlot 12.3. For inhibition experiments (FlAsH, fluorescein, ReAsH, or As2O3), PTP samples (50 nM) were preincubated with the compound of interest (or control) for 120 min at 37 °C prior to initiation of the PTP assays. The PTP activities of the solutions were then measured when they were assayed at 37 °C with pNPP as described above at a pNPP concentration equal to the previously determined KM of the enzyme; 50% inhibitory-concentration (IC50) values were estimated by fitting the inhibition data to a four-parameter logistic equation for IC50 determination in SigmaPlot 12.3. For FlAsH-inhibition time-dependence experiments, pNPP (at a final concentration equal to the previously determined KM of the enzyme) was incubated with FlAsH [or DMSO, 1% (v/v)] in PTP buffer at 37 °C, and PTP reactions were initiated by adding enzyme (50 nM). The change in absorbance at 405 nm was then measured continuously at 37 °C.
For inhibition experiments using whole cell lysates, total PTP activities of lysates from cells expressing the PTP of interest were measured using pNPP as described above. To prepare lysates, 100 mL aliquots of BL21(DE3)-CodonPlus Escherichia coli cells expressing the PTP of interest (as described in Expression and Purification of Shp2 and Shp1 in the Supporting Information) were removed from larger cultures, pelleted, and stored at −80 °C. The pellets were then resuspended in PTP buffer, lysed with a French press, and clarified by centrifugation. Lysates were incubated with FlAsH-EDT2 (concentrations ranging from 0.78125 to 25 μM) or DMSO [1% (v/v)] for 120 min at 37 °C and assayed with 10 mM pNPP.
For FlAsH-inhibition assays using 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) as a substrate, PTP samples (50 nM) were incubated with DMSO [0.5% (v/v)] or FlAsH (concentrations ranging from 62.5 nM to 1 μM) for 150 min at 30 °C in PTP buffer. Reactions were initiated by the addition of DiFMUP (20 μM; total reaction volume of 200 μL), and the fluorescence (excitation at 360 nm, emission at 450 nm) of the resulting solutions was measured continuously on a SpectraMax plate reader (Molecular Devices) at 30 °C. For time-dependence experiments, PTP samples (50 nM) were incubated with FlAsH (1 μM) or DMSO for 15–240 min at 30 °C. The PTP activities of the solutions were then assayed with DiFMUP as described above. IC50 values were estimated as described above.
Phosphatase Activity and Inhibition Assays with Phosphopeptide
PTP assays using phosphopeptide as a substrate were conducted in a total volume of 150 μL, containing reaction buffer [50 mM 3,3-dimethyl glutarate (pH 7.0), 125 mM NaCl, and 1 mM EDTA], enzyme (diluted to 50 nM in reaction buffer), and phosphopeptide substrate (100 μM, DADEpYLIPQQG, Calbiochem). Reactions were initiated by the addition of the phosphopeptide after incubation of the enzyme with FlAsH for 150 min at room temperature, and the change in absorbance at 282 nm was measured continuously.21
Mass Spectrometry-Based FlAsH-Induced Cysteine-Protection Assay22
Samples for liquid chromatography–tandem mass spectrometry (LC–MS/MS) were prepared by incubating wild-type or C333P Shp2 (100 μg, 2.7 μM) with either DMSO or FlAsH (27 μM) for 30 min in Shp2 catalytic domain storage buffer [50 mM 3,3-dimethyl glutarate (pH 7.0), 1 mM EDTA, and 1 mM TCEP]. Samples were then incubated with iodoacetic acid (IAA, 50 mM) for 30 min to modify free cysteine residues.16 IAA labeling was quenched with TCEP (100 mM), and the samples were precipitated with trifluoroacetic acid and assessed via sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Coomassie-stained gel slices were cut into 1 mm × 1 mm pieces and treated with trypsin. Digested samples dissolved in acetonitrile and trifluoroacetic acid were injected onto a custom packed 2 cm × 100 μm C18 Magic 5 μm particle trap column. Labeled peptides were then eluted and sprayed from a custom packed emitter (75 μm × 25 cm, C18 Magic 3 μm particle) with a linear gradient from 95% solvent A (0.1% formic acid in water) to 35% solvent B (0.1% formic acid in acetonitrile) in 60 min at a flow rate of 300 nL/min on a Waters Nano Acquity UPLC system. Data dependent acquisitions were performed on a Q Exactive mass spectrometer (Thermo Scientific) according to an experiment in which full MS scans from m/z 300 to 1750 were acquired at a resolution of 70000 followed by 12 MS/MS scans acquired under HCD fragmentation at a resolution of 35000 with an isolation width of 1.2 Da. Raw data files were processed with Mascot Distiller (version 2.5) prior to searching with Mascot Server (version 2.4) against a SwissProt Human database containing the construct sequences. Search parameters utilized were fully tryptic with two missed cleavages, parent mass tolerances of 10 ppm, and fragment mass tolerances of 0.05 Da. Variable modifications of acetyl (protein N-terminus), pyroglutamic for N-terminal glutamine, oxidation of methionine, and carboxymethyl cysteine were considered. Search results were used to create spectral libraries for the Skyline software (University of Washington, Seattle, WA), which was used to quantitate selected peptides using precursor intensity data from extracted ion chromatograms. Intensities for peptides identified in the FlAsH-treated protein sample were compared to the corresponding peptides identified in the DMSO-treated sample, and their resulting intensity ratios were calculated. Intensity ratios were then divided by the average of all non-cysteine-containing peptide ratios to determine normalized relative abundance values. Peptides that were detected three or more times in the wild-type Shp2 experiments were included in the abundance analysis.
Results and Discussion
Selective Shp2 Inhibition by the Organic Biarsenical Compound FlAsH-EDT2
The organic biarsenical compounds FlAsH-EDT219 and ReAsH-EDT223 (henceforth FlAsH and ReAsH, respectively, for the sake of simplicity) make strong and specific interactions with the tetracysteine peptide motif CCXXCC, and these biarsenicals have been used for labeling engineered tetracysteine-tagged proteins in a wide variety of applications.24 (See Figure 2A for the structures of FlAsH, ReAsH, and FlAsH’s parent fluorophore, fluorescein.) Wild-type PTP domains, including Shp2’s catalytic domain, however, contain no cysteine-rich motifs, and biarsenicals have not previously been shown to substantially inhibit the activity of any wild-type PTP.1,20,25 We were therefore surprised to observe that the ability of the wild-type mouse Shp2 catalytic domain to dephosphorylate the small-molecule substrate pNPP is strongly inhibited in the presence of the most widely used biarsenical FlAsH. After incubation with 1 μM FlAsH, the PTP activity of Shp2 is almost undetectable, whereas the PTP activities of catalytic domains from a range of other PTPs representing four different subfamilies1 are not substantially affected or only moderately so (Figure 1A; subfamily NT1, PTP1B and TCPTP; subfamily NT5, PTPH1; subfamily NT7, FAP-1; and subfamily R7, HePTP). In addition to the strong inhibition of Shp2, only the catalytic domain of Shp1, which shares 59% PTP-domain identity with Shp2 and is a fellow member of the NT2 subfamily,26 is more than 50% inhibited at 1 μM FlAsH. These findings led us to investigate the dose responses of Shp2 and Shp1 in the presence of FlAsH. Consistent with our initial screen, the activity of Shp2 dropped dramatically in a dose-dependent manner when it was preincubated with FlAsH (Figure 1B). From these data, a Shp2/FlAsH 50% inhibitory-concentration (IC50) value of 74 nM and a Shp1/FlAsH IC50 of approximately 700 nM could be estimated. Note that FlAsH so potently inhibits Shp2 that the IC50 value approaches the concentration of enzyme in the assay (50 nM enzyme, the lowest concentration that can be routinely used with pNPP at pH 7.0). FlAsH’s inhibition of Shp2 is therefore essentially stoichiometric, suggesting that the observed IC50 value is highly dependent on the assay conditions chosen.
Figure 2.
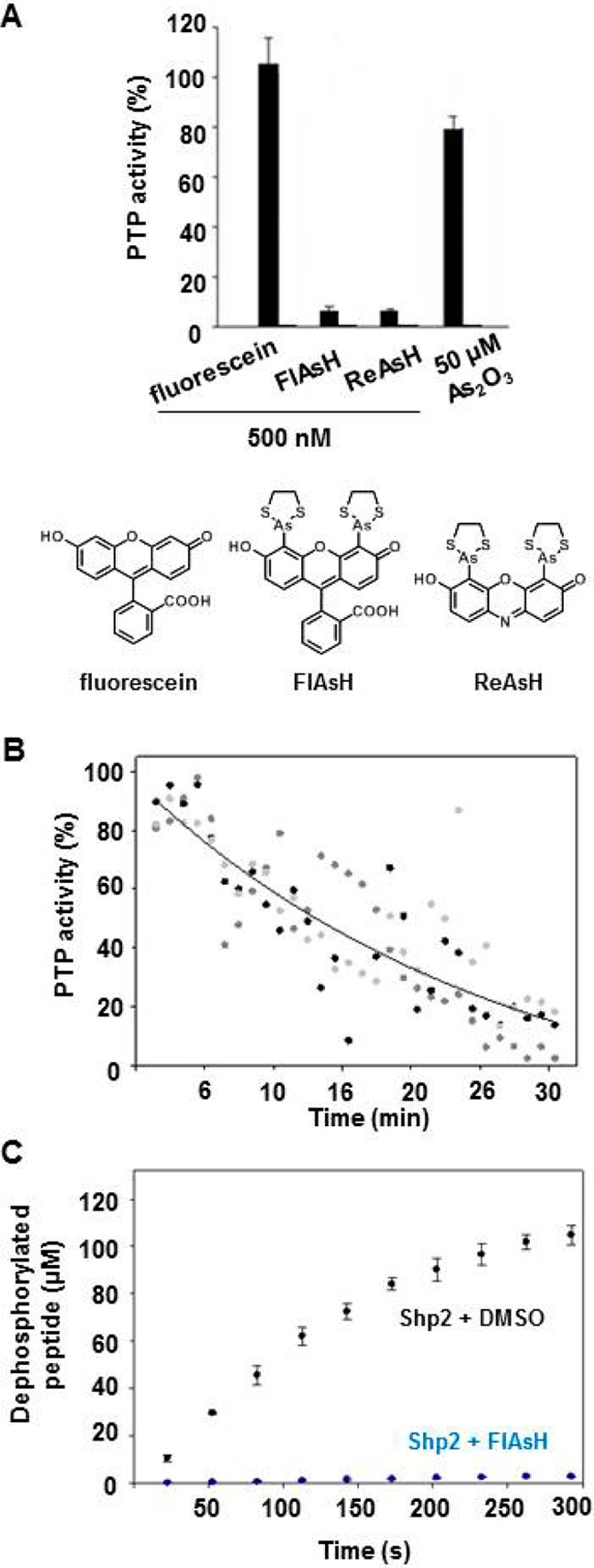
FlAsH-induced inhibition of Shp2 requires the presence of organic arsenical moieties, is time-dependent, and is substrate-independent. (A) After incubation (120 min) with the indicated compounds, Shp2’s PTP activity was measured with pNPP and normalized to no-inhibitor controls. (B) Shp2’s rates of dephosphorylating pNPP in the presence of 1 μM FlAsH over 1 min windows were normalized to a DMSO-only control. The results of three independent experiments (grayscale) were averaged, and the averaged data (not shown) were fit as a single-variable exponential decay. (C) Phosphatase activity was measured by continuous absorbance at 282 nm21 with a phosphopeptide substrate (DADEpYLIPQQG) at pH 7.0 after incubation (150 min) with FlAsH (225 nM).
Figure 1.
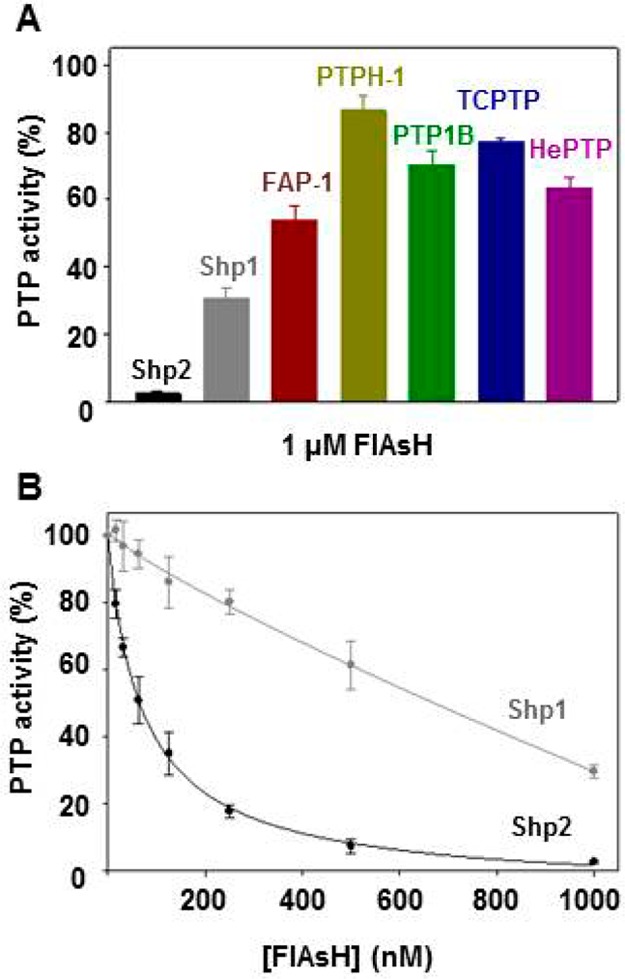
Shp2 is potently and selectively inhibited by FlAsH. (A and B) After incubation (120 min) at the indicated FlAsH concentrations at pH 7.0, phosphatase activities of the indicated PTPs were measured with pNPP and normalized to a DMSO-only control for the corresponding enzyme.
All of the PTPs investigated here contain a strictly conserved cysteine residue within their active sites,1 and it is possible that biarsenicals could inhibit PTP activity through covalent engagement of active-site cysteines. The remarkable Shp2 selectivity that FlAsH displays, however, is not consistent with targeting of a widely conserved PTP feature, leading us to hypothesize that the compound may be acting on Shp2 allosterically, not at the highly conserved PTP active site.
Characterization of Selective Shp2 Inhibition by FlAsH
To more fully understand the nature of selective Shp2 inhibition by FlAsH, we performed Michaelis–Menten kinetics on the enzyme after incubation with an intermediate FlAsH concentration (225 nM). We found that FlAsH-induced Shp2 inhibition manifests exclusively as a decrease in the enzyme’s catalytic rate constant (kcat), whereas Shp2’s Michaelis constant for pNPP (KM) is unaffected by FlAsH (Table 1).
Table 1. Michaelis–Menten Kinetics of the Shp2 Catalytic Domain in the Absence and Presence of FlAsH (225 nM).
| enzyme | kcat (s–1) | KM (pNPP, mM) |
|---|---|---|
| Shp2 without FlAsH | 3.5 ± 0.36 | 3.2 ± 0.33 |
| Shp2 with FlAsH | 0.60 ± 0.066 | 3.5 ± 0.68 |
To further characterize FlAsH’s mechanism of action, we investigated the structure–activity relationships of Shp2 inhibition. We found that FlAsH’s potency requires the presence of the compound’s arsenical ethanedithiol moieties, as shown by the lack of Shp2 inhibition induced by fluorescein, the non-arsenic-containing compound from which FlAsH is synthesized (Figure 2A).18 By contrast, a biarsenical compound that does not derive from fluorescein (ReAsH23) inhibits Shp2 with potency that is comparable to that of FlAsH (Figure 2A). Shp2 is not substantially inhibited, however, by the inorganic As(III) compound arsenic trioxide even at a concentration (50 μM) 100 times greater than the concentration at which FlAsH and ReAsH inhibit the enzyme almost to background levels (500 nM).
We next measured the time dependence of FlAsH-induced Shp2 inhibition and found that the degree of inhibition increases over time, requiring approximately 30 min at 1 μM FlAsH to achieve maximal inhibition (Figure 2B). To further ensure that the observed sensitivity of Shp2 to biarsenicals is not an artifact of the use of the colorogenic small-molecule substrate pNPP in the enzymatic assays, we tested the ability of FlAsH to inhibit Shp2’s phosphatase activity on the phosphopeptide substrate, DADEpYLIPQQG.21,27 We found that strong FlAsH-induced Shp2 inhibition is also observed in this phosphopeptide-based assay (Figure 2C), demonstrating that the effects of FlAsH on Shp2 activity are not substrate-dependent. Together, these results, coupled with the compound structure–activity data mentioned above, establish FlAsH and ReAsH as bona fide inhibitors of Shp2 activity. Moreover, the data suggest that that these biarsenicals target the enzyme through the compounds’ well-characterized covalent, arsenic-based mechanism of action, presumably by bonding to cysteine residues in the Shp2 catalytic domain.
Identification of a Potential Biarsenical-Binding Site on the Shp2 Catalytic Domain
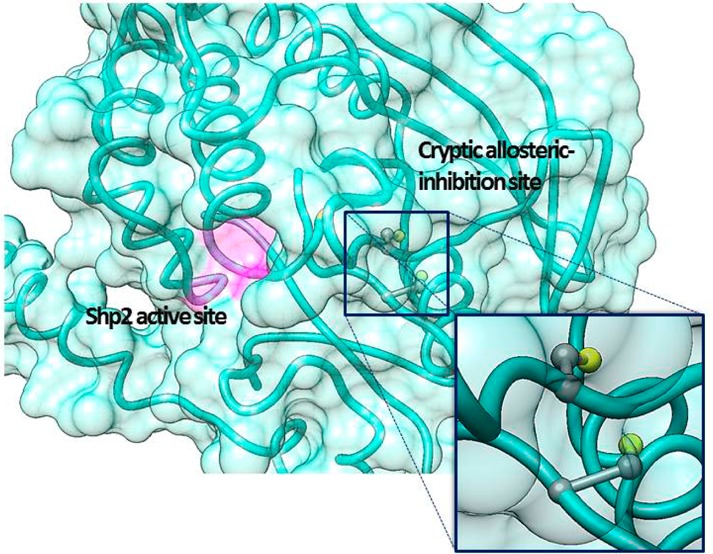
In an attempt to identify a putative biarsenical-binding allosteric site on Shp2, we asked whether its catalytic domain contains any cysteine-rich motifs that are not present in other PTP domains, as biarsenical compounds are known to bind to a small set of naturally occurring proteins that contain unusually cysteine-rich peptide motifs,28,29 in addition to proteins that contain engineered tetracysteine sequences.24 An initial inspection of Shp2’s catalytic-domain primary sequence, however, offers few clues about the position of its uniquely sensitive biarsenical-binding site.1 The entire Shp2 PTP domain contains only six cysteine residues (C259, C318, C333, C367, C459, and C486), which are not closely spaced in the protein’s primary sequence. (C459 is the active-site cysteine residue, completely conserved among classical PTPs, that is integral to the PTP catalytic mechanism.) Intriguingly, however, C333 of Shp2 is found at a position that is occupied by proline in almost all other classical PTP domains [Shp1 is the exception (Figure 3A)]. C333 lies within motif 4 of the PTP domain, which helps to constitute part of the core structure “behind” the enzyme’s active site (Figure 3B).1 The side chain of Shp2’s C333 residue is also close in space to C367’s side chain (5.7 Å from Cβ to Cβ), and it has been shown previously that these two “backdoor” cysteines can form a disulfide bond when the Shp2 catalytic domain is exposed to oxidants.22,30 Thus, we hypothesized that the C333–C367 interface could constitute a Shp2-specific binding site for organic biarsenicals, with C333 acting as the critical specificity element and C367 providing a second non-Shp2-specific cysteine residue that would be necessary for strong binding. Although the side chains of both C333 and C367 are buried in Shp2 crystal structures (Figure 3B),26,31 the earlier finding of Hansen and co-workers that the electrophilic reagent ABDF can target the conserved equivalent of C367 in a PTP catalytic domain (C121 in PTP1B) provides support for the idea that the buried C333 and C367 residues could be accessible, at least transiently, to small molecules in solution.16
Figure 3.
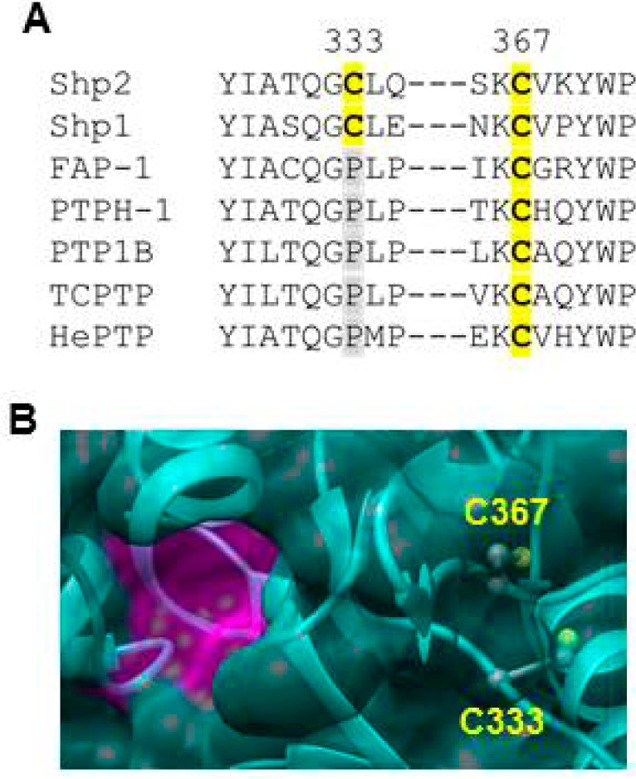
Shp2’s backdoor cysteines constitute a buried allosteric site. (A) Partial primary sequence alignment (human Shp2 numbering) of the PTPs that appear in Figure 1A. (B) Three-dimensional structure of the Shp2 catalytic domain (Protein Data Bank entry 3B7O).26 The PTP active site is colored magenta. The enzyme’s solvent-accessible surface is shown but rendered transparently so that the buried C333 and C367 side chains (colored by element) can be visualized.
Desensitization of the Shp2 Catalytic Domain by Site-Directed Mutagenesis
To investigate whether a buried cysteine residue (C333) could indeed represent a determinant for targeting an enzyme with high selectivity, we used site-directed mutagenesis to convert Shp2’s unusual cysteine to the amino acid residue found at the corresponding position in most other classical PTPs, proline (C333P Shp2). The proline mutation had no substantial effect on the in vitro catalytic competency of the Shp2 catalytic domain in the absence of the biarsenical compound FlAsH (for wild-type Shp2, kcat = 3.5 ± 0.36 s–1 and KM for pNPP = 3.2 ± 0.33 mM; for C333P Shp2, kcat = 3.1 ± 0.28 s–1 and KM for pNPP = 3.6 ± 0.62 mM). Strikingly, however, the FlAsH sensitivity of Shp2 activity is strongly dependent on the presence of cysteine 333. Inhibition assays revealed that mutation of cysteine 333 to proline is sufficient to completely abolish the Shp2 catalytic domain’s unusual FlAsH sensitivity (Figure 4A): in agreement with our findings for wild-type PTPs that contain a proline residue at position 333 (Figure 1A), C333P Shp2 activity is not substantially affected at FlAsH concentrations that inhibit the nonmutated Shp2 catalytic domain to background. The strong desensitization conferred by the C333P mutation is observed regardless of the biarsenical compound or the substrate used in the inhibition assays (Figure S1 of the Supporting Information). These results are consistent with the hypothesis that C333 and C367 constitute a biarsenical-binding site, with the Shp-specific C333 acting as the key specificity determinant. (Mutation of C367 yielded poorly expressing and inactive protein, an observation that comports with previous findings.22)
Figure 4.
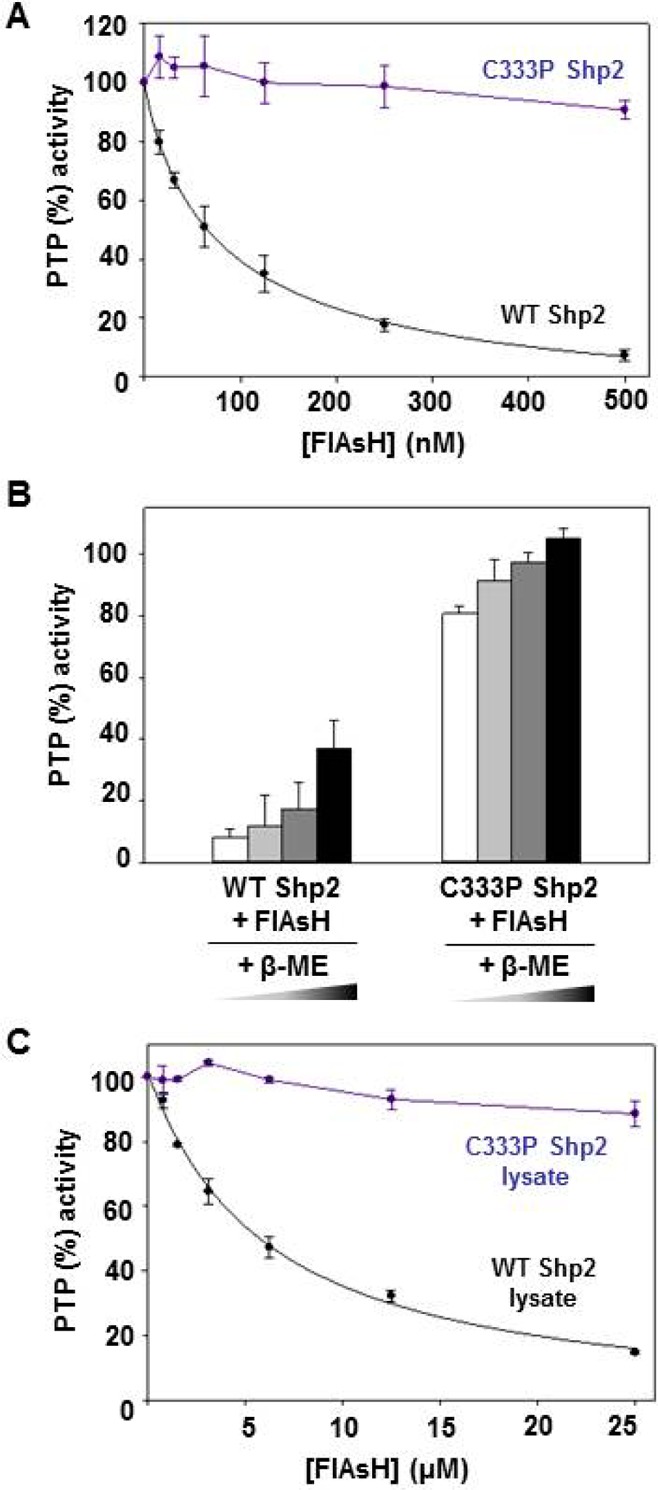
Backdoor cysteine 333 is required for the FlAsH sensitivity of Shp2. (A) After incubation with the indicated FlAsH concentrations, PTP activities of the catalytic domains of wild-type and C333P Shp2 were measured with pNPP and normalized to DMSO-only controls. (B) After incubation with 500 nM FlAsH and varying concentrations of β-mercaptoethanol (β-ME) (white bars, 0 mM; light gray bars, 250 μM; dark gray bars, 500 μM; black bars, 1 mM), PTP activities of the catalytic domains of wild-type and C333P Shp2 were measured with pNPP and normalized to no-FlAsH controls. (C) Target-specific inhibition of Shp2 in a complex proteomic mixture. After incubation with the indicated FlAsH concentrations, PTP activities of lysates of cells expressing wild-type or C333P Shp2 were measured with pNPP and normalized to DMSO-only controls.
In an attempt to further gauge the specificity of FlAsH-mediated PTP inhibition and its strong dependence on the presence of C333, we asked whether FlAsH could effectively target Shp2 in the presence of free thiols in solution. We found that, although the thiol-containing reagent β-mercaptoethanol (β-ME) modestly reduces the potency of FlAsH, the biarsenical compound nevertheless potently inhibits Shp2 in the presence of β-ME (Figure 4B). Importantly, C333’s role in determining the level of Shp2’s inhibitor sensitivity remains operative in the presence of β-ME (Figure 4B). Additionally, we have investigated whether FlAsH can target Shp2 in the context of the competing proteins (and their attendant free cysteines) in a complex proteome. Upon measuring the dose-dependent inhibition of total PTP activity from lysates of E. coli that overexpress either wild-type Shp2 or C333P Shp2, we find that the activity of wild-type Shp2 lysate is inhibited in a dose-dependent manner, with an apparent IC50 of 6 μM, whereas the activity of C333P Shp2-expressing lysate is unaffected at concentrations up to 25 μM (Figure 4C). Collectively, these data support the hypothesis that C333 plays a critical role in determining the inhibitor sensitivity of Shp2, even in the presence of competing molecules that reduce the apparent potency of a compound that is capable of targeting the putative 333/367 allosteric site.
FlAsH-Induced Protection of Cysteines 333 and 367
To further evaluate the accessibility of C333 and C367 and gather more direct evidence for a putative interaction between FlAsH and the C333–C367 interface, we conducted a cysteine-protection assay in which Shp2’s free cysteines were labeled with iodoacetic acid (IAA) in the presence or absence of FlAsH.22 Proteolysis, followed by identification and quantification of Shp2-derived peptides, revealed that the peptides containing C333 and C367 were strongly and specifically protected from carboxymethylation by the presence of FlAsH (Figure 5A,B). By contrast, when we conducted the equivalent experiment on C333P Shp2, we found that the C367 peptide was not protected (Figure 5C). (The peptide that contains amino acid 333 does not contain cysteine in the C333P Shp2 experiment.) It is important to note that no protection of Shp2’s active-site cysteine, C459, was observed in the protection assays conducted with either the wild-type or C333P Shp2 domains, providing further evidence that biarsenical compounds exert their inhibitory effects at an allosteric site, not the PTP active site. The cysteine-protection assays, taken together with the C333P-inhibition assays described above, suggest that FlAsH directly associates with C333/C367, that the association requires the presence of the Shp-specific and buried cysteine residue C333, and that C333 acts as the specificity element that affords the Shp2 catalytic domain its sensitivity to FlAsH.
Figure 5.

Protection of C333 and C367 by FlAsH. (A) Sequence of the Shp2 catalytic-domain construct used in the cysteine-protection assays, with the sequences of peptides identified in the MS experiments (1–11) highlighted. Wild-type (B) or C333P (C) Shp2 (2.7 μM) was incubated with DMSO or FlAsH (27 μM), followed by addition of iodoacetic acid (50 mM). The labeled proteins were trypsinized, and the abundances of the resulting peptides were quantitated by LC–MS/MS. Relative abundances indicate the normalized intensities of the indicated peptides in a FlAsH-treated sample as compared to its no-FlAsH control. Peptides containing positions 333 and 367 are colored orange. Peptides containing other carboxymethylated cysteines are colored blue. Other non-cysteine-containing peptides are colored gray.
Inhibition and Desensitization of Full-Length Shp2
The full-length Shp2 protein (Shp2-FL) is substantially more complex than the isolated Shp2 catalytic domain used in the selectivity-determining experiments described above. Shp2-FL contains two SH2 domains to the N-terminal side of its PTP domain, one of which binds to the PTP domain and autoinhibits its enzymatic activity.32,33 The inherent in vitro activity of Shp2-FL is therefore far lower than that of the isolated catalytic domain, and for reasons of experimental efficiency, the latter enzyme is often used in Shp2-inhibitor-discovery efforts, as described above. The use of the isolated PTP domain, however, leaves open the possibility that an observed Shp2-inhibition event is an artifact arising from the truncation of the protein. This concern is particularly acute for a small-molecule allosteric modulator of Shp2 activity, as the regulation of Shp2’s activity by its SH2 domain is itself allosteric; how the two allosteric events (autoinhibition and allosteric-site targeting) might interact is unclear.
To test whether Shp2’s allosteric site can be targeted in the context of the full-length enzyme, we expressed human Shp2-FL and assayed its activity in the presence of FlAsH. In agreement with the results on the isolated PTP domain, we found that Shp2-FL undergoes strong inhibition upon being incubated with FlAsH (Figure 6A). Dose-dependence experiments revealed a FlAsH IC50 value of 295 nM when Shp2-FL was assayed at a concentration of 50 nM (Figure 6B). (The lower activity of Shp2-FL with respect to the isolated catalytic domain necessitated a switch of substrate to a small-molecule PTP substrate more efficient than pNPP; see Materials and Methods and the legend of Figure 6 for details.) Also in agreement with previous observations, the inhibition of Shp2-FL is time-dependent, albeit with a rate of inhibition slower than that of the Shp2 catalytic domain (Figure 6B), as preincubation for approximately 150 min was required for full inhibition (compare with Figure 2B). Importantly, the sensitivity of Shp2-FL to FlAsH is abolished by mutation of cysteine 333 to proline (C333P Shp2-FL), suggesting that the mechanism of inhibition of the full-length protein is identical to that of the isolated catalytic domain (Figure 6A,B).
Figure 6.
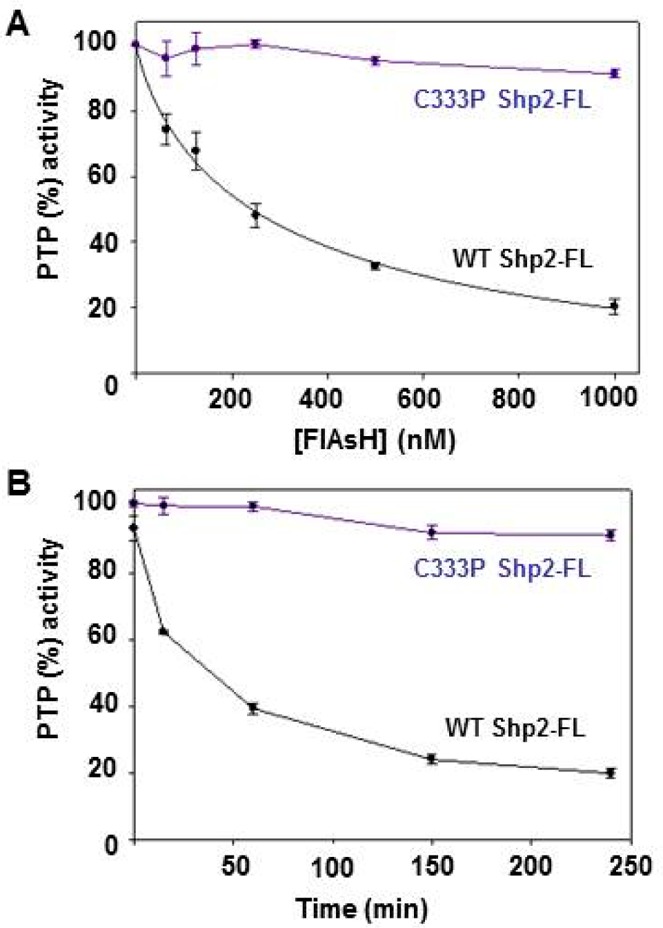
FlAsH inhibits full-length Shp2 in a dose- and time-dependent manner. PTP activities of full-length wild-type and C333P Shp2 were measured with DiFMUP at pH 7.0 after incubation with the indicated FlAsH concentrations for 150 min (A) or with FlAsH (1 μM) for the indicated time intervals (B). Data points represent the rates of dephosphorylation over a 10 min time window (centered at the indicated time point) normalized to a DMSO-only control for the corresponding enzyme.
Conclusion and Outlook
Our data establish the presence of a unique biarsenical-reactive allosteric site on the catalytic domain of the oncogenic PTP Shp2 and provide the first demonstration that a nonconserved cysteine residue can be targeted for selective PTP inhibition. The unique biarsenical sensitivity of Shp2 among PTPs may provide a means for studying the signaling roles of the enzyme and its clinically relevant mutants with high specificity in living cells. Moreover, given the potential importance of Shp2 inhibitors in anticancer pharmaceutical development, its newly unveiled allosteric site may represent a target for Shp2-directed drug-discovery efforts that range beyond the decidedly nonclinically ideal biarsenical compounds used in the study presented here. Nonconserved cysteine residues have previously been targeted to achieve selective inhibition in enzyme families beyond the PTPs, notably, the protein kinases.34,35 The striking feature of Shp2’s novel allosteric site is that its key selectivity determinant, C333, is completely buried in Shp2 crystal structures.26,31 Although we do not currently understand the molecular mechanism by which C333 participates in biarsenical binding, one plausible hypothesis is that, in solution, the Shp2 enzyme can sample (at least) two conformational states: an insensitive state in which the allosteric site is buried and a sensitive state in which the site is exposed.30 Binding of a biarsenical by the sensitive state would then preclude reversion to the insensitive state and hold the protein in the sensitive, and inactivated, state. Small changes in the kinetics or thermodynamics of this structural transition could potentially lead to significant changes in the accessibility of the allosteric site, a hypothesis that is consistent with our observation that not all C333-containing PTPs are equally sensitive to biarsenicals (e.g., Shp1). The speculative nature of our sampling hypothesis notwithstanding, recent NMR studies of a DNA-binding protein have demonstrated that transient, and therefore invisible to crystallography, protein conformational states can possess radically altered ligand binding properties with respect to their corresponding ground-state structures.36 As more examples of cryptic ligand-binding sites are revealed, and as our understanding of protein dynamics develops, our notion of what constitutes a druggable ligand-binding site may expand significantly, opening promising new avenues for drug discovery.
Acknowledgments
We thank Jamie Oh for cloning of the Shp2 catalytic-domain expression vector and Ben Neel and Nickolay Chirgadze (Princess Margaret Cancer Center, University Health Network) for their generous gift of the Shp2-FL expression vector. We also thank Ben Neel, Xiaonan Wang, and Minerva Fernandez for helpful conversations. LC–MS/MS experiments were conducted by the University of Massachusetts Medical School’s Proteomics & Mass Spectrometry Facility.
Glossary
Abbreviations
- PTP
protein tyrosine phosphatase
- Shp2
Src-homology-2-domain-containing PTP 2
- FlAsH
fluorescein arsenical hairpin binder
- pNPP
p-nitrophenyl phosphate
- pY
phosphotyrosine.
Supporting Information Available
Figure S1 and experimental details for the cloning, mutagenesis, expression, and purification of the PTP constructs used in this study. This material is available free of charge via the Internet at http://pubs.acs.org.
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health via Grant R15GM071388. We also gratefully acknowledge the Henry Dreyfus Teacher-Scholar Awards Program (A.C.B.) and Amherst College for funding.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Andersen J. N.; Mortensen O. H.; Peters G. H.; Drake P. G.; Iversen L. F.; Olsen O. H.; Jansen P. G.; Andersen H. S.; Tonks N. K.; Møller N. P. (2001) Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol. 21, 7117–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks W. J.; Elson A.; Harroch S.; Pulido R.; Stoker A.; den Hertog J. (2013) Protein tyrosine phosphatases in health and disease. FEBS J. 280, 708–730. [DOI] [PubMed] [Google Scholar]
- Hardy S.; Julien S. G.; Tremblay M. L. (2012) Impact of oncogenic protein tyrosine phosphatases in cancer. Anti-Cancer Agents Med. Chem. 12, 4–18. [DOI] [PubMed] [Google Scholar]
- Chan G.; Kalaitzidis D.; Neel B. G. (2008) The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 27, 179–192. [DOI] [PubMed] [Google Scholar]
- Mohi M. G.; Neel B. G. (2007) The role of Shp2 (PTPN11) in cancer. Curr. Opin. Genet. Dev. 17, 23–30. [DOI] [PubMed] [Google Scholar]
- Chan R. J.; Feng G. S. (2007) PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood 109, 862–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia M.; Niemeyer C. M.; Fragale A.; Song X. L.; Buechner J.; Jung A.; Hahlen K.; Hasle H.; Licht J. D.; Gelb B. D. (2003) Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 34, 148–150. [DOI] [PubMed] [Google Scholar]
- Bentires-Alj M.; Paez J. G.; David F. S.; Keilhack H.; Halmos B.; Naoki K.; Maris J. M.; Richardson A.; Bardelli A.; Sugarbaker D. J.; Richards W. G.; Du J. Y.; Girard L.; Minna J. D.; Loh M. L.; Fisher D. E.; Velculescu V. E.; Vogelstein B.; Meyerson M.; Sellers W. R.; Neel B. G. (2004) Activating mutations of the Noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 64, 8816–8820. [DOI] [PubMed] [Google Scholar]
- Barr A. J. (2010) Protein tyrosine phosphatases as drug targets: Strategies and challenges of inhibitor development. Future Med. Chem. 2, 1563–1576. [DOI] [PubMed] [Google Scholar]
- Sobhia M. E.; Paul S.; Shinde R.; Potluri M.; Gundam V.; Kaur A.; Haokip T. (2012) Protein tyrosine phosphatase inhibitors: A patent review (2002–2011). Expert Opin. Ther. Pat. 22, 125–153. [DOI] [PubMed] [Google Scholar]
- He R.; Zeng L. F.; He Y.; Zhang S.; Zhang Z. Y. (2013) Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J. 280, 731–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L. F.; Zhang R. Y.; Yu Z. H.; Li S.; Wu L.; Gunawan A. M.; Lane B. S.; Mali R. S.; Li X.; Chan R. J.; Kapur R.; Wells C. D.; Zhang Z. Y. (2014) Therapeutic potential of targeting the oncogenic SHP2 phosphatase. J. Med. Chem. 57, 6594–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaskovich M. A. (2009) Drug discovery and protein tyrosine phosphatases. Curr. Med. Chem. 16, 2095–2176. [DOI] [PubMed] [Google Scholar]
- Perron M. D.; Chowdhury S.; Aubry I.; Purisima E.; Tremblay M. L.; Saragovi H. U. (2014) Allosteric noncompetitive small molecule selective inhibitors of CD45 tyrosine phosphatase suppress T-cell receptor signals and inflammation in vivo. Mol. Pharmacol. 85, 553–563. [DOI] [PubMed] [Google Scholar]
- Krishnan N.; Koveal D.; Miller D. H.; Xue B.; Akshinthala S. D.; Kragelj J.; Jensen M. R.; Gauss C.-M.; Page R.; Blackledge M.; Muthuswamy S. K.; Peti W.; Tonks N. K. (2014) Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 10, 558–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen S. K.; Cancilla M. T.; Shiau T. P.; Kung J.; Chen T.; Erlanson D. A. (2005) Allosteric inhibition of PTP1B activity by selective modification of a non-active site cysteine residue. Biochemistry 44, 7704–7712. [DOI] [PubMed] [Google Scholar]
- Wiesmann C.; Barr K. J.; Kung J.; Zhu J.; Erlanson D. A.; Shen W.; Fahr B. J.; Zhong M.; Taylor L.; Randal M.; McDowell R. S.; Hansen S. K. (2004) Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 11, 730–737. [DOI] [PubMed] [Google Scholar]
- Adams S. R.; Tsien R. Y. (2008) Preparation of the membrane-permeant biarsenicals FlAsH-EDT2 and ReAsH-EDT2 for fluorescent labeling of tetracysteine-tagged proteins. Nat. Protoc. 3, 1527–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin B. A.; Adams S. R.; Tsien R. Y. (1998) Specific covalent labeling of recombinant protein molecules inside live cells. Science 281, 269–272. [DOI] [PubMed] [Google Scholar]
- Zhang X. Y.; Bishop A. C. (2007) Site-specific incorporation of allosteric-inhibition sites in a protein tyrosine phosphatase. J. Am. Chem. Soc. 129, 3812–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. Y.; Maclean D.; Thieme-Sefler A. M.; Roeske R. W.; Dixon J. E. (1993) A continuous spectrophotometric and fluorimetric assay for protein tyrosine phosphatase using phosphotyrosine-containing peptides. Anal. Biochem. 211, 7–15. [DOI] [PubMed] [Google Scholar]
- Chen C. Y.; Willard D.; Rudolph J. (2009) Redox regulation of SH2-domain-containing protein tyrosine phosphatases by two backdoor cysteines. Biochemistry 48, 1399–1409. [DOI] [PubMed] [Google Scholar]
- Adams S. R.; Campbell R. E.; Gross L. A.; Martin B. R.; Walkup G. K.; Yao Y.; Llopis J.; Tsien R. Y. (2002) New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: Synthesis and biological applications. J. Am. Chem. Soc. 124, 6063–6076. [DOI] [PubMed] [Google Scholar]
- Pomorski A.; Krezel A. (2011) Exploration of biarsenical chemistry: Challenges in protein research. ChemBioChem 12, 1152–1167. [DOI] [PubMed] [Google Scholar]
- Zhang X. Y.; Chen V. L.; Rosen M. S.; Blair E. R.; Lone A. M.; Bishop A. C. (2008) Allele-specific inhibition of divergent protein tyrosine phosphatases with a single small molecule. Bioorg. Med. Chem. 16, 8090–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr A. J.; Ugochukwu E.; Lee W. H.; King O. N. F.; Filippakopoulos P.; Alfano I.; Savitsky P.; Burgess-Brown N. A.; Muller S.; Knapp S. (2009) Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 136, 352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. Y.; Thieme-Sefler A. M.; Maclean D.; McNamara D. J.; Dobrusin E. M.; Sawyer T. K.; Dixon J. E. (1993) Substrate specificity of the protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 90, 4446–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroffekova K.; Proenza C.; Beam K. G. (2001) The protein-labeling reagent FLASH-EDT2 binds not only to CCXXCC motifs but also non-specifically to endogenous cysteine-rich proteins. Pfluegers Arch. 442, 859–866. [DOI] [PubMed] [Google Scholar]
- Wang T.; Yan P.; Squier T. C.; Mayer M. U. (2007) Prospecting the proteome: Identification of naturally occurring binding motifs for biarsenical probes. ChemBioChem 8, 1937–1940. [DOI] [PubMed] [Google Scholar]
- Parsons Z. D.; Gates K. S. (2013) Thiol-dependent recovery of catalytic activity from oxidized protein tyrosine phosphatases. Biochemistry 52, 6412–6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof P.; Pluskey S.; Dhe-Paganon S.; Eck M. J.; Shoelson S. E. (1998) Crystal structure of the tyrosine phosphatase SHP-2. Cell 92, 441–450. [DOI] [PubMed] [Google Scholar]
- Barford D.; Neel B. G. (1998) Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure 6, 249–254. [DOI] [PubMed] [Google Scholar]
- Neel B. G.; Gu H. H.; Pao L. (2003) The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28, 284–293. [DOI] [PubMed] [Google Scholar]
- Cohen M. S.; Zhang C.; Shokat K. M.; Taunton J. (2005) Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 308, 1318–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Sabnis Y.; Zhao Z.; Zhang T.; Buhrlage S. J.; Jones L. H.; Gray N. S. (2013) Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng S. R.; Kalodimos C. G. (2013) Allosteric inhibition through suppression of transient conformational states. Nat. Chem. Biol. 9, 462–465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.