

Abstract
The circulating protein transthyretin (TTR) can unfold, oligomerize, and form highly structured amyloid fibrils that are deposited in tissues, causing organ damage and disease. This pathogenic process is caused by a heritable TTR point mutation in cases of familial TTR-related amyloidosis or wild-type TTR in cases of age-associated amyloidosis (previously called senile systemic amyloidosis). The TTR amyloid cascade is hypothesized to begin with the dissociation of the TTR native tetrameric structure into folded but unstable monomeric TTR subunits. Unfolding of monomeric TTR initiates an oligomerization process leading to aggregation and fibril formation. Numerous proteostatic mechanisms for regulating the TTR amyloid cascade exist. Extracellular chaperones provide an innate defense against misfolded proteins. Clusterin (CLU), a plasma protein, has the capacity to recognize exposed hydrophobic regions of misfolded proteins, shielding them from aggregation. We have previously demonstrated that CLU is associated with the amyloid fibrils in cardiac tissues from patients with TTR amyloidosis. In this study, we have used tetrameric and monomeric TTR structural variants to determine the ability of CLU to inhibit TTR amyloid fibril formation. Using circular dichroism spectroscopy, we determined that CLU preferentially stabilizes monomeric TTR and generates increasingly stable conformations under acid stress. Moreover, studies using surface plasmon resonance showed a direct interaction of CLU with high-molecular weight TTR oligomers. The interactions of CLU with monomeric and aggregated TTR proceed in a cooperative manner in the presence of diflunisal, a small molecule drug used to stabilize TTR tetramers.
Protein misfolding and aggregation are recognized as critical processes in the pathogenesis of a wide range of human diseases. In particular, tissue deposition of aberrantly folded and self-associated proteins as highly organized β-sheet structured amyloid fibrils is the hallmark of a diverse group of diseases known as the amyloidoses. More than 25 proteins have been identified as being amyloidogenic; these proteins form the insoluble amyloid fibrils that are deposited either locally at a single site or systemically at multiple sites throughout the body.1,2 In the systemic amyloidoses, the majority of precursor proteins are derived from plasma, including transthyretin (TTR), immunoglobulin light chains, fibrinogen, and apolipoprotein AI. The TTR-associated forms of amyloidosis include inherited and acquired types; in familial TTR-related amyloidosis (ATTRm), a point mutation in the TTR gene results in expression of an amyloidogenic variant, and in senile systemic amyloidosis (SSA), wild-type (wt) TTR forms the amyloid fibril deposits.3,4 SSA is now more precisely termed ATTRwt.
At present, the widely accepted model of amyloid fibril formation involves the structural destabilization and conformational change of a natively folded protein, with consequential exposure of hydrophobic regions normally buried within the core of the protein.5−7 The misfolded or unfolded state increases the propensity for protein self-aggregation. Likely efficacious approaches to stabilize the native protein fold utilize pharmacological chaperones to raise the free energy barrier of unfolding and prevent amyloid fibril formation.8−10
The homotetrameric TTR transport protein is rich in β-sheet structure, containing two binding pockets for thyroxine located at the interfaces of its four subunits. In its ligand-bound state, TTR has increased quaternary structural stability. Many small polycyclic molecules of diverse structures are capable of binding to TTR in the thyroxine-binding pockets and stabilizing the tetramer.11−17 One such compound is diflunisal, a nonsteroidal anti-inflammatory drug recently shown to slow progression of ATTRm polyneuropathy in a randomized multicenter phase III clinical trial.18−24
In plasma, the majority of tetrameric TTR circulates ligand-free, leaving it vulnerable to amyloid formation.25 TTR amyloidogenesis begins with the rate-limiting step of tetramer dissociation.26−33 Single-amino acid changes in the subunits of the protein destabilize the tetramer, promoting dissociation to dimers and monomers.26−29 Further unfolding of the monomers generates multiple non-native state conformations and initiates TTR oligomerization to form high-molecular weight (HMW) species. As tetramer dissociation is the rate-limiting step in TTR fibrillation, a nucleation phase is absent from the TTR amyloid cascade.34 Oligomerization continues until protofibrils are formed; several protofibrils bundle together to form a mature amyloid fibril. In addition to the amyloid fibril protein, accessory molecules are thought to play a role in amyloid formation kinetics and stability, such as glycosaminoglycans, serum amyloid P component, and extracellular chaperones.35−38
Clusterin (CLU), also termed apolipoprotein J, is a ubiquitous protein that functions as an extracellular chaperone.39 CLU has remarkable conformational adaptability attributed to three large molten globule domains, three amphipathic regions, and two coiled-coil α-helices.40 This molecular structure of CLU is responsible for the unique high-affinity, low-specificity binding of the chaperone, allowing it to inhibit the precipitation of slowly aggregating partially unfolded proteins.41,42 Interestingly, CLU chaperone activity increases under mildly acidic conditions as its structural equilibrium shifts from multimers toward the chaperone-active heterodimeric form, increasing regions of solvent-exposed hydrophobicity.43 Analyses of CLU complexes have indicated that the activity of the protein is dependent on both stoichiometry and structure, i.e., specific misfolded protein to CLU concentration ratios, as well as certain misfolded protein conformations.44,45
Previously, we have shown that CLU is a component of TTR amyloid deposits in cardiac tissues from patients with ATTRwt and ATTRm.46 In the study presented here, we examined the stabilizing effect of CLU on wild-type and variant forms of TTR that differ in sequence, structure, and stability during amyloid formation. Using circular dichroism (CD) spectroscopy, we tested whether CLU affects structural transitions of tetrameric and monomeric forms of TTR under stress conditions. Additionally, we employed surface plasmon resonance (SPR) detection to determine the ability of CLU to recognize oligomeric forms of TTR. These biophysical, structural, and interaction analyses permit direct observation and characterization of the chaperoning interactions between CLU and TTR. Finally, we report the results of a novel approach of stabilizing TTR amyloidogenic precursor proteins using a combination of CLU and diflunisal.
Experimental Procedures
Expression and Purification of Recombinant Transthyretin
Recombinant TTR (rTTR) was expressed and purified using the method described by Kingsbury et al.47 Modification of the expression vector for rTTR variant synthesis was achieved via site-directed mutagenesis using the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Specifically, sequences were modified for substitutions of leucine to proline at position 55 (L55P) to generate an unstable tetrameric variant of rTTR. In addition, F87M and L110M mutations were introduced to generate an engineered rTTR subunit that reportedly does not form tetramers, termed monomeric rTTR (monoTTR, F87M/L110M).30
Serum Clusterin Immunoaffinity Purification
A mouse hybridoma cell line producing the G7 monoclonal IgG1 against human serum CLU was received as a generous gift from L. A. Aarden (The University of Amsterdam, Amsterdam, The Netherlands). An immunoaffinity column was generated using covalent coupling of the G7 anti-human CLU monoclonal IgG1 to cyanogen bromide (CNBr)-activated Sepharose 4B resin (Sigma, St. Louis, MO). All column procedures were performed at 4 °C and protocols adapted from previously described methods48 and personal communications from M. R. Wilson (The University of Wollongong, Wollongong, Australia). A multichannel peristaltic pump system using microcassettes and Masterflex tubing #6424-14 (Cole-Parmer, Vernon Hills, IL) was set to a flow rate of 0.5 mL/min (6 rpm) to deliver serum. One hundred milliliters of pooled human serum collected from healthy male individuals (Bioreclamation Inc., Westbury, NY) was quickly thawed at 37 °C and diluted 1:1 into PBS containing cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail Tablets (Roche, Indianapolis, IN) and 5 mM EDTA. The diluted serum was chilled on ice for 10 min, filtered, degassed, and then applied to the G7 column at a flow rate of 0.5 mL/min (6 rpm). The G7 column was washed extensively with PBS by gravity flow until the absorbance at 280 nm of the eluate was ≤0.01. Removal of ApoA-I bound to CLU was accomplished by washing with 0.5% Triton X-100 and extensive rinsing. Next, a pre-elution washing step using 0.2 M sodium acetate and 0.5 M NaCl (pH 5.0) was performed with extensive rinsing for stringent purification of CLU. Serum CLU was eluted using 0.2 M glycine and 0.5 M NaCl (pH 2.5) and monitored by absorbance measurements at 280 nm. Purified CLU fractions were pooled and dialyzed against PBS (pH 7.4).
Acid-Mediated Transthyretin Oligomer and Amyloid Fibril Formation
It has been demonstrated that, at physiologic concentrations, wt TTR tetramer is resistant to dissociation between pH 7.0 and 5.0.49 We used acid-mediated aggregation assays for generation of wt, L55P, and monomeric (F87M/L110M) rTTR oligomers and amyloid fibrils described herein. For oligomer formation, wt rTTR (3.6 μM) was dialyzed at 4 °C against ddH2O for 24 h and then against 10 mM HCl (pH 2.0) for 96 h. Production of oligomers was initiated by addition of NaCl adjusted to a concentration of 100 mM at 25 °C.50 Neutralization of the oligomerization reaction was achieved using a 1:6 dilution of 0.5 M Na2HPO4 (pH 7.4). For amyloid fibril formation, wt and L55P tetrameric variants were dialyzed in separate assays against 10 mM Na2HPO4 and 100 mM KCl (pH 7.1) overnight at 4 °C. Monomeric rTTR was buffer exchanged rapidly into 10 mM Na2HPO4 and 100 mM KCl (pH 7.1) using Econo-Pac 10DG columns (Bio-Rad, Hercules, CA) to preserve protein stability. Recombinant TTR protein solutions were quantified by a bicinchoninic acid assay and diluted to 7.2 μM. Treatments of rTTR with diflunisal at 100 M excess or CLU or bovine serum albumin (BSA) at 7.2 μM (equimolar ratio with respect to rTTR) were conducted for 2 h at room temperature and neutral pH. Subsequently, rTTR solutions were diluted 1:1 into 200 mM potassium acetate, 100 mM potassium chloride, and 2 mM EDTA (pH 5.0) and incubated at 37 °C for up to 14 days.
Circular Dichroism Spectroscopy
CD spectra were recorded using a Jasco J-815 spectropolarimeter equipped with thermoelectric temperature controllers (Jasco Inc.). Far-UV (193–250 nm, 1 nm bandwidth) data were collected using a 1.0 mm path length quartz cell, smoothed with the Jasco noise reduction routine, normalized to protein concentration, and presented as mean residue molar ellipticity (MRE) for individual proteins. For protein mixtures, ellipticity data are presented as CD signals in millidegrees. CD melting and stability data were recorded at 220 nm while samples were heated at a constant rate from 25 to 98 °C. Melting temperatures (Tm) were determined from peak positions in the first derivative of the CD melting curves.51
Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis (SDS–PAGE) Analysis
Samples were cross-linked for 2 min with 5 μL of 25% glutaraldehyde, and the reaction was quenched with 5 μL of 7% NaBH4; 12.5 μL of 6× SDS reducing Laemmli load buffer was added to each reaction solution and the mixture boiled for 5 min. Samples were analyzed using 10% SDS–PAGE developed for 2 h using GelCode blue Coomassie blue G-250 reagent (Thermo Fisher, Rockford, IL).
Surface Plasmon Resonance
A ProteOn XPR36 surface plasmon resonance (SPR) apparatus and sensor chips (Bio-Rad) were used for this study. Wild-type tetramers and HMW oligomers of rTTR were each separately immobilized on a GLH high-capacity sensor chip for studies of diflunisal and Congo red binding and on a GLM medium-capacity sensor chip for analysis of CLU binding. The rTTR ligands were immobilized using an amine coupling reaction; 2 μM rTTR ligand samples were diluted in acetate buffer (pH 4.0) and injected over the activated sensor chip surface for 5 min at a flow rate of 30 μL/min. A reference surface was always prepared in parallel using the same immobilization procedure in the absence of ligand. Final ligand immobilization levels achieved for GLH surfaces were as follows: native rTTR tetramer, 11000 resonance units (RU); 24 h HMW oligomers, 5100 RU. For GLM surfaces, the levels were as follows: native rTTR tetramer, 5800 RU; 24 h HMW oligomers, 4300 RU. In this system, 1 RU = 1 pg of protein/mm2. Diflunisal, Congo red, and CLU analytes were injected over immobilized rTTR ligands; diflunisal and Congo red samples were injected for 1–2 min at a flow rate of 100 μL/min, and CLU was injected for 5 min at a flow rate of 50 μL/min. PBS (pH 7.4) and 0.005% Tween 20 were used as running buffer and analyte diluent. Sensorgrams were double referenced to blank ligand surfaces and blank analyte buffer injections; data were modeled using the conservative Langmuir 1:1 (analyte:ligand) interaction model.
Congo Red Binding
Insoluble rTTR amyloid fibrils were collected by centrifugation at 25000g for 5 min, and the supernatant was removed. For polarized light microscopy, pelleted fibrils were suspended in low-melt agarose (Thermo Fisher), formalin-fixed, paraffin-embedded, serially sectioned to thicknesses of 5 μm, and processed for Congo red staining. Deparaffinized sections were placed in an alkaline 80% alcohol/NaCl mixture for 20 min, stained with alkaline Congo red for 20 min, rinsed in ethanol and xylene, and observed by light microscopy using bright and polarized views.
For determination of the amount of bound Congo red in solution, pelleted fibrils were resuspended in 10 μM Congo red, 150 mM NaCl, and 5 mM KH2PO4 (pH 7.4) to a final volume of 500 μL. Spectrophotometric scanning in the wavelength (λ) range between 350 and 700 nm was performed using a Cary 300 UV–vis spectrophotometer (Agilent Technologies, Santa Clara, CA) for quantification of binding of Congo red to amyloid structures in suspension. A red-shift of the Congo red spectrum in the wavelength range near 540 nm indicated the presence of amyloid, and bound Congo red concentrations were determined as previously described.52,53
Results
Secondary and Quaternary Structure, and Thermal Stability of Transthyretin Correspond to Mutational Status
TTR amyloidogenesis is promoted by tetramer-destabilizing amino acid substitutions. To investigate the effects of CLU and diflunisal on the steps of TTR unfolding and aggregation, we utilized rTTR, wt and two variants. The mutant proteins included L55P, a highly amyloidogenic variant found in ATTRm patients with early onset familial amyloid polyneuropathy, and F87M/L110M (monoTTR), an engineered variant that is present mostly in monomeric form at neutral pH.30 Purified rTTR proteins were rich in β-sheet content as assessed by far-UV CD (Figure 1A). The observed CD spectra after pH 5.0 buffer adjustment (Figure 1A) did not differ from spectra observed at pH 7.0 (data not shown). When quaternary structures were analyzed by chemical cross-linking and gel electrophoresis, wt and L55P at 3.6 μM occurred mainly as 56 kDa forms corresponding to tetrameric TTR; while minor amounts of dimeric TTR (∼30 kDa) were also observed, little to no monomeric (14 kDa) protein was noted (Figure 1B, lanes 1 and 3). The most abundant form of cross-linked monoTTR appeared at an electrophoretic migration end point consistent with TTR monomer (14 kDa); minor amounts of dimer (∼30 kDa) were also noted (Figure 1B, lane 2).
Figure 1.

Structural and thermal stability analyses of wild-type transthyretin (TTR), variant TTR, and clusterin (CLU) proteins monitored by circular dichroism (CD) and electrophoretic migration. (A) Far-UV CD spectra of wtTTR (black), L55P TTR (green), and monomeric F87M/L110M TTR (monoTTR, red). (B) Glutaraldehyde cross-linked rTTR samples with 25 μL of 3.57 μM protein loaded into each lane and analyzed by SDS–PAGE with Coomassie staining. (C) rTTR thermal stability determined by continuous CD monitoring at 220 nm while the samples were heated at a rate of 2 °C/min. Far-UV CD spectra of monomeric rTTR (inset of panel C) and CLU (inset of panel D) recorded at 25 °C before thermal stress (solid lines) and at 98 °C (dashed lines) after heating. Protein concentrations were 3.6 μM in 5 mM Na2HPO4, 100 mM potassium acetate, and 50 mM potassium chloride (pH 5.0).
CD analysis was used to assess the thermal stabilities of wt, L55P, and monomeric rTTR upon heating of the proteins from 25 to 98 °C (Figure 1C). A pH of 5.0 was chosen to ensure both L55P tetramer dissociation and monomer unfolding. These conditions also preserve CLU functionality.54 Far-UV CD spectra were obtained for monoTTR and CLU at 25 °C (solid lines) and heated to 98 °C (dashed lines, Figure 1C,D, inset in each panel). The native β-sheet rich structure of monoTTR and α-helix structure of CLU are partially lost upon heating of the proteins to 98 °C. A wavelength of 220 nm was chosen for monitoring changes in secondary structure during thermal unfolding.
The thermal stabilities of rTTR proteins corresponded to the presence of amino acid substitutions and quaternary structure (Figure 1C); wt was most stable, L55P intermediate, and monoTTR least stable. For wt TTR [Figure 1C (black circles)], no loss of secondary structure was observed upon protein heating; this was consistent with a Tm reported to be >100 °C.55 L55P TTR [Figure 1C (green diamonds)] began to unfold at 65 °C; however, the unfolding was incomplete as no plateau was reached before 100 °C. MonoTTR [Figure 1C (red triangles)] displayed a complete transition with a Tm of 60 °C. CLU showed a single sigmoidal transition with a Tm at 48 °C (Figure 1D).
Clusterin Preferentially Stabilizes TTR Monomeric Structures Forming Thermally Resistant Complexes
Next, we tested whether CLU affects structural transitions of tetrameric and monomeric forms of rTTR upon heating. Data from the observed thermal unfolding of equimolar rTTR/CLU mixtures were used to generate individual melting curves and compared to the calculated summed values (Figure 2). The summed melting curves (filled black circles) are the additive spectral changes of each protein, independently acquired and generated by spectral composite computation. For wtTTR/CLU mixtures, summed (filled circles) and observed (open circles) thermal unfolding profiles exhibited nearly identical melt transitions (Figure 2A). The L55P TTR/CLU summed and observed melting comparison showed a small but distinct deviation in the CD signal at 220 nm (Figure 2B). For the monoTTR/CLU mixture, the summed (black circles) and observed (red triangles) melting data showed a considerable divergence beginning at 45 °C (Figure 2C), leading to a loss of secondary structure in the monoTTR/CLU mixture. This result was substantially smaller than the calculated sum of CD signals from individual proteins, suggesting a distinct stabilization effect of CLU on the secondary structure of monoTTR.
Figure 2.

Comparison of thermal unfolding data for equimolar mixtures of transthyretin (TTR) variants with clusterin (CLU). Secondary structural changes of each rTTR/CLU mixture during heating at a rate of 2 °C/min were obtained by continuous monitoring of the CD signal at 220 nm. Observed melting curves for equimolar mixtures of (A) wt, (B) L55P, and (C) monoTTR with CLU are shown as open black circles, green diamonds, and red triangles, respectively; each data set was compared to the corresponding sum of melting curves from the individual proteins (filled black circles). (D) Data from a direct comparison of observed melting curves for equimolar mixtures of CLU and wt, L55P, or monomeric rTTR are shown as open black circles, green diamonds, or red triangles, respectively.
Direct comparisons of observed melting data for TTR/CLU mixtures were also examined (Figure 2D). The wtTTR/CLU (black circles) and L55P TTR/CLU (green diamonds) mixtures showed similar unfolding of CLU at a Tm of 48 °C; the monoTTR/CLU mixture (red triangles) exhibited less change in secondary structure for this low-temperature transition. Two distinct transitions were evident in the melting curves of L55P TTR/CLU and monoTTR/CLU mixtures; however, only in the monoTTR/CLU sample was there a completed second transition demonstrated by a plateau in the curve above 90 °C. The higher-Tm transition in the L55P TTR/CLU mixture corresponded to the L55P tetramer melt transition observed in Figure 1C.
To compare the thermal unfolding of monoTTR, CLU, and the monoTTR/CLU mixture, the data were scaled together (Figure 3). CLU (filled blue squares) and the monoTTR/CLU mixture (open red triangles) showed identical low-Tm transitions corresponding to CLU unfolding [Tm = 48 °C (vertical dashed line)]. Interestingly, the monoTTR/CLU mixture had a unique Tm transition above 80 °C that was not observed in individual samples of monoTTR or CLU. The higher-Tm transition of the monoTTR/CLU mixture indicated retention of secondary structure by the proteins in the mixture (vertical dashed line). Midpoint transition temperatures determined from peak positions in the first derivatives of the CD data further illustrated the presence of a high-Tm transition for the monoTTR/CLU mixture (Figure 3B). Apparent Tm values were 48 °C for CLU, 60 °C for monoTTR, and 47 and ∼80 °C and higher for the monoTTR/CLU mixture. The unique Tm transition above 80 °C was shown to be a rate-dependent process with a shifted melting transition and greater structural change observed under slower thermal stress conditions (Figure 3C). Reversibility assessment indicated that unfolding of L55P TTR and monoTTR was irreversible (data not shown). Interestingly, while unfolding of the L55P TTR/CLU mixture was partially reversible, results from our studies of the monoTTR/CLU mixture indicated a completely reversible unfolding–folding phenomenon.
Figure 3.
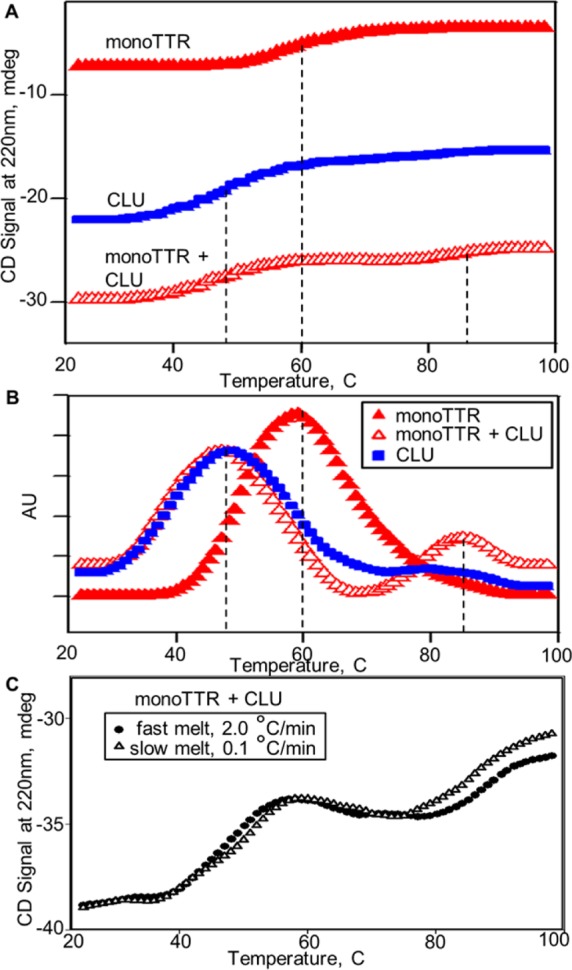
Effect of clusterin on thermal unfolding of transthyretin (TTR) monomers. (A) Thermal unfolding profiles for monoTTR, CLU, and an equimolar mixture of the proteins were constructed from CD data recorded at 220 nm with samples heated continuously at a rate of 2 °C/min. The apparent melting temperatures (Tm) for samples, indicated by vertical dashed lines, were determined from peak maxima obtained from first-derivative calculations (B) of the melting data. CLU alone (blue squares) and the monoTTR/CLU mixture (open red triangles) exhibit similar lower-temperature structural transitions (Tm values of 48 and 47 °C, respectively) and a plateau at ∼60 °C. MonoTTR alone (filled red triangles) exhibits a single structural transition with a Tm of 60 °C and a plateau at ∼80 °C. The monoTTR/CLU mixture also displays a second unique, higher-temperature structural transition (>80 °C). (C) Comparison of monoTTR/CLU mixture unfolding curves recorded at fast (2 °C/min) and slow (0.1 °C/min) heating rates, showing a rate-dependent shift in the high-temperature (>80 °C) melting transition.
Oligomeric β-Sheet Rich Structures of Transthyretin Are Bound by Clusterin in a Stable Manner
We tested the ability of CLU to directly interact with oligomeric forms of rTTR. Wild-type rTTR oligomers were generated at pH 2.0 by rapid aggregation initiated upon addition of NaCl after rTTR unfolding; by 60 min, all tetrameric rTTR was transformed into oligomers with a molecular weight of >200 kDa [high-molecular weight (HMW)] and monomers (Figure 4A). Wild-type rTTR secondary structural changes during oligomerization were monitored using CD (Figure 4B). A loss of native β-sheet content, as seen in the wtTTR native sample (black line), and a transition to random-coil conformation occurred at pH 2.0 (wtTTR acid denatured, dashed red line). A subsequent transition to β-sheet structure and accumulation of β-sheet content over time were observed, indicating conformational conversion of the rTTR during oligomerization. Light microscopic examination of precipitated rTTR after a 14 day incubation under acidic conditions showed Congo red binding and amyloid fibril specific apple-green birefringence when viewed under standard and polarized light, respectively (Figure 4C,D).
Figure 4.
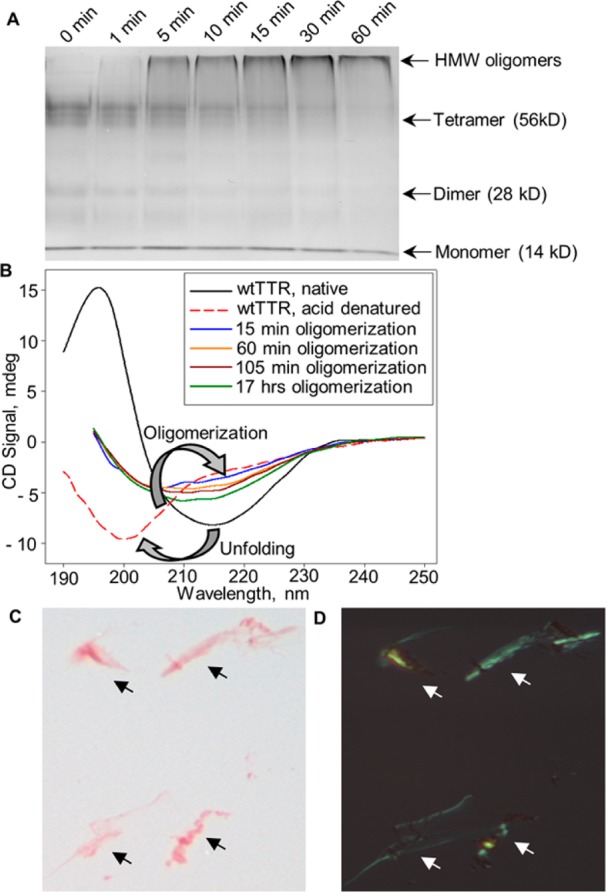
Transthyretin (TTR) high-molecular weight (HMW) oligomers and amyloid fibril synthesis in vitro. (A) wt rTTR samples, removed at various time points during the acid-mediated oligomer formation assay, were cross-linked with glutaraldehyde and analyzed by SDS–PAGE. (B) CD spectra of samples removed at timed intervals (15, 60 and 105 min, and 17 h) from a single continuous reaction mixture showing secondary structural transitions during rapid oligomerization. (C and D) Light microscopy of rTTR amyloid fibrils, formed after 2 weeks in the acid-mediated assay, stained with Congo red, and viewed under bright (C) and polarized (D) light. Arrows indicate wt rTTR amyloid fibrils.
SPR biosensor detection was used to characterize surfaces to which the previously described native or oligomeric forms of wt rTTR had been covalently bound. We used diflunisal and Congo red to determine the structural nature of rTTR-modified surfaces (Figure 5). The native rTTR surface demonstrated substantial diflunisal binding (0–60 s injection) and negligible Congo red binding (0–120 s injection), indicating intact TTR tetrameric structure (Figure 5A,B, gray lines) as measured by a change in response units (RU) in the SPR sensorgrams. An observed decrease in the level of diflunisal binding (Figure 5A, black line) with a corresponding marked increase in the level of Congo red binding (Figure 5B, black line) of HMW TTR surfaces was also measured. Serum-purified CLU was passed over the native and HMW TTR surfaces (panels C and D of Figure 5, respectively). Concentration-dependent binding of CLU to the HMW TTR surface was observed during the injection (0–300 s) phase (Figure 5D). No CLU binding took place with the native TTR surface (Figure 5C). It is important to note that the CLU binding sensorgrams demonstrate a concentration-dependent increase in magnitude that was consistent with the increased accumulated mass of CLU during the injection phase. In addition, the dissociation phase beginning at 300 s (vertical dashed line) was quite slow, indicating that the CLU–HMW TTR interactions at the SPR chip surface were more stable than interactions of TTR with diflunisal and Congo red.
Figure 5.

Characterization of tetrameric and oligomeric transthyretin (TTR) states using diflunisal and Congo red, with analysis of clusterin–transthyretin (CLU–TTR) interactions by surface plasmon resonance (SPR). Sensorgrams of (A) diflunisal and (B) Congo red analyte injections over surface-bound wt native TTR (gray lines) or soluble HMW TTR oligomers (black lines). CLU analyte injections at 200 (blue line) and 400 nM (red line) over (C) native and (D) HMW oligomeric wt TTR surfaces are shown. Analyte injection periods begin at 0 s with vertical dashed lines marking the end of injection periods. CLU–HMW TTR sensorgram data were fit using a conservative Langmuir 1:1 (analyte:ligand) interaction model (D, black lines).
Clusterin Prevents Transthyretin Amyloid Fibril Formation and Demonstrates Cooperative Inhibition in the Presence of Diflunisal
Finally, we investigated the ability of CLU, diflunisal, and a combination of the two to inhibit wt, L55P, and monomeric rTTR amyloid fibril formation under mildly acidic conditions. Interestingly, monoTTR in the presence of diflunisal, at neutral pH incubated for 2 h, showed enhanced dimerization and forced tetramer formation (Figure 6). After a 10 day incubation period, amyloid fibril formation reaction end points were measured using the quantitative Congo red binding spectral shift method.53 As wt TTR tetramer is stable at pH 5.0, the spectral absorbance characteristics of wt rTTR reactions were nearly identical to those of Congo red alone, indicating no fibril formation. However, L55P and monomeric rTTR end point reactions showed significant amyloid formation as indicated by increased amounts of Congo red bound in solution (Figure 7A,B). For L55P, a one-way analysis of variance (ANOVA) was used to test differences among treatments with diflunisal, CLU, a combination of the two, and BSA as a negative control for the reaction (Figure 7A; F = 46.77; p = 0.0004). Tukey post hoc comparisons of the treatments indicate that diflunisal (n = 2; no bound Congo red indicated), CLU [n = 2; M = 0.52; 95% confidence interval (CI) from −0.76 to 1.81], and diflunisal with CLU (n = 2; no bound Congo red indicated) yielded bound Congo red concentrations significantly lower than those with untreated L55P (n = 2; M = 1.57; 95% CI from 0.54 to 2.59) and L55P TTR with BSA (n = 2; M = 1.77; 95% CI from −1.35 to 4.89). For the monomer, a one-way ANOVA was used to test differences among treatments with diflunisal, CLU, a combination of the two, and BSA (Figure 7B; F = 407.9; p < 0.0001). Tukey post hoc comparisons of the treatments indicate that CLU (n = 3; M = 1.54; 95% CI from 1.27 to 1.81), and diflunisal with CLU (n = 3; M = 1.04; 95% CI from 0.92 to 1.15) resulted in amounts of bound Congo red significantly smaller than those with untreated monomer (n = 3; M = 2.64; 95% CI from 2.42 to 2.87) or monomer with BSA (n = 3; M = 2.82; 95% CI from 2.74 to 2.89). Additionally, amounts of bound Congo red in the combination treatments of monomer with diflunisal and CLU were significantly smaller than those for the treatment with only CLU (p < 0.001).
Figure 6.

Quaternary structural analysis of the F87M/L110M monomeric (monoTTR) rTTR variant treated with diflunisal (Dif.). Samples of F87M/L110M rTTR treated with diflunisal (left lane) and alone (middle lane), and wt rTTR alone (right lane) were cross-linked and analyzed by SDS–PAGE with Coomassie staining.
Figure 7.

Quantitative analysis of transthyretin (TTR) amyloid fibrils formed in vitro in the presence of diflunisal (Dif.) and clusterin (CLU). Concentrations of amyloid-bound Congo red in (A) L55P TTR and (B) monoTTR fibril synthesis reaction mixtures pretreated with Dif., CLU, Dif. and CLU, or BSA, after a 10 day incubation period at 37 °C and pH 5.0. Corresponding wt rTTR treatments showed no fibril formation as wt tetramer is stable at pH 5.0 (data not shown). Data were background subtracted with respect to amount of Congo red bound to negative control in each treatment. **p < 0.005, and ***p < 0.001.
Discussion
The inherent plasticity and unstructured nature of most proteins require macromolecular assistance to prevent aggregation and ensure proper functioning. In vivo proteostasis networks are normally in place to maintain a properly folded proteome, influence the rate of proteome folding, prevent aggregation, and mediate degradation allowing efficient protein turnover.56,57 Recently, extracellular chaperones have been proposed as integral components of a quality control system facilitating removal of circulating misfolded proteins and, as such, may play a significant role in the pathophysiology of TTR amyloidosis and other protein deposition diseases.38,58 CLU was the first extracellular chaperone to be identified, and extensive investigations over the past decade have shown that it has a chaperoning function similar to that of small heat shock proteins such as αB-crystallin.38−40,44,45,59,60 We have identified CLU as a component of TTR amyloid deposits in cardiac tissues from patients with ATTRwt (SSA) and ATTRm,46 and others have reported co-aggregation of CLU with variant TTR in a cerebrospinal fluid matrix.61
Herein, we report the first biophysical studies detailing the stabilizing effect of CLU on TTR unfolding and aggregation. Our CD data revealed that CLU has the capacity to stabilize the secondary structure of monomeric TTR during protein unfolding. Indeed, CLU preferentially bound to and stabilized monomeric TTR (mono, F87M/L110M). Equimolar mixtures of CLU and TTR were chosen within normal physiological concentrations as proamyloid effects of CLU have been reported under conditions in which the amyloid precursor is present in molar excess.44
Comparative analyses between the summed and observed CD spectral profiles for equimolar mixtures of monomeric TTR and CLU (monoTTR/CLU) showed a stabilization effect not predicted by the summed melting curve (Figure 2). A delay in the higher-Tm transition observed for the L55P TTR/CLU mixture was evident in the divergence from the summed protein melting curve. This indicated a small but substantial stabilization effect during L55P unfolding, although it was not nearly as robust as that observed for monomeric TTR, because of rate-limiting tetramer destabilization. The disassembly of the L55P tetramer into monomers that interact with CLU may account for this observation. As for the mixtures of the wt TTR tetramer with CLU, no such difference between the summed and observed spectral changes was detected, thus providing evidence of a preferential CLU stabilization effect on monomeric TTR. Slight fluctuations at the 25 °C baseline for all profiles were within experimental error, possibly because of minor protein concentration differences. Moreover, low-temperature unfolding transitions were identical in the CD spectra of CLU versus the equimolar mixtures of TTR tetramer and CLU (tetramer/CLU). Hence, it appears that in the wtTTR/CLU mixture, CLU and tetrameric TTR unfold independently.
The equimolar monoTTR/CLU mixture showed deviation of the CD signal at lower temperatures (CLU alone melt transition) with less unfolding during this early transition compared to that of CLU mixtures with wt and L55P TTR (Figure 2D). The secondary structure in the monoTTR/CLU mixture was preserved at the higher temperatures as demonstrated by the melt transition above 80 °C (Figure 3); these data indicate significant stabilization of TTR monomer by the presence of CLU. As CLU and monoTTR unfold simultaneously, chaperone interactions help to stabilize secondary structures and generate a unique molecular complex with a Tm higher than that of either individual protein. These data suggest that the structural stability in the mixture of the two proteins is greater than that of either CLU or monoTTR alone. The stabilization of unfolding TTR monomer by CLU supports a mechanism whereby CLU, acting in a chaperoning capacity, interacts with TTR monomer and prevents a conformational conversion to HMW oligomeric species. A mutual stabilization effect of these proteins on one another is possible under thermal stress conditions, although the reversible nature of unfolding and additional BSA controls presented in Figure 7 further supports the chaperoning model of CLU. The chaperoning interactions of CLU with TTR observed in Figure 3 likely occurred at lower temperatures as we detected the loss of those protein structures at the higher-temperature melt transition.
To confirm that CLU binds directly to misfolded and aggregated forms of TTR, but not to tetramer, we used SPR. SPR biosensor chip surfaces, prepared with HMW oligomeric wt TTR, exhibited an increased level of Congo red binding with a concomitant decrease in the level of diflunisal binding (Figure 5). This indicated that partially unfolded and oligomeric forms of TTR, i.e., amyloidogenic structures, were present and could be detected by small molecule binding. CLU bound, in a concentration-dependent manner, to unfolded and oligomeric HMW TTR that was coupled to the biosensor surfaces; a slow dissociation phase indicated that this was a stable association between CLU and HMW TTR. These data provide evidence that CLU has the capacity to bind higher-order oligomeric species of TTR.
By using rTTR variants with differing structures, stabilities, and propensities to form amyloidogenic oligomers and fibrils, we were able to test the effects of CLU and diflunisal on aggregation and fibrillogenesis, while comparing the differences between monomeric and tetrameric forms of TTR. Studies of L55P, pretreated with diflunisal, demonstrated the capacity of the small molecule to stabilize tetrameric TTR and inhibit fibril formation as shown by the absence of observable Congo red binding (Figure 7A). CLU treatment of L55P significantly inhibited fibril formation as was indicated by the decreased level of bound Congo red in solution, and the diflunisal/CLU mixture completely eliminated binding of Congo red to L55P. As anticipated, the propensity of monomeric TTR to form fibrils was stronger than that of L55P. Interestingly, despite enhanced dimerization and forced tetramerization of monomeric TTR by diflunisal (Figure 6), these quaternary structural changes did not inhibit amyloid formation over the tested time interval. However, CLU pretreatments significantly inhibited monomeric TTR fibril formation, while the combination of CLU and diflunisal showed significantly greater decreases in the level of fibril formation (Figure 7B), exhibiting a novel cooperative effect.
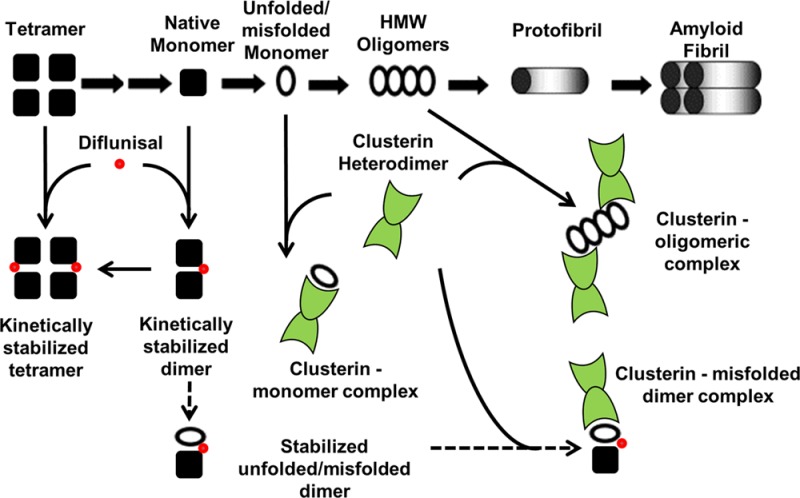
Wild-type TTR and more than 100 TTR variants are amyloidogenic; i.e., these proteins can all form fibrils that are similar in ultrastructure and composition. While it is possible that other mechanisms unique to different TTR variants exist, it is widely accepted that TTR amyloid fibril formation is dependent on dissociation of the native tetramer to monomer. This initial rate-limiting step of tetramer dissociation, believed to be a common stage in TTR amyloid pathogenesis despite the amino acid sequence heterogeneity of TTR variants, is the basis of our interpretation of the experimental data presented in this report and in defining a role for CLU in the TTR amyloid hypothesis. Our data support a mechanism whereby both unfolded/misfolded TTR monomer and HMW oligomeric species are chaperoned by CLU to divert these amyloid precursors off the pathway of amyloid fibril formation (Figure 8, thick solid arrows). The observed cooperative effect of the combination of CLU and diflunisal on inhibiting amyloid fibril formation may be due to the enhancement of CLU chaperoning capacity in the presence of diflunisal, given that diflunisal did not inhibit monoTTR amyloid fibril formation. This has yet to be determined, but a diflunisal-stabilized TTR intermediate could explain this observed phenomenon, thereby enhancing CLU proteostatic functions to promote clearance (Figure 8, dashed arrows). The complex milieu of in vivo amyloid pathology is difficult to precisely reproduce in experimental models. The biological meaning of interactions of CLU with TTR in amyloidosis is unclear and warrants further investigation. Potential neuroprotective mechanisms have been suggested along with evidence that CLU influences the aggregation of TTR in culture.62 Our biochemical experiments support a solution-based interaction model for the chaperoning activity of CLU in the circulation and in tissues rich in unfolded TTR.
Figure 8.
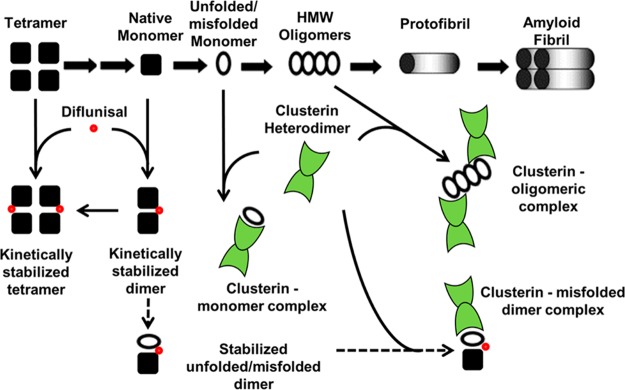
Transthyretin (TTR) amyloid fibril formation cascade and a possible model of clusterin (CLU) chaperoning interactions with unfolded TTR and diflunisal. The promiscuous chaperoning capacity of CLU and its ability to recognize regions of exposed hydrophobicity afford multiple points of interaction with unfolded/misfolded TTR along the amyloid fibril formation pathway. Shown are potential pathways for CLU stabilization of TTR monomers and oligomers, diverting these precursors off the pathway of TTR amyloid fibril formation (thick solid arrows). The influence of diflunisal on monomeric TTR dimerization and forced tetramerization indicates the potency of binding of small molecules to variant TTR. A cooperative effect of CLU and diflunisal on preventing amyloid fibril formation, hypothetically, through a diflunisal-stabilized TTR intermediate allowing for enhanced CLU chaperoning capacity (dashed arrows), is also shown.
The nonsteroidal anti-inflammatory drug diflunisal has been shown to stabilize TTR tetramers in vitro(18) and to slow the progression of polyneuropathy in patients with ATTRm amyloidosis.24 The activity of diflunisal in vivo may depend upon its ability to enhance the chaperoning activity of endogenous CLU, as well as its direct effect on stabilization of TTR tetramers (Figure 8). One might also propose a novel combinational approach to ATTRm or ATTRwt (SSA) treatment involving both diflunisal and CLU to augment the chaperoning and removal of misfolded and aggregated proteins. Small molecule activation of innate proteostasis networks may also bolster extracellular chaperone activity to further enhance pharmacological chaperone therapies.
Acknowledgments
We thank Clarissa Koch for her contributions in the culturing of hybridoma cell lines and synthesis of recombinant proteins, Drs. David A. Harris and Brian R. Fluharty for assistance with the surface plasmon resonance detection system, and Dr. Olga Gursky for thoughtful discussions about the circular dichroism analyses.
Glossary
Abbreviations
- TTR
transthyretin
- CLU
clusterin
- ATTRm
familial TTR-related amyloidosis
- ATTRwt
wild-type TTR-related amyloidosis
- SSA
senile systemic amyloidosis
- CD
circular dichroism
- SPR
surface plasmon resonance
- Tm
melting temperature
- wt
wild-type
- monoTTR
monomeric TTR
- HMW
high-molecular weight
- rTTR
recombinant TTR
- RU
resonance units
- Dif.
diflunisal.
Support for this study was provided by grants from the National Institutes of Health (RO1 AG031804 to L.H.C.), the Walk for an Angel Fund, the Gerry Foundation, and the Young Family Amyloid Research Fund.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Picken M. M. (2010) Amyloidosis: Where Are We Now and Where Are We Heading?. Arch. Pathol. Lab. Med. 134, 545–551. [DOI] [PubMed] [Google Scholar]
- Westermark P.; Benson M. D.; Buxbaum J. N.; Cohen A. S.; Frangione B.; Ikeda S.-I.; Masters C. L.; Merlini G.; Saraiva M. J.; Sipe J. D. (2007) A primer of amyloid nomenclature. Amyloid 14, 179–183. [DOI] [PubMed] [Google Scholar]
- Ruberg F. L.; Berk J. L. (2012) Transthyretin (TTR) Cardiac Amyloidosis. Circulation 126, 1286–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors L. H.; Doros G.; Sam F.; Badiee A.; Seldin D. C.; Skinner M. (2011) Clinical features and survival in senile systemic amyloidosis: Comparison to familial transthyretin cardiomyopathy. Amyloid 18, 157–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F.; Dobson C. M. (2006) Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 75, 333–366. [DOI] [PubMed] [Google Scholar]
- Invernizzi G.; Papaleo E.; Sabate R.; Ventura S. (2012) Protein aggregation: Mechanisms and functional consequences. Int. J. Biochem. Cell Biol. 44, 1541–1554. [DOI] [PubMed] [Google Scholar]
- Rodrigues J. R.; Simões C. J. V.; Silva C. G.; Brito R. M. M. (2010) Potentially amyloidogenic conformational intermediates populate the unfolding landscape of transthyretin: Insights from molecular dynamics simulations. Protein Sci. 19, 202–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello J. P.; Petäjä-Repo U. E.; Bichet D. G.; Bouvier M. (2000) Pharmacological chaperones: A new twist on receptor folding. Trends Pharmacol. Sci. 21, 466–469. [DOI] [PubMed] [Google Scholar]
- Conn P.; Leaños-Miranda A.; Janovick J. (2002) Protein Origami: Therapeutic Rescue of Misfolded Gene Products. Mol. Interventions 2, 308–316. [DOI] [PubMed] [Google Scholar]
- Cohen F. E.; Kelly J. W. (2003) Therapeutic approaches to protein-misfolding diseases. Nature 426, 905–909. [DOI] [PubMed] [Google Scholar]
- Miroy G.; Lai Z.; Lashuel H.; Peterson S.; Strang C.; Kelly J. (1996) Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc. Natl. Acad. Sci. U.S.A. 93, 15051–15056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baures P. W.; Oza V. B.; Peterson S. A.; Kelly J. W. (1999) Synthesis and evaluation of inhibitors of transthyretin amyloid formation based on the non-steroidal anti-inflammatory drug, flufenamic acid. Bioorg. Med. Chem. 7, 1339–1347. [DOI] [PubMed] [Google Scholar]
- Hammarström P.; Wiseman R. L.; Powers E. T.; Kelly J. W. (2003) Prevention of Transthyretin Amyloid Disease by Changing Protein Misfolding Energetics. Science 299, 713–716. [DOI] [PubMed] [Google Scholar]
- Buxbaum J.; Reixach N. (2009) Transthyretin: The servant of many masters. Cell. Mol. Life Sci. 66, 3095–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira N.; Saraiva M. J.; Almeida M. R. (2011) Natural polyphenols inhibit different steps of the process of transthyretin (TTR) amyloid fibril formation. FEBS Lett. 585, 2424–2430. [DOI] [PubMed] [Google Scholar]
- Ferreira N.; Saraiva M. J.; Almeida M. R. (2012) Natural polyphenols as modulators of TTR amyloidogenesis: In vitro and in vivo evidences towards therapy. Amyloid 19, 39–42. [DOI] [PubMed] [Google Scholar]
- Sant’Anna R. O.; Braga C. A.; Polikarpov I.; Ventura S.; Lima L. M.; Foguel D. (2013) Inhibition of Human Transthyretin Aggregation by Non-Steroidal Anti-Inflammatory Compounds: A Structural and Thermodynamic Analysis. Int. J. Mol. Sci. 14, 5284–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klabunde T.; Petrassi H. M.; Oza V. B.; Raman P.; Kelly J. W.; Sacchettini J. C. (2000) Rational design of potent human transthyretin amyloid disease inhibitors. Nat. Struct. Mol. Biol. 7, 312–321. [DOI] [PubMed] [Google Scholar]
- Adamski-Werner S. L.; Palaninathan S. K.; Sacchettini J. C.; Kelly J. W. (2004) Diflunisal Analogues Stabilize the Native State of Transthyretin. Potent Inhibition of Amyloidogenesis. J. Med. Chem. 47, 355–374. [DOI] [PubMed] [Google Scholar]
- Miller S. R.; Sekijima Y.; Kelly J. W. (2004) Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab. Invest. 84, 545–552. [DOI] [PubMed] [Google Scholar]
- Tojo K.; Sekijima Y.; Kelly J. W.; Ikeda S.-I. (2006) Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci. Res. 56, 441–449. [DOI] [PubMed] [Google Scholar]
- Kingsbury J. S.; Laue T. M.; Klimtchuk E. S.; Théberge R.; Costello C. E.; Connors L. H. (2008) The Modulation of Transthyretin Tetramer Stability by Cysteine 10 Adducts and the Drug Diflunisal: Direct Analysis by Fluorescence-Detected Analytical Ultracentrifugation. J. Biol. Chem. 283, 11887–11896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk J. L.; Suhr O. B.; Sekijima Y.; Yamashita T.; Heneghan M.; Zeldenrust S. R.; Ando Y.; Ikeda S.-I.; Gorevic P.; Merlini G.; Kelly J. W.; Skinner M.; Bisbee A. B.; Dyck P. J.; Obici L. (2012) The Diflunisal Trial: Study accrual and drug tolerance. Amyloid 19, 37–38. [DOI] [PubMed] [Google Scholar]
- Berk J. L.; Suhr O. B.; Obici L.; Sekijima Y.; Zeldenrust S. R.; Yamashita T.; Heneghan M. A.; Gorevic P. D.; Litchy W. J.; Wiesman J. F.; Nordh E.; Corato M.; Lozza A.; Cortese A.; Robinson-Papp J.; Colton T.; Rybin D. V.; Bisbee A. B.; Ando Y.; Ikeda S.-I.; Seldin D. C.; Merlini G.; Skinner M.; Kelly J. W.; Dyck P. J. (2013) Repurposing Diflunisal for Familial Amyloid Polyneuropathy: A Randomized Clinical Trial. JAMA, J. Am. Med. Assoc. 24, 2658–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S. M.; Connelly S.; Fearns C.; Powers E. T.; Kelly J. W. (2012) The Transthyretin Amyloidoses: From Delineating the Molecular Mechanism of Aggregation Linked to Pathology to a Regulatory-Agency-Approved Drug. J. Mol. Biol. 421, 185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel H. A.; Lai Z.; Kelly J. W. (1998) Characterization of the transthyretin acid denaturation pathways by analytical ultracentrifugation: Implications for wild-type, V30M, and L55P amyloid fibril formation. Biochemistry 37, 17851–17864. [DOI] [PubMed] [Google Scholar]
- Quintas A.; Saraiva M. J. M.; Brito R. M. M. (1999) The Tetrameric Protein Transthyretin Dissociates to a Non-native Monomer in Solution: A Novel Model for Amyloidogenesis. J. Biol. Chem. 274, 32943–32949. [DOI] [PubMed] [Google Scholar]
- Quintas A.; Vaz D. C.; Cardoso I.; Saraiva M. J. M.; Brito R. M. M. (2001) Tetramer Dissociation and Monomer Partial Unfolding Precedes Protofibril Formation in Amyloidogenic Transthyretin Variants. J. Biol. Chem. 276, 27207–27213. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Buxbaum J. N.; Kelly J. W. (2001) The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc. Natl. Acad. Sci. U.S.A. 98, 14943–14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Smith C. S.; Petrassi H. M.; Hammarström P.; White J. T.; Sacchettini J. C.; Kelly J. W. (2001) An Engineered Transthyretin Monomer that Is Nonamyloidogenic, Unless It Is Partially Denatured. Biochemistry 40, 11442–11452. [DOI] [PubMed] [Google Scholar]
- Sebastião M. P.; Lamzin V.; Saraiva M. J.; Damas A. M. (2001) Transthyretin stability as a key factor in amyloidogenesis: X-ray analysis at atomic resolution. J. Mol. Biol. 306, 733–744. [DOI] [PubMed] [Google Scholar]
- Shinohara Y.; Mizuguchi M.; Matsubara K.; Takeuchi M.; Matsuura A.; Aoki T.; Igarashi K.; Nagadome H.; Terada Y.; Kawano K. (2003) Biophysical Analyses of the Transthyretin Variants, Tyr114His and Tyr116Ser, Associated with Familial Amyloidotic Polyneuropathy. Biochemistry 42, 15053–15060. [DOI] [PubMed] [Google Scholar]
- Hurshman Babbes A. R.; Powers E. T.; Kelly J. W. (2008) Quantification of the Thermodynamically Linked Quaternary and Tertiary Structural Stabilities of Transthyretin and Its Disease-Associated Variants: The Relationship between Stability and Amyloidosis. Biochemistry 47, 6969–6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurshman A. R.; White J. T.; Powers E. T.; Kelly J. W. (2004) Transthyretin Aggregation under Partially Denaturing Conditions Is a Downhill Polymerization. Biochemistry 43, 7365–7381. [DOI] [PubMed] [Google Scholar]
- Alexandrescu A. T. (2005) Amyloid accomplices and enforcers. Protein Sci. 14, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgault S.; Solomon J. P.; Reixach N.; Kelly J. W. (2011) Sulfated Glycosaminoglycans Accelerate Transthyretin Amyloidogenesis by Quaternary Structural Conversion. Biochemistry 50, 1001–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mold M.; Shrive A. K.; Exley C. (2012) Serum Amyloid P Component Accelerates the Formation and Enhances the Stability of Amyloid Fibrils in a Physiologically Significant Under-Saturated Solution of Amyloid-β42. J. Alzheimer’s Dis. 29, 875–881. [DOI] [PubMed] [Google Scholar]
- Wilson M. R.; Yerbury J. J.; Poon S. (2008) Potential roles of abundant extracellular chaperones in the control of amyloid formation and toxicity. Mol. BioSyst. 4, 42–52. [DOI] [PubMed] [Google Scholar]
- Humphreys D. T.; Carver J. A.; Easterbrook-Smith S. B.; Wilson M. R. (1999) Clusterin Has Chaperone-like Activity Similar to That of Small Heat Shock Proteins. J. Biol. Chem. 274, 6875–6881. [DOI] [PubMed] [Google Scholar]
- Wilson M. R.; Easterbrook-Smith S. B. (2000) Clusterin is a secreted mammalian chaperone. Trends Biochem. Sci. 25, 95–98. [DOI] [PubMed] [Google Scholar]
- Poon S.; Easterbrook-Smith S. B.; Rybchyn M. S.; Carver J. A.; Wilson M. R. (2000) Clusterin Is an ATP Independent Chaperone with Very Broad Substrate Specificity that Stabilizes Stressed Proteins in a Folding-Competent State. Biochemistry 39, 15953–15960. [DOI] [PubMed] [Google Scholar]
- Poon S.; Treweek T. M.; Wilson M. R.; Easterbrook-Smith S. B.; Carver J. A. (2002) Clusterin is an extracellular chaperone that specifically interacts with slowly aggregating proteins on their off-folding pathway. FEBS Lett. 513, 259–266. [DOI] [PubMed] [Google Scholar]
- Poon S.; Rybchyn M. S.; Easterbrook-Smith S. B.; Carver J. A.; Pankhurst G. J.; Wilson M. R. (2002) Mildly Acidic pH Activates the Extracellular Molecular Chaperone Clusterin. J. Biol. Chem. 277, 39532–39540. [DOI] [PubMed] [Google Scholar]
- Yerbury J. J.; Poon S.; Meehan S.; Thompson B.; Kumita J. R.; Dobson C. M.; Wilson M. R. (2007) The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 21, 2312–2322. [DOI] [PubMed] [Google Scholar]
- Wyatt A. R.; Yerbury J. J.; Wilson M. R. (2009) Structural Characterization of Clusterin-Chaperone Client Protein Complexes. J. Biol. Chem. 284, 21920–21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene M. J.; Sam F.; Soo Hoo P. T.; Patel R. S.; Seldin D. C.; Connors L. H. (2011) Evidence for a Functional Role of the Molecular Chaperone Clusterin in Amyloidotic Cardiomyopathy. Am. J. Pathol. 178, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsbury J. S.; Klimtchuk E. S.; Théberge R.; Costello C. E.; Connors L. H. (2007) Expression, purification, and in vitro cysteine-10 modification of native sequence recombinant human transthyretin. Protein Expression Purif. 53, 370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M.; Easterbrook-Smith S. (1992) Clusterin binds by a multivalent mechanism to the Fc and Fab regions of IgG. Biochim. Biophys. Acta 1159, 319–326. [DOI] [PubMed] [Google Scholar]
- Lai Z.; Colon W.; Kelly J. W. (1996) The Acid-Mediated Denaturation Pathway of Transthyretin Yields a Conformational Intermediate That Can Self-Assemble into Amyloid. Biochemistry 35, 6470–6482. [DOI] [PubMed] [Google Scholar]
- Lindgren M.; Sörgjerd K.; Hammarström P. (2005) Detection and Characterization of Aggregates, Prefibrillar Amyloidogenic Oligomers, and Protofibrils Using Fluorescence Spectroscopy. Biophys. J. 88, 4200–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John D. M.; Weeks K. M. (2000) Van’t Hoff enthalpies without baselines. Protein Sci. 9, 1416–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk W. E.; Pettegrew J. W.; Abraham D. J. (1989) Quantitative evaluation of congo red binding to amyloid-like proteins with a β-pleated sheet conformation. J. Histochem. Cytochem. 37, 1273–1281. [DOI] [PubMed] [Google Scholar]
- Klunk W. E., Jacob R. F., Mason R. P., and Ronald W. (1999) Quantifying amyloid by congo red spectral shift assay. In Methods in Enzymology, pp 285–305, Academic Press, San Diego. [DOI] [PubMed] [Google Scholar]
- Hochgrebe T.; Pankhurst G. J.; Wilce J.; Easterbrook-Smith S. B. (2000) pH-Dependent Changes in the in Vitro Ligand-Binding Properties and Structure of Human Clusterin. Biochemistry 39, 1411–1419. [DOI] [PubMed] [Google Scholar]
- Shnyrov V. L.; Villar E.; Zhadan G. G.; Sanchez-Ruiz J. M.; Quintas A.; Saraiva M. J. M.; Brito R. M. M. (2000) Comparative calorimetric study of non-amyloidogenic and amyloidogenic variants of the homotetrameric protein transthyretin. Biophys. Chem. 88, 61–67. [DOI] [PubMed] [Google Scholar]
- Powers E. T.; Morimoto R. I.; Dillin A.; Kelly J. W.; Balch W. E. (2009) Biological and Chemical Approaches to Diseases of Proteostasis Deficiency. Annu. Rev. Biochem. 78, 959–991. [DOI] [PubMed] [Google Scholar]
- Lindquist S. L.; Kelly J. W. (2012) Chemical and Biological Approaches for Adapting Proteostasis to Ameliorate Protein Misfolding and Aggregation Diseases-Progress and Prognosis. Cold Spring Harbor Perspect. Biol. 3, 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt A. R.; Yerbury J. J.; Dabbs R. A.; Wilson M. R. (2012) Roles of Extracellular Chaperones in Amyloidosis. J. Mol. Biol. 421, 499–516. [DOI] [PubMed] [Google Scholar]
- Wyatt A. R.; Wilson M. R. (2010) Identification of human plasma proteins as major clients for the extracellular chaperone clusterin. J. Biol. Chem. 285, 3532–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt A. R.; Yerbury J. J.; Berghofer P.; Greguric I.; Katsifis A.; Dobson C. M.; Wilson M. R. (2011) Clusterin facilitates in vivo clearance of extracellular misfolded proteins. Cell. Mol. Life Sci. 68, 3919–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo E. P. C.; Pereira H. M.; Garratt R. C.; Kelly J. W.; Foguel D.; Palhano F. L. (2011) Dissecting the Structure, Thermodynamic Stability, and Aggregation Properties of the A25T Transthyretin (A25T-TTR) Variant Involved in Leptomeningeal Amyloidosis: Identifying Protein Partners That Co-Aggregate during A25T-TTR Fibrillogenesis in Cerebrospinal Fluid. Biochemistry 50, 11070–11083. [DOI] [PubMed] [Google Scholar]
- Magalhães J.; Saraiva M. J. (2011) Clusterin Overexpression and Its Possible Protective Role in Transthyretin Deposition in Familial Amyloidotic Polyneuropathy. J. Neuropathol. Exp. Neurol. 70, 1097–1106. [DOI] [PubMed] [Google Scholar]