

Abstract
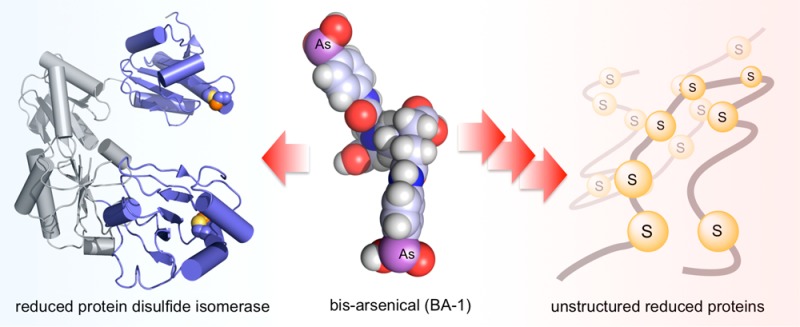
The renewed use of arsenicals as chemotherapeutics has rekindled interest in the biochemistry of As(III) species. In this work, simple bis- and tris-arsenical derivatives were synthesized with the aim of exploiting the chelate effect in the inhibition of thiol-disulfide oxidoreductases (here, Quiescin sulfhydryl oxidase, QSOX, and protein disulfide isomerase, PDI) that utilize two or more CxxC motifs in the catalysis of oxidative protein folding. Coupling 4-aminophenylarsenoxide (APAO) to acid chloride or anhydride derivatives yielded two bis-arsenical prototypes, BA-1 and BA-2, and a tris-arsenical, TA-1. Unlike the monoarsenical, APAO, these new reagents proved to be strong inhibitors of oxidative protein folding in the presence of a realistic intracellular concentration of competing monothiol (here, 5 mM reduced glutathione, GSH). However, this inhibition does not reflect direct inactivation of QSOX or PDI, but avid binding of MVAs to the reduced unfolded protein substrates themselves. Titrations of reduced riboflavin-binding protein with MVAs show that all 18 protein −SH groups can be captured by these arsenicals. With reduced RNase, addition of substoichiometric levels of MVAs is accompanied by the formation of Congo Red- and Thioflavin T-positive fibrillar aggregates. Even with Kd values of ∼50 nM, MVAs are ineffective inhibitors of PDI in the presence of millimolar levels of competing GSH. These results underscore the difficulties of designing effective and specific arsenical inhibitors for folded enzymes and proteins. Some of the cellular effects of arsenicals likely reflect their propensity to associate very tightly and nonspecifically to conformationally mobile cysteine-rich regions of proteins, thereby interfering with folding and/or function.
The finding that administration of arsenic trioxide is remarkably effective in the treatment of acute promyelocytic leukemia1 has stimulated renewed interest in the potential of arsenicals for the treatment of a variety of other types of cancer. A range of organic and inorganic arsenicals has recently been synthesized to deliver arsenic species to cells.2−5 The prototypical reagent, arsenic trioxide, yields arsonous acid in aqueous solution, and this reagent can bind up to three thiols, as depicted in Figure 1B.6 It is this coordination of thiols to As(III) species that is believed to underlie both the toxicity and the clinical potential of arsenicals. While arsenite (arsonous acid has a first pK of 9.2) binds many monothiols comparatively weakly, bis- or tris-mercaptans, in which the sulfhydryl groups can attain a preferred coordination geometry around the arsenic,5−8 can lead to tighter binding via the chelate effect. Indeed a large number of enzymes with redox-active dithiol motifs have been shown to be inhibited by arsenite with Kd values typically in the low micromolar range.5,7,9 In addition to arsenite, a number of monoalkyl and monoaryl As(III) species (including those previously studied in this laboratory; Figure 1A,C) have been shown to strongly coordinate with proteins and enzymes containing two or more juxtaposed and accessible thiol groups.5,7,10 A number of these proteins contain cysteine pairs contributed by redox-active disulfide motifs, including thioredoxins, their cognate reductases, dihydrolipoyl dehydrogenase, and glutathione reductase.5,7,10−14
Figure 1.

Arsenicals and their coordination to sulfhydryl species. (A) Some of the monoarsenicals discussed in this work. (B) Coordination of arsonous acid to a trithiol containing motif. (C) Coordination of an alkyl or aryl arsenical to a dithiol. (D) Structure of the bis- and tris-arsenicals used in this work. BA-2 consists of a mixture of regioisomers (see the text).
These data encouraged us to explore the interaction between the monoarsenicals and the enzymes that participate in oxidative protein folding because many of these proteins employ multiple catalytically essential redox-active disulfide motifs (frequently, a pair of cysteines separated by two amino acids, abbreviated here as CxxC). Figure 2A depicts an in vitro model of oxidative folding in which insertion of the correct disulfide pairings into reduced riboflavin binding protein (RfBP1) generates an apoprotein capable of rapid and stoichiometric binding of riboflavin with complete quenching of the strong fluorescence of the free vitamin.15 Here, the initial disulfide generation is catalyzed by Quiescin-sulfhydryl oxidase (QSOX), and mispairings are corrected by protein disulfide isomerase (PDI) in its reduced form.15 Figure 2B depicts the two redox-active CxxC motifs in QSOX;16−19 the first is contained in a highly oxidizing thioredoxin domain20 that collects reducing equivalents from client unfolded protein substrates (here, reduced RfBP) prior to their delivery to a second CxxC motif housed in the helix-rich ERV domain containing the bound FAD cofactor. Figure 2C shows that the outermost a and a′ thioredoxin domains of PDI contains two CxxC motifs.21,22 In their reduced states, the N-terminal cysteine residue of each CxxC motif can generate mixed disulfide intermediates, with previously mispaired cysteine residues in the client protein initiating iterative shuffling of disulfide connectivities. Alternatively, PDI can cycle between reduced and oxidized states, promoting iterative reduction and reoxidation within the client protein until the native pairings emerge.21,23,24 However, in the simplest of oxidative folding systems depicted in Figure 2A, efficient recovery of functional RfBP could be achieved with nanomolar QSOX and micromolar levels of reduced PDI in aerobic solution.15 With this folding system, we previously found that arsenite, MMA and PSAO (Figure 1A), profoundly attenuated the ability of reoxidized RfBP to rebind riboflavin.8 However, the effect did not reflect the direct inhibition of QSOX or PDI but was largely due to the unexpected ability of these monoarsenicals to capture the reduced unfolded conformationally mobile RfBP.8
Figure 2.
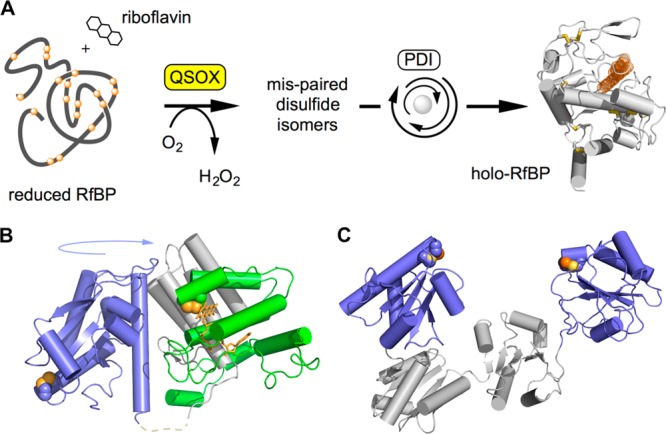
Oxidative protein folding catalyzed by QSOX and reduced PDI. (A) An assay for oxidative folding used in this work. QSOX inserts disulfides into reduced RfBP. Mispaired disulfides are corrected iteratively by PDI, and the fluorescence of free riboflavin is quenched on binding to active apo-RfBP. (B) Structure of an open conformation of QSOX from Trypanosoma brucei. CxxC motifs in the thioredoxin (blue) and ERV (green) domains are shown by solid yellow spheres. These CxxC motifs are brought together during catalysis by a large-scale rotation involving a flexible interdomain linking region (dashed line). Vertebrate QSOXs appear to be mechanistically identical, although they have an additional redox-inactive thioredoxin domain of unknown function. (C) The two CxxC motifs in the a and a′ domains in one (of multiple) conformation of human PDI (PDB 4EL1) are highlighted.
Here, we explore the synthesis and characterization of simple bis- and tris-arsenical prototypes of reagents that we envisaged might lead to more effective inhibition of enzymes with multiple CxxC motifs. For example, during QSOX catalysis, a flexible interdomain linker (shown dotted in Figure 2B) allows the two catalytically essential CxxC motifs to share a mixed disulfide bond18,20 and hence they are candidates for capture by small bis-arsenicals. Similarly, the flexibility of PDI proteins22,25−27 and recent studies supporting intraprotein redox communication27,28 suggest that their CxxC motifs might be captured by multivalent arsenicals. The fluorescent bis-arsenicals pioneered by Tsien and co-workers feature arsenoxide functional groups spaced ∼5 Å apart on the same edge of either a xanthene or phenoxazine ring system. These FlAsH and ReAsH derivatives have proved to be widely useful in labeling conformationally mobile tetracysteine tags in a range of cellular contexts,29−32 but they are structurally unsuited for bridging independently folded CxxC-containing domains.
While we have recently developed a strategy for attaching arsenicals to flexible polypeptide chains,33 here we explore the design and implementation of simpler reagents that might serve as prototypes for membrane-permeant derivatives. The three reagents tested here (Figure 1D) prove to be straightforward to synthesize and show promise as inhibitors of enzymes with multiple redox-active disulfide motifs. Nevertheless, we found that their dominant effect is again to interfere with oxidative protein folding by a particularly avid binding to the disordered reduced substrates of these folding systems.
Materials and Methods
Materials
Guanidine hydrochloride (GuHCl), bovine pancreatic ribonuclease A (RNase A), bovine pancreatic insulin, thioflavin T, ethylenediaminetetraacetic dianhydride, 1,3,5 benzenetricarbony trichloride, and 3,3′,4,4′-biphenyltetracarboxylic dianhydride were purchased from Sigma-Aldrich. 5,5′-Dithiobis-2-nitrobenzoic acid (DTNB), dithiothreitol (DTT), and tris(2-carboxyethyl) phosphine hydrochloride (TCEP) were obtained from Gold Biotechnology. Size-exclusion PD-10 columns were purchased from GE Healthcare, and Congo Red was purchased from MP Biomedicals.
Synthesis of APAO and PSAO
All arsenicals were handled with appropriate care in view of the toxicity of As(III) species. 4-Aminophenyl arsenoxide (APAO) was synthesized as described previously.13 APAO was succinylated to yield the more water-soluble and experimentally tractable analogue PSAO as described by Cline et al.34 All arsenicals were quantitated as described previously33 via titration with a standardized solution of DTT following the spectrum of the solution at 300 nm.
Synthesis of BA-1:2,2′-(Ethane-1,2-diylbis((2-((4-arsinephenyl)amino)-2-oxoethyl)azanediyl))diacetic Acid
APAO (156 mg, 0.39 mmol) was dissolved in 5 mL of dry DMF in a round-bottomed flask followed by the addition of ethylenediaminetetraacetic dianhydride (100 mg, 0.39 mmol; Figure S1A). After stirring overnight, the product was purified by reverse-phase HPLC using a linear gradient of 0.1% formic acid in water to 90% acetonitrile containing 1% formic acid over 30 min at 1 mL/min on a Phenomenex C18 column (10 mm, 300 Å, 250 × 10.00 mm). The fractions containing the product were collected and lyophilized. 1H NMR (CD3OD methanol-d4) δ (ppm) 3.30 (4H), 3.85 (4H), 3.95 (4H), 7.39 (4H), 7.49 (4H); 13C NMR δ (ppm) 50.6, 51.6, 54.8, 55.0, 56.7, 70.0, 107.9, 120.2, 126.8, 143.9, 147.6, 171.9; mass calcd, 658.02; found, 659.03 [M + H]+.
Synthesis of Synthesis of BA-2:3′,4-Bis((4-arsinephenyl)carbamoyl)-[1,1′-biphenyl]-3,4′-dicarboxylic Acid
APAO (201 mg, 1.01 mmol) was dissolved in dry DMF in a round-bottomed flask followed by the addition of 100 mg (0.34 mmol) of 3,3′,4,4′-biphenyltetracarboxylic dianhydride to the reaction mixture, and the mixture was stirred overnight (Figure S1B). The reaction was monitored by TLC (pure methanol), and the final product was separated as a yellow solid from the reaction mixture by trituration with ethyl acetate. 1H NMR (CD3OD) δ (ppm) 7.52 (4H), 7.66 (1H), 7.74 (4H), 7.78 (1H), 7.86 (1H), 7.88 (1H), 8.07 (2H); 13C NMR δ (ppm) 121.17, 129.83, 129.22, 129.13, 127.34, 127.54, 131.74, 132.34, 141.39, 142.52, 144.04, 158.21, 168.09, 170.68; mass calcd, 695.97; found, 696.93 [M + H]+. BA-2 was a mixture of regioisomers (3,3′, 3,4′, and 4,4′) that could not be readily separated chromatographically or by crystallization and was therefore used as-is.
Synthesis of TA-1:1,3,5-((Benzenetricarbonyltris(azanediyl)) Tris(benzene-4,1-diyl))triarsineous Acid
APAO (298 mg, 1.48 mmol) was dissolved in 5 mL of acetone in a dry round-bottomed flask followed by 100 mg (0.37 mmol) of 1,3,5 benzenetricarbonyl trichloride. The reaction mixture was allowed to stir overnight, and the insoluble product was removed by filtration and washed with acetone to remove excess APAO. 1H NMR (CD3OD) δ (ppm) 7.59 (6H), 7.79 (6H), 8.53(3H); 13C NMR δ (ppm) 121.82, 124.26, 131.96, 133.18, 137.29, 167.12; mass calcd, 758.92; found, 759.89 [M + H]+.
Methods
All absorption spectra were collected on an Agilent 8453 UV/vis spectrophotometer, and the data were analyzed using ChemStation software. Experiments were performed at least in duplicate in 50 mM potassium phosphate buffer containing 1 mM EDTA, pH 7.5, 25 °C, unless otherwise noted. NMR was performed on a Bruker Avance DX 400 spectrometer. Mass spectroscopy was done on a Q-TOF Ultima LC-MS/MS instrument. Stock solutions of DTT, GSH, and TCEP were standardized using DTNB.35 PDI was purified as in Rancy et al.15 Avian QSOX36 and chicken riboflavin binding protein (RfBP)37 were prepared as described previously and were generous gifts from Drs. Karen Hoober and Harold B. White, III, respectively.
Preparation of Reduced Proteins
PDI was incubated with a 40-fold molar excess of DTT at 25 °C for 1 h in 50 mM potassium phosphate buffer containing 1 mM EDTA, pH 7.5. The reduced enzyme was freed from excess reductant by size-exclusion chromatography using a PD-10 column equilibrated with 50 mM phosphate buffer without DTT and stored at −20 °C until further use. Bovine pancreatic RNase A was incubated for 2 h at 37 °C with an 80-fold molar excess DTT in 100 mM Tris buffer, pH 8.0, containing 1 mM EDTA and 6 M GuHCl. The reduced, denatured protein was separated from excess reductant and GuHCl using a PD-10 column equilibrated with 10 mM sodium acetate buffer containing 1 mM EDTA, pH 4.0. The reduced protein solution was stored anaerobically under nitrogen at 4 °C. Lyophilized RfBP was incubated for 2 h at 37 °C with an 180-fold molar excess of DTT in 100 mM Tris buffer, pH 8.0, containing 1 mM EDTA and 6 M GuHCl. The reduced, denatured protein was applied to a PD-10 column equilibrated with same buffer without the reductant and stored anaerobically at 4 °C. Reduced protein concentrations were determined using the following molar absorptivities at 280 nm: RNase, 9300 M–1 cm–1; RfBP, 49 000 M–1 cm–1; and PDI, 56 400 M–1 cm–1. Thiol titers were determined using DTNB (with a molar absorptivity of the TNB thiolate of 14 150 M–1 cm–1).38
Oxidative Protein Folding Assay Using RfBP
The refolding assay was performed by incubating 1 μM reduced and denatured chicken RfBP (18 μM free thiols) with 30 nM QSOX, 30 μM reduced PDI, and arsenicals in the presence of 0.8 μM free riboflavin.15 The loss of riboflavin fluorescence over time was monitored for 60 min (excitation 450 nm and emission 530 nm, with 2 and 16 nm slit widths, respectively; under these conditions, photobleaching of riboflavin was insignificant15). Control reactions were performed under the same conditions in the absence of arsenicals.
Oxidative Protein Folding Assay Using RNase
Reduced unfolded RNase (10 μM protein, 80 μM free thiols) was added to a mixture of 5 μM reduced PDI in 0.4 mL of a redox buffer composed of 1 mM/0.2 mM or 5 mM/1 mM reduced/oxidized glutathione, respectively, in 50 mM Tris buffer, pH 7.5, containing 1 mM EDTA, with or without 10 μM As(III) species. Aliquots (60 μL) were removed every 4 min and mixed with 60 μL of 2 mM cCMP solution, and the RNase activity followed the hydrolysis of cCMP to CMP at 296 nm for 2 min.15
Binding of Arsenicals to Reduced PDI
The binding of arsenicals to PDI was assessed as before8 by the increase in absorbance at 300 nm. Spectra were recorded 20 min after each addition, and the data were fit to a binding equation that accounts for the depletion of ligand upon protein binding.8 Here, the starting absorbance was fixed, and the final absorbance, Kd, and stoichiometry were variables.
Turbidometric Insulin Reductase Assay
A solution of 50 μM insulin solution in 50 mM phosphate buffer, pH 7.5, contained 1 mM EDTA, the arsenicals as needed, and either 100 μM TCEP or 5 mM GSH as reducing agents, followed rapidly by the addition of 1 μM PDI. The increase in turbidity was followed at 600 nm, and the lag phase calculated as described earlier.33 Control reaction used the same conditions in the absence of arsenicals.
O2 Electrode Assays
QSOX (30 nM) was assayed at 25 °C with 5 mM TCEP or 5 mM GSH as oxidase substrates in a Clark-type oxygen electrode in 50 mM phosphate buffer, pH 7.5, containing 1 mM EDTA in the absence or presence of arsenicals.
Thioflavin T Assay
Reduced RNase (30 μM) was mixed with 5 μM thioflavin T in the presence of either 5 mM GSH or 1 mM TCEP (to suppress the formation of any adventitious protein disulfides over extended incubation periods) in a total volume of 200 μL. The increase in fluorescence (excitation, 450 nm; emission, 485 nm) was monitored over time in a PerkinElmer fusion plate reader in the presence or absence of arsenicals.
Congo Red Assay
Reduced RNase (20 μM) was incubated with 10 μM Congo Red dye and arsenical compounds for 30 min in the absence or presence of 5 mM GSH. The spectrum of each reaction mixture was subtracted from the spectrum of Congo Red alone to more clearly observe the spectral shift at 540 nm.
Transmission Electron Microscopy
Imaging was performed using a Zeiss LIBRA 120 transmission electron microscope equipped with a Gatan Ultrascan 1000 2k × 2k CCD camera at the Delaware Biotechnology Institute. Reduced RNase (50 μM) and arsenicals were incubated overnight in the presence of 5 mM GSH or 1 mM TCEP (to suppress adventitious disulfide bond formation). Aliquots (4 μL) were placed on TEM grids (400 copper mesh with a Formvar-carbon film FCF400Cu from Electron Microscopy Sciences). The excess liquid was wicked with filter paper, and the grid was washed three times with water before staining with 1% uranyl acetate. The grid was again wicked with filter paper and air-dried for 1 h before recording the image.
Simulation of Competition between Dithiols and Glutathione for an Arsenical Center
The partition between dithiols and GSH complexes of monoarsenical MMA was simulated using COPASI39 using the following equations with Kd values for complexation of MMA by GSH calculated from the data of Spuches et al.6
Kd values for hypothetical dithiol complexes of MMA ranged from 10–5 to 10–12 M (see the text). Appropriate forward and reverse rate constants were chosen to give these Kd values for MMA, and the simulation was run until equilibrium was reached from starting concentrations of 10 μM MMA, 10 μM dithiol, and 5 mM GSH. The percentage of MMA complexed with the dithiol was then plotted as a function of the Kd of the MMA–dithiol complexes.
Results and Discussion
Synthesis of Bis- and Tris-arsenicals
The synthesis and characterization of bis- and tris-arsenicals (BA-1, BA-2, and TA-1; Figures 1 and S1) are described in Materials and Methods. These derivatives are readily synthesized by coupling 4-aminophenyl arsenoxide with commercially available anhydrides or acyl chlorides in a one-pot reaction. The compounds were stored at 4 °C. Stock solutions were prepared in phosphate buffer or methanol and stored at 4 °C prior to use. Concentrations were determined gravimetrically and confirmed via titration with a standardized solution of DTT following the increase in absorbance in the near UV region of the spectrum that accompanies coordination of the arsenoxide species with thiols.6,8 Where appropriate, the effectiveness of the arsenicals was compared at a constant concentration of 10 μM total arsenic (corresponding to 5 μM BA-1 and BA-2 and 3.33 μM TA-1).
Bis- and Tris-arsenicals Inhibit Oxidative Protein Folding in Vitro
Figure 3A shows the decline in fluorescence as free riboflavin binds to apo-RfBP formed during the oxidation driven by 30 nM QSOX in aerobic solution with non-native pairings addressed with 30 μM reduced PDI.15 In contrast to the control incubation, the inclusion of 10 μM As(III) species (either as 5 μM bis-arsenicals BA-1 and BA-2 or 3.33 μM TA-1) leads to the almost complete inhibition of riboflavin binding, as shown by the fluorescence monitored with time in Figure 3A. The data are also depicted in the bar diagram plotting the decrease in fluorescence at 60 min normalized to the percent of the control incubation. Under these conditions, the multivalent arsenicals are comparably as effective as a 10 μM concentration of monothiols MMA and PSAO. However, when oxidative folding mixtures are supplemented with a concentration of GSH that could be encountered intracellularly (5 mM; Figure 3B), the monoarsenicals are notably less effective at inhibiting oxidative folding, whereas BA-1 and BA -2 and TA-1 still inhibit the rebinding of riboflavin almost completely. Although the monothiol GSH binds arsenicals relatively weakly,6 the relatively high concentration of intracellular GSH lowers the concentration of arsenic species available to bind protein targets (a more quantitative discussion is presented later). In summary, these data show that the multivalent arsenicals are much more effective at inhibiting oxidative folding than their monoarsenical counterparts in the presence of competing glutathione.
Figure 3.
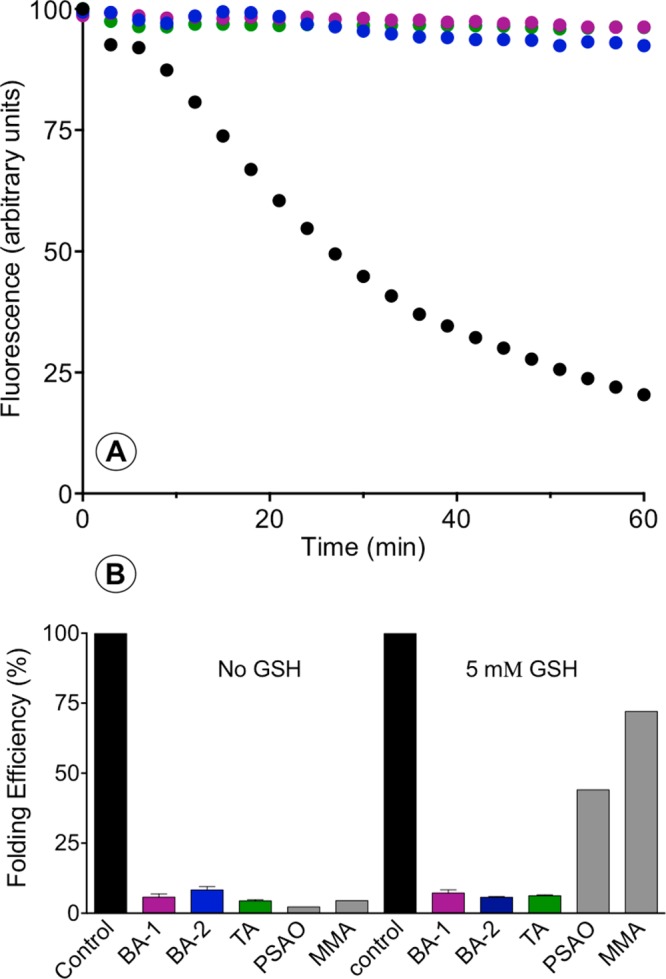
Inhibition of the oxidative folding of reduced RfBP in the presence of bis- and tris-arsenicals. (A) Oxidative protein folding monitored by the loss of riboflavin fluorescence as apo-RfBP is generated by the combined action of 30 nM QSOX and 30 μM reduced PDI in aerobic buffer, pH 7.5 (see Materials and Methods). BA-1, BA-2, and TA-1 (at an aggregate As(III) concentration of 10 μM) strongly suppress oxidative folding under these conditions. (B) Extent of riboflavin binding as a percentage of that observed in the control recorded at 60 min in the absence of arsenical. Data for PSAO and MMA are taken from Ramadan et al.8 These experiments were then repeated with the additional presence of 5 mM GSH, and the data are summarized in panel B.
Effect of Bis- and Tris-arsenicals on Oxidative Refolding of RNase
In a widely used model for oxidative protein folding, reduced unfolded RNase is added to a glutathione redox buffer containing PDI to accelerate the oxidation of cysteine residues and the correction of mispaired disulfides (Figure 4A).40 Samples are then removed and assayed discontinuously for RNase enzymatic activity (see Materials and Methods). In this system, GSSG, and not the oxidase QSOX, drives disulfide bond generation in the client protein. The multivalent arsenicals are rather comparable in inhibiting regain of RNase activity using redox buffer of either 1 mM GSH paired with 0.2 mM GSSG (where the redox poise is approximately −170 mV41) or 5 mM GSH and 1 mM GSSG (poised at approximately −190 mV). Following recovery of approximately 25% of active RNase, oxidative refolding of the remaining protein is largely inhibited after about 8 min (Figure 4B,C). Here, the activity recovered before cessation of refolding was somewhat greater when a higher concentration of competing GSH was employed (Figure 4C). In contrast, PSAO gave minimal inhibition of the refolding of RNase with either 5 or 1 mM GSH (Figure 4B,C).
Figure 4.
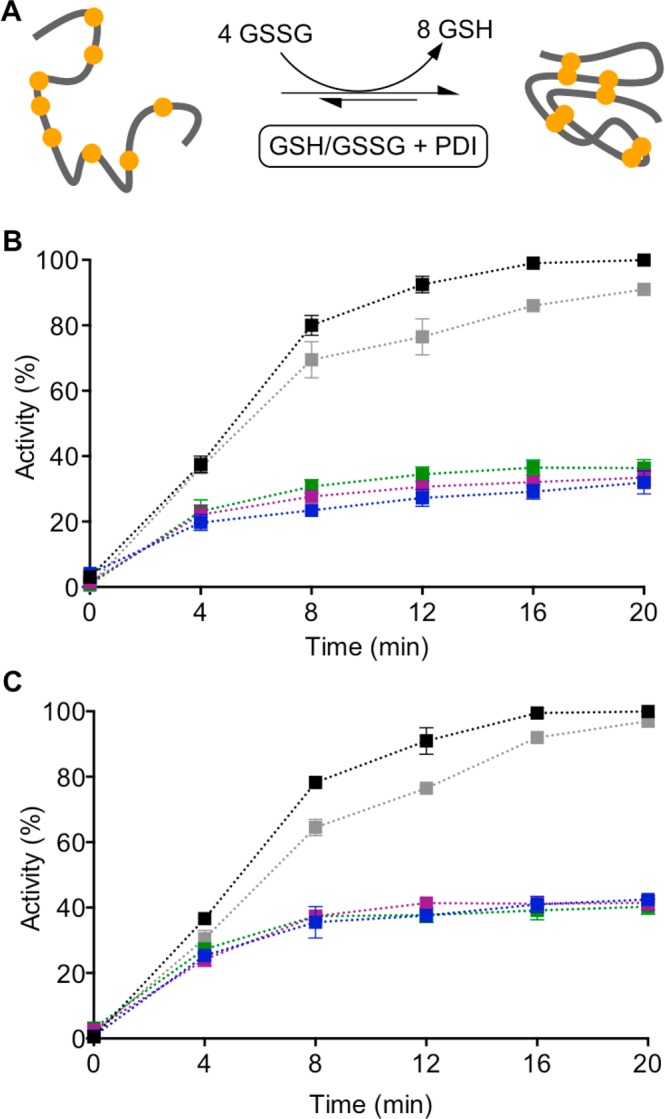
Inhibition of the oxidative refolding of RNase by arsenicals. (A) Schematic representation of the refolding of RNase in the presence of a redox buffer and PDI. Reduced and denatured RNase (10 μM; 80 μM thiols) was incubated with 5 μM reduced PDI and a redox buffer containing either 1 mM GSH and 0.2 mM GSSG (B) or 5 mM GSH and 1 mM GSSG (C). The colors used are as follows: control (no arsenical), black; 5 μM BA-1, blue; 5 μM BA-2, pink; 3.33 μM TA-1, green; and 10 μM PSAO, gray.
Bis- and Tris-arsenicals as Inhibitors of QSOX
The above data showed strong inhibition of the refolding of a protein with complex disulfide connectivity (RfBP; 9 disulfides with 34 million possible combinations) in the presence of PDI and QSOX and marked inhibition of a much simpler refolding system involving RNase with only 4 disulfides using PDI in the presence of a glutathione redox buffer. These data suggested that QSOX was not the major target of these multivalent arsenicals. To address this issue, we investigated the effects of BA-1, BA-2, and TA-1 on QSOX activity. We utilized two alternate substrates of the oxidase; the first is GSH at 5 mM, approximating intracellular GSH concentrations both in the cytosol and in the endoplasmic reticulum.21 QSOX activity is not significantly inhibited in the presence of this thiol substrate under the conditions of the assay (Figure 5; see Materials and Methods). In the absence of GSH that can compete for free As(III) (see later), use of a noncoordinating model phosphine substrate of QSOX (TCEP) reveals modest inhibition of the oxidase (Figure 5). These data further demonstrate that the inhibition of oxidative folding of the refolding of RfBP in the presence of 5 mM GSH (Figure 3) cannot be principally due to QSOX. Having ruled out a major effect on the first enzyme of oxidative folding, we next examine the effects of these multivalent arsenicals on PDI.
Figure 5.

Inhibition of QSOX reactivity by bis- and tris-arsenicals. The activity of 30 nM avian QSOX was evaluated in the oxygen electrode assay (see Materials and Methods) 100–400 s after the addition of enzyme to a solution containing either 5 mM GSH (left) or TCEP (right) as substrates and 5 μM BA-1 and BA-2 and 3.33 μM TA-1. Data are expressed as percentages of a control assayed with either GSH or TCEP (left bar).
Interaction between Multivalent Arsenicals and PDI
Figure 6A depicts the standard reductase assay8,42 used to assess the potency of the multivalent arsenical inhibitors for PDI. Reduction of PDI drives isomerase-catalyzed reduction of the interchain disulfides of insulin with consequent accumulation of the isolated weakly soluble isolated B-chain. After a reproducible lag phase, a threshold concentration is reached, leading to the appearance of B-chain aggregates with the onset of light scattering. A marked delay in aggregation time is evident with all three multivalent arsenicals when the non-As(III)-coordinating reductant TCEP was used (Figure 6B). The times for the onset of detectable turbidity are summarized for BA-1, BA-2, and TA-1 in Figure 6D. In contrast, the monovalent arsenical PSAO was ineffective under these conditions. We considered whether the apparent inhibition of the isomerase observed in Figure 6B might be an artifact of sequestration and solubilization of the reduced B-chain by the polar arsenical. However, these arsenicals do not delay the development of light scattering when insulin is reduced in the presence of 5 mM TCEP alone (see Figure S2) and hence they inhibit PDI activity directly under these conditions. Although substantial inhibition of PDI is observed using TCEP (Figure 6B,D), replacement of this noncoordinating reductant by 5 mM GSH results in only marginal lengthening of aggregation times using BA-1, BA-2, and TA-1 (Figure 6C,E). Thus, although multivalent arsenicals are more potent inhibitors of PDI than the monoarsenicals, they are still ineffective in the presence of millimolar levels of GSH (see later).
Figure 6.

Effect of multivalent arsenicals on the reductase activity of PDI. (A) Schematic of the assay. Porcine insulin (50 μM) in 50 mM phosphate buffer, pH 7.5, containing 1 mM EDTA) was mixed with either 100 μM TCEP (B) or 5 mM GSH (C) in the absence or presence of 10 μM of As(III) in BA-1, BA-2, and TA-1. Time zero corresponds to the addition of 1 μM PDI (see Materials and Methods). The onset of turbidity was observed at 600 nm and is expressed in bar graph form in panels D and E (for B and C, respectively; see Materials and Methods).
These data prompted examination of the dissociation constants for the multivalent arsenicals benchmarked against the behavior of the monoarsenical PSAO.8 It should be noted that the CxxC motifs of the a and a′ thioredoxin domains of mammalian PDI have very similar active site sequences and essentially identical redox potentials.43 As might be expected, there was no evidence for heterogeneity in binding when PSAO was titrated with PDI containing these two reduced CxxC motifs (Kd = 1.1 μM8). Prior to determination of Kd values, we assessed the stoichiometry of binding between the BA-1 and reduced PDI. Figure 7 shows a representative titration using relatively high protein concentration to facilitate an accurate quantitation of binding stoichiometry; at 10 μM PDI, we observe a unit stoichiometry for bis-arsenical binding. These data show that two reduced CxxC motifs are captured by a single molecule of BA-1, raising issues concerning the sterics of their interaction. The distance between the a and a′ CxxC motifs in the crystal structure of human PDI shown in Figure 2C is about 40 Å, compared to a likely maximal interarsenic distance of ∼20 Å for a fully extended BA-1 molecule (Figure 1D). However, multiple experimental and computational approaches show that the outermost a and a′ domains in human PDI are highly mobile, allowing the CxxC motifs to communicate directly via an interdomain mixed disulfide.27,28,44,45 Hence, cross-linking these domains via a single bis-arsenical is certainly feasible and consistent with the stoichiometry observed in Figure 7. Titration experiments were then conducted with 1 μM PDI to permit an estimation of the dissociation constant between the isomerase and BA-1. As observed in the inset to Figure 7, the increase in absorbance can be fit to a Kd of 54 ± 29 nM with a stoichiometry of 0.90 ± 0.08. BA-2 shows a comparable Kd of 56 ± 36 nM with a stoichiometry of 1.0 ± 0.11 (Figure S3). The sizable uncertainties encountered in Kd values reflect the difficulties in conducting absorbance titrations for tightly binding ligands at relatively low protein concentrations. However, both Kd values are some 20-fold lower than that for the monoarsenical PSAO, consistent with the expression of a very modest chelate effect with these bis-arsenicals. We note later that, even at Kd values of ∼50 nM, these bis-arsenicals bind insufficiently tightly to render them effective inhibitors of PDI at the GSH concentrations that prevail intracellularly. A titration of 1 μM reduced PDI with the trisarsenical, TA-1, shows a Kd of 100 nM ± 32 nM with a stoichiometry of 0.89 ± 0.08 per reduced PDI molecule. Evidently, the third arsenical site goes unused in these experiments.
Figure 7.

Interaction between BA-1 and reduced PDI. The main figure shows a spectrophotometric determination of the net stoichiometry of binding of BA-1 to 10 μM reduced PDI (see Materials and Methods). The inset repeats the titration using 1 μM reduced PDI to provide an estimate of the dissociation constant. The solid curve is fit to a Kd of 54 nM ± 29 nM with a stoichiometry of 0.90 ± 0.08 molecules of the bis-arsenical BA-1 (see Materials and Methods).
Binding of Multivalent Arsenicals to Reduced RfBP
The experiments in Figures 3 and 4 show that oxidative folding is strongly inhibited by the multivalent inhibitors studied here, although Figures 5 and 6 show that neither QSOX nor PDI are significantly inactivated by these arsenicals in the presence of millimolar levels of glutathione. Figure 8 confirms that these multivalent arsenicals do indeed bind avidly to reduced unfolded RfBP. This protein has 18 free thiols and hence a stoichiometry of ∼4.5 molecules of BA-1 and BA-2 would be expected per reduced RfBP. Figure 8 shows that both arsenical moieties in BA-1 and BA-2 can each capture a pair of reduced protein thiols. The tris-arsenical also shows a stoichiometry comparable to that of the bisarsenicals, suggesting that, as observed with reduced PDI (Figure S3), one arsenic site remains unused. Presumably, the steric requirements for coordinating 6 cysteine peptide thiols around a single molecule of TA-1 preclude a significant contribution of this binding mode. In all cases, the sharp end point of the titrations in Figure 8 suggests that the multivalent arsenical reagents bind very tightly to reduced RfBp. This conclusion is reinforced by the finding that these reagents interfere with the oxidative refolding of reduced RfBP even in the presence of 5 mM competing GSH. In contrast, the monoarsenicals are ineffective at inhibiting oxidative folding when challenged with 5 mM glutathione (Figure 3B), consistent with the expectation that they would bind more weakly to reduced RfBP. In the next section, we describe our attempts to extend these studies to reduced RNase.
Figure 8.

Titration of reduced RfBP with multivalent arsenicals. Reduced RfBP was prepared as in Materials and Methods, and 1 μM of the protein (18 μM thiols) was titrated with increasing concentration of the arsenicals. The increase in absorbance at 300 nm was recorded 20 min after each addition for BA-1, BA-2, and TA-1. All data sets gave sharp end points shown by the dotted lines. If all arsenical moieties in these MVAs were captured by the 18 −SH groups in reduced RfBP, then the stoichiometry for the bis-arsenical would be 4.5, and that for the tris-arsenical, 3.0.
The Interaction of Multivalent Arsenicals with Reduced RNase
Although we were able to conduct detailed titrations of reduced RfBP using BA-1, BA-2, and TA-1, comparable experiments with rRNase proved to be unworkable because of the consistent onset of light scattering as the titration progressed. We had previously noted that exposing reduced RNase to the monoarsenical MMA led to the formation of filamentous deposits, although this effect was not observed with PSAO or arsenite under comparable conditions.8 Thus, such aggregation is not a universal consequence of thiol coordination by arsenicals. In contrast, all three MVAs promoted formation of RNase aggregates that were insoluble and readily detected using thioflavin T fluorescence measurements (Figure 9A).46 Importantly, these aggregates were still formed in the presence of 5 mM GSH (Figure 9B), whereas they do not appear when solutions of reduced RNase and GSH are subject to MMA treatment.8 These data again show that MVAs bind more tightly to reduced unfolded proteins than the monoarsenicals studied earlier. Spectrophotometric experiments with Congo Red46 are also consistent with an association of unfolded reduced RNase into structures with significant β-sheet content. Figure 9C shows the expected red shift that occurs in the presence of multivalent arsenicals; again, these effects are also observed in the presence of GSH (Figure 9D). Finally, the formation of irregular fibrillar like structure of these aggregates was confirmed by TEM47 (Figure S4). Overall, it appears that these MVAs are effective at promoting beta-rich structures in reduced RNase.
Figure 9.
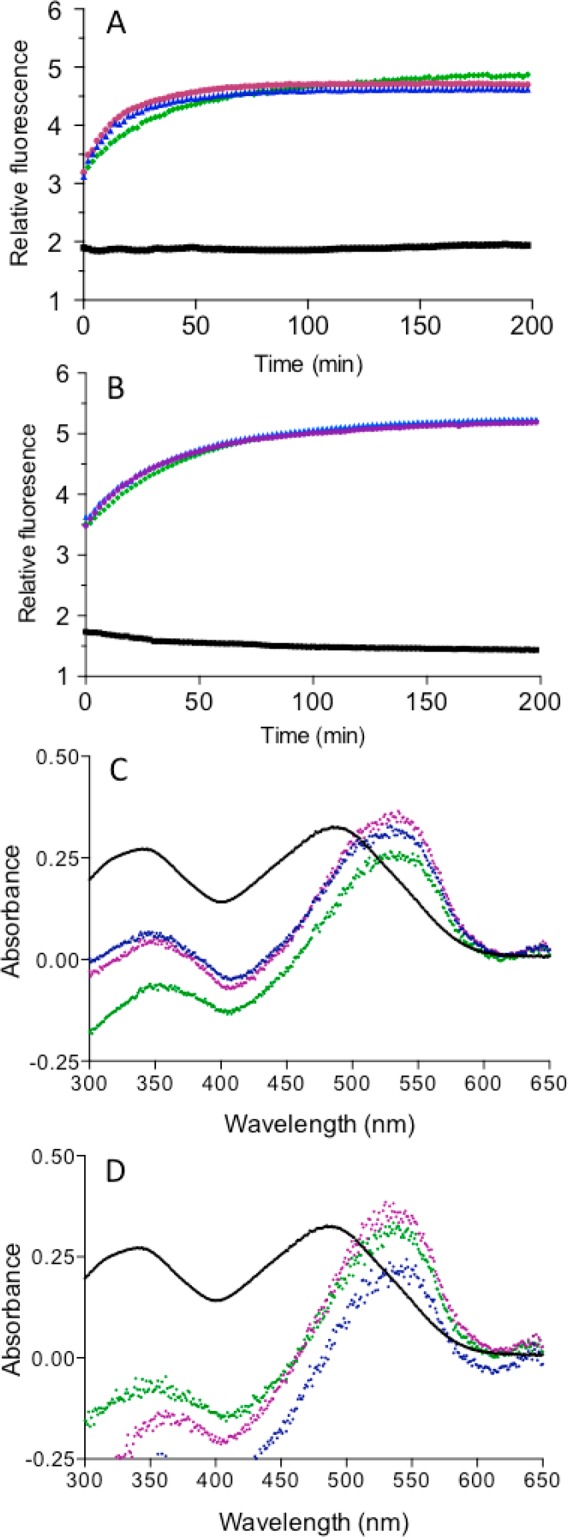
Monitoring MVA-induced aggregation of reduced RNase with Thioflavin T and Congo Red. Reduced RNase (30 μM) was mixed with 5 μM thioflavin T dye and MVAs in the absence (A) or presence (B) of 5 mM GSH. The increase in fluorescence was monitored over time (exciting at 450 nm with emission at 485 nm; see Materials and Methods). No significant increase in fluorescence was observed without the inclusion of the multivalent arsenicals in both cases. Control (no arsenical), black squares; 5 μM BA-1, pink circles; 5 μM BA-2, blue triangles; and 3.33 μM TA-1, green diamonds. (C, D) Congo Red spectral shift assay using 20 μM reduced RNase and 10 μM Congo Red dye and MVAs after 30 min in 50 mM phosphate buffer, pH 7.5, containing 1 mM EDTA in the absence (C) or presence of 5 mM GSH (D). Control (no arsenical), black; 5 μM BA-1, pink; 5 μM BA-2, blue; and 3.33 μM TA-1, green.
Conclusions
This work has explored the design and implementation of small molecule multivalent-arsenicals on redox-active enzymes containing multiple CxxC motifs. The three reagents, BA-1, BA-2, and TA- 1, were readily synthesized by coupling 4-aminophenyl arsenoxide with commercially available anhydrides or acyl chlorides in a one-pot reaction. These compounds were able to inhibit the oxidative folding pathways of RfBP and RNase even in the presence of 5 mM GSH. The principal targets of these compounds were reduced unfolded protein substrates and not the CxxC motifs of QSOX or PDI. A striking difference between the prior work with monoarsenicals8 and the current study is that the multivalent arsenicals appear to bind avidly to reduced unfolded proteins so that they cannot be effectively displaced by millimolar levels of GSH that would prevail intracellularly. The studies of Wilcox and colleagues6 now permit a quantitative framework to predict the competition between a given dithiol and 5 mM GSH for an arsenical center (Figure 10A). At the right is shown the Kd values for GSH calculated from their data. The competition with a dithiol is shown at the left (green panel), with calculations for the percentage of MMA complexed by this CxxC surrogate shown in Figure 10B. At a Kd of 10–6 M for MMA–dithiol, only 1% of this complex will be present, with the balance of MMA bound to glutathione. Indeed, a Kd value of 6 nM would be required to secure 50% of MMA in a dithiol complex; a Kd of 10 pM would be needed to sequester 97% of the MMA.
Figure 10.
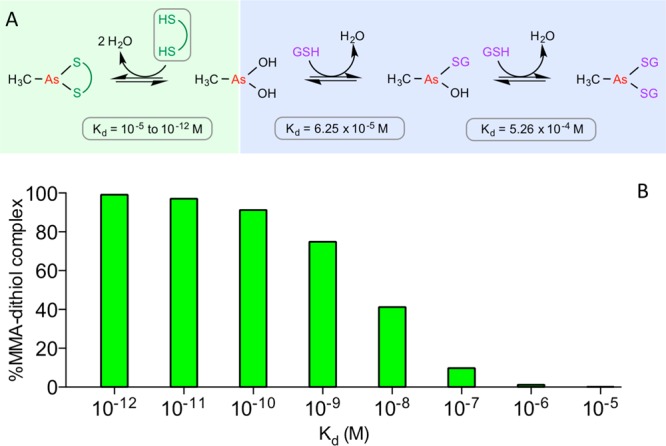
Simulated interaction of MMA with dithiols in the presence of competing GSH. (A) Complexation between 10 μM MMA and 10 μM dithiols (green colored region) and the competing coordination of MMA by 5 mM GSH (blue area; Kd values were calculated from ref (6)). (B) Illustration of how the avidity of dithiol binding to MMA influences the percentage of arsenical complexed with dithiol in competition with binding to GSH at 5 mM.
These data provide a rationalization for a number of observations. While the multivalent arsenicals do bind significantly more tightly to reduced PDI than do the monoarsenicals, the lower Kd of ∼60 nM is still insufficient to allow reduced PDI to bind more than 15% of the arsenicals in the presence of 5 mM GSH. Under these conditions, the coordination of reduced PDI to the monoarsenical PSAO (Kd = 1.1 μM) is <1% of the total. Given these thermodynamic restraints, the observations that multivalent arsenicals remain effective inhibitors of oxidative protein folding of reduced RfBP, even in the presence of 5 mM GSH, suggests that they bind with subnanomolar Kd values to the reduced protein. Furthermore, the failure of 5 mM GSH to suppress, or reverse, the aggregation of reduced RNase induced by low concentrations of multivalent arsenicals is readily rationalized by an avid association with these reagents and is in contrast to the behavior of the monoarsenical MMA.8 The very tight binding of bis-arsenicals to cysteine residues in unstructured proteins likely contributes to the significant intracellular background fluorescence encountered when the arsenical reagents FlAsH and ReAsH are used to follow tetracysteine tagged-proteins within mammalian cells.32,48,49
In summary, the prevailing intracellular concentration of GSH requires a target dithiol motif to bind with a Kd of less that about 10 nM to be significantly populated with the As(III) species (Figure 10). A recent comprehensive tabulation of binding affinities between arsenicals and a wide range of folded proteins and enzymes records no examples of such tight binding, with the bulk of the measurements showing Kd values above 1 μM.7 Clearly, the design of arsenical reagents that exhibit high selectivity in the context of the multiplicity and aggregate abundance of intracellul thiols will constitute a major challenge. Extracellularly, this requirement is greatly relaxed because the prevailing concentration of GSH and other low molecular weight monothiols is likely to be considerably less than 100 μM.50 Hence, at this GSH concentration, a Kd of 1 μM would lead to ∼60% sequestration of a dithiol by the arsenical (compared to ∼1% at 5 mM GSH; Figure 10). Hence, multivalent arsenicals may still find utility as inhibitors of extracellular oxidoreductases with two or more catalytically active dithiol motifs.
Acknowledgments
We thank Drs. Joel Schneider and Joe Fox for advice on the syntheses of arsenical derivatives and Shannon Modla and Samuel Jacobs for acquiring the transmission electron microscope images.
Glossary
Abbreviations
- APAO
4-aminophenylarsonous acid
- BA-1
2,2′-(ethane-1,2-diylbis((2-((4-arsinephenyl)amino)-2-oxoethyl)azanediyl))diacetic acid
- BA-2
3′,4-bis((4-arsinephenyl)carbamoyl)-[1,1′-biphenyl]-3,4′-dicarboxylic acid
- DTNB
5, 5′-dithiobis-2-nitrobenzoic acid
- DTT
dithiothreitol
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- GuHCl
guanidine hydrochloride
- TA-1
1,3,5-((benzenetricarbonyltris(azanediyl)) tris(benzene-4,1-diyl))triarsonous acid
- PDI
protein disulfide isomerase
- QSOX
quiescin-sulfhydryl oxidase
- RNase
ribonuclease A
- RfBP
riboflavin binding protein
- TCEP
tris(2-carboxyethyl)phosphine
Supporting Information Available
Synthetic schemes for the arsenicals BA-1, BA-2, and TA-1; data for the interaction between the arsenicals and PDI; and transmission electron microscopy studies of reduced RNase treated with MVAs in the absence and presence of competing GSH. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† (D.R.) Qatar Science & Technology Park, Qatar Foundation, Doha, Qatar.
This work was supported in part by the National Institutes of Health (grant GM26643 to C.T.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wang Z. Y.; Chen Z. (2008) Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 111, 2505–2515. [DOI] [PubMed] [Google Scholar]
- Wu J.; Henderson C.; Feun L.; Van Veldhuizen P.; Gold P.; Zheng H.; Ryan T.; Blaszkowsky L. S.; Chen H.; Costa M.; Rosenzweig B.; Nierodzik M.; Hochster H.; Muggia F.; Abbadessa G.; Lewis J.; Zhu A. X. (2010) Phase II study of darinaparsin in patients with advanced hepatocellular carcinoma. Invest. New Drugs 28, 670–676. [DOI] [PubMed] [Google Scholar]
- Swindell E. P.; Hankins P. L.; Chen H.; Miodragovic D. U.; O’Halloran T. V. (2013) Anticancer activity of small-molecule and nanoparticulate arsenic(III) complexes. Inorg. Chem. 52, 12292–12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miodragovic E. U.; Quentzel J. A.; Kurutz J. W.; Stern C. L.; Ahn R. W.; Kandela I.; Mazar A.; O’Halloran T. V. (2013) Robust structure and reactivity of aqueous arsenous acid-platinum(II) anticancer complexes. Angew. Chem., Int. Ed. 52, 10749–10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nidhubhghaill O. M.; Sadler P. J. (1991) The structure and reactivity of arsenic compounds—biological activity and drug design. Struct. Bonding (Berlin, Ger.) 78, 129–190. [Google Scholar]
- Spuches A. M.; Kruszyna H. G.; Rich A. M.; Wilcox D. E. (2005) Thermodynamics of the As(III)–thiol interaction: arsenite and monomethylarsenite complexes with glutathione, dihydrolipoic acid, and other thiol ligands. Inorg. Chem. 44, 2964–2972. [DOI] [PubMed] [Google Scholar]
- Shen S. W.; Li X. F.; Cullen W. R.; Weinfeld M.; Le X. C. (2013) Arsenic Binding to Proteins. Chem. Rev. 113, 7769–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan D.; Rancy P. C.; Nagarkar R. P.; Schneider J. P.; Thorpe C. (2009) Arsenic(III) species inhibit oxidative protein folding in vitro. Biochemistry 48, 424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb J. (1966) Enzymes and Metabolic Inhibitors; Academic Press: New York. [Google Scholar]
- Ramadan D.; Cline D. J.; Bai S.; Thorpe C.; Schneider J. P. (2007) Effects of As(III) binding on beta-hairpin structure. J. Am. Chem. Soc. 129, 2981–2988. [DOI] [PubMed] [Google Scholar]
- Lin S.; Cullen W. R.; Thomas D. J. (1999) Methylarsenicals and arsinothiols are potent inhibitors of mouse liver thioredoxin reductase. Chem. Res. Toxicol. 12, 924–930. [DOI] [PubMed] [Google Scholar]
- Styblo M.; Serves S. V.; Cullen W. R.; Thomas D. J. (1997) Comparative inhibition of yeast glutathione reductase by arsenicals and arsenothiols. Chem. Res. Toxicol. 10, 27–33. [DOI] [PubMed] [Google Scholar]
- Stevenson K. J.; Hale G.; Perham R. N. (1978) Inhibition of pyruvate dehydrogenase multienzyme complex from Escherichia coli with mono- and bifunctional arsenoxides. Biochemistry 17, 2189–2192. [DOI] [PubMed] [Google Scholar]
- Knowles F. C. (1985) Reactions of lipoamide dehydrogenase and glutathione-reductase with arsonic acids and arsonous acids. Arch. Biochem. Biophys. 242, 1–10. [DOI] [PubMed] [Google Scholar]
- Rancy P. C.; Thorpe C. (2008) Oxidative protein folding in vitro: a study of the cooperation between quiescin-sulfhydryl oxidase and protein disulfide isomerase. Biochemistry 47, 12047–12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raje S.; Thorpe C. (2003) Inter-domain redox communication in flavoenzymes of the quiescin/sulfhydryl oxidase family: role of a thioredoxin domain in disulfide bond formation. Biochemistry 42, 4560–4568. [DOI] [PubMed] [Google Scholar]
- Heckler E. J.; Alon A.; Fass D.; Thorpe C. (2008) Human quiescin-sulfhydryl oxidase, QSOX1: probing internal redox steps by mutagenesis. Biochemistry 47, 4955–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon A.; Grossman I.; Gat Y.; Kodali V. K.; DiMaio F.; Mehlman T.; Haran G.; Baker D.; Thorpe C.; Fass D. (2012) The dynamic disulphide relay of quiescin sulphydryl oxidase. Nature 488, 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodali V. K.; Thorpe C. (2010) Oxidative protein folding and the quiescin-sulfhydryl oxidase family of flavoproteins. Antioxid. Redox Signaling 13, 1217–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel B. A.; Kodali V. K.; Thorpe C. (2014) Going through the barrier: coupled disulfide exchange reactions promote efficient catalysis in Quiescin sulfhydryl oxidase. J. Biol. Chem. 289, 5274–5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatahet F.; Ruddock L. W. (2009) Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signaling 11, 2807–2850. [DOI] [PubMed] [Google Scholar]
- Tian G.; Xiang S.; Noiva R.; Lennarz W. J.; Schindelin H. (2006) The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell 124, 61–73. [DOI] [PubMed] [Google Scholar]
- Schwaller M.; Wilkinson B.; Gilbert H. F. (2003) Reduction–reoxidation cycles contribute to catalysis of disulfide isomerization by protein-disulfide isomerase. J. Biol. Chem. 278, 7154–7159. [DOI] [PubMed] [Google Scholar]
- Wilkinson B.; Gilbert H. F. (2004) Protein disulfide isomerase. Biochim. Biophys. Acta 1699, 35–44. [DOI] [PubMed] [Google Scholar]
- Tian G.; Kober F. X.; Lewandrowski U.; Sickmann A.; Lennarz W. J.; Schindelin H. (2008) The catalytic activity of protein-disulfide isomerase requires a conformationally flexible molecule. J. Biol. Chem. 283, 33630–33640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatahet F.; Ruddock L. W. (2007) Substrate recognition by the protein disulfide isomerases. FEBS J. 274, 5223–5234. [DOI] [PubMed] [Google Scholar]
- Yang S.; Wang X.; Cui L.; Ding X.; Niu L.; Yang F.; Wang C.; Wang C. C.; Lou J. (2014) Compact conformations of human protein disulfide isomerase. PLoS One 9, e103472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K.; Iemura S.; Kamiya Y.; Ron D.; Kato K.; Natsume T.; Nagata K. (2013) Ero1-alpha and PDIs constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J. Cell. Biol. 202, 861–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams S. R.; Campbell R. E.; Gross L. A.; Martin B. R.; Walkup G. K.; Yao Y.; Llopis J.; Tsien R. Y. (2002) New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J. Am. Chem. Soc. 124, 6063–6076. [DOI] [PubMed] [Google Scholar]
- Griffin B. A.; Adams S. R.; Tsien R. Y. (1998) Specific covalent labeling of recombinant protein molecules inside live cells. Science 281, 269–272. [DOI] [PubMed] [Google Scholar]
- Pomorski A.; Krezel A. (2011) Exploration of biarsenical chemistry—challenges in protein research. ChemBioChem 12, 1152–1167. [DOI] [PubMed] [Google Scholar]
- Scheck R. A.; Schepartz A. (2011) Surveying protein structure and function using bis-arsenical small molecules. Acc. Chem. Res. 44, 654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapra A.; Thorpe C. (2013) An arsenical-maleimide for the generation of new targeted biochemical reagents. J. Am. Chem. Soc. 135, 2415–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline D. J.; Thorpe C.; Schneider J. P. (2003) Effects of As(III) binding on alpha-helical structure. J. Am. Chem. Soc. 125, 2923–2929. [DOI] [PubMed] [Google Scholar]
- Cline D. J.; Redding S. E.; Brohawn S. G.; Psathas J. N.; Schneider J. P.; Thorpe C. (2004) New water-soluble phosphines as reductants of peptide and protein disulfide bonds: reactivity and membrane permeability. Biochemistry 43, 15195–15203. [DOI] [PubMed] [Google Scholar]
- Hoober K. L.; Joneja B.; White H. B. III; Thorpe C. (1996) A sulfhydryl oxidase from chicken egg white. J. Biol. Chem. 271, 30510–30516. [DOI] [PubMed] [Google Scholar]
- Miller M. S.; White H. B. III. (1986) Isolation of avian riboflavin-binding protein. Methods Enzymol. 122, 227–234. [DOI] [PubMed] [Google Scholar]
- Riddles P. W.; Blakeley R. L.; Zerner B. (1979) Ellmans reagent: 5,5′-dithiobis(2-nitrobenzoic acid)—re-examination. Anal. Biochem. 94, 75–81. [DOI] [PubMed] [Google Scholar]
- Hoops S.; Sahle S.; Gauges R.; Lee C.; Pahle J.; Simus N.; Singhal M.; Xu L.; Mendes P.; Kummer U. (2006) COPASI—a COmplex PAthway SImulator. Bioinformatics 22, 3067–3074. [DOI] [PubMed] [Google Scholar]
- Lyles M. M.; Gilbert H. F. (1991) Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: dependence of the rate on the composition of the redox buffer. Biochemistry 30, 613–619. [DOI] [PubMed] [Google Scholar]
- Gilbert H. F. (1995) Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 251, 8–28. [DOI] [PubMed] [Google Scholar]
- Holmgren A. (1979) Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J. Biol. Chem. 254, 9627–9632. [PubMed] [Google Scholar]
- Chambers J. E.; Tavender T. J.; Oka O. B.; Warwood S.; Knight D.; Bulleid N. J. (2010) The reduction potential of the active site disulfides of human protein disulfide isomerase limits oxidation of the enzyme by Ero1alpha. J. Biol. Chem. 285, 29200–29207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Chen S.; Wang X.; Wang L.; Wallis A. K.; Freedman R. B.; Wang C. C. (2010) Plasticity of human protein disulfide isomerase: evidence for mobility around the X-linker region and its functional significance. J. Biol. Chem. 285, 26788–26797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng L.; Rasmussen M. I.; Chailyan A.; Houen G.; Hojrup P. (2014) Probing the structure of human protein disulfide isomerase by chemical cross-linking combined with mass spectrometry. J. Proteomics 108, 1–16. [DOI] [PubMed] [Google Scholar]
- Ban T.; Hamada D.; Hasegawa K.; Naiki H.; Goto Y. (2003) Direct observation of amyloid fibril growth monitored by thioflavin T fluorescence. J. Biol. Chem. 278, 16462–16465. [DOI] [PubMed] [Google Scholar]
- Nilsson M. R. (2004) Techniques to study amyloid fibril formation in vitro. Methods 34, 151–160. [DOI] [PubMed] [Google Scholar]
- Hoffmann C.; Gaietta G.; Zurn A.; Adams S. R.; Terrillon S.; Ellisman M. H.; Tsien R. Y.; Lohse M. J. (2010) Fluorescent labeling of tetracysteine-tagged proteins in intact cells. Nat. Protoc. 5, 1666–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroffekova K.; Proenza C.; Beam K. G. (2001) The protein-labeling reagent FLASH-EDT2 binds not only to CCXXCC motifs but also non-specifically to endogenous cysteine-rich proteins. Pfluegers Arch. 442, 859–866. [DOI] [PubMed] [Google Scholar]
- Smith C. V.; Jones D. P.; Guenthner T. M.; Lash L. H.; Lauterburg B. H. (1996) Compartmentation of glutathione: Implications for the study of toxicity and disease. Toxicol. Appl. Pharmacol. 140, 1–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.