

Abstract
Despite two decades of research, the structure–function relationships of endogenous, physiological forms of α-synuclein (αSyn) are not well understood. Most in vitro studies of this Parkinson’s disease-related protein have focused on recombinant αSyn that is unfolded and monomeric, assuming that this represents its state in the normal human brain. Recently, we have provided evidence that αSyn exists in considerable part in neurons, erythrocytes, and other cells as a metastable multimer that principally sizes as a tetramer. In contrast to recombinant αSyn, physiological tetramers purified from human erythrocytes have substantial α-helical content and resist pathological aggregation into β-sheet rich fibers. Here, we report the first method to fully purify soluble αSyn from the most relevant source, human brain. We describe protocols that purify αSyn to homogeneity from nondiseased human cortex using ammonium sulfate precipitation, gel filtration, and ion exchange, hydrophobic interaction, and affinity chromatographies. Cross-linking of the starting material and the partially purified chromatographic fractions revealed abundant αSyn multimers, including apparent tetramers, but these were destabilized in large part to monomers during the final purification step. The method also fully purified the homologue β-synuclein, with a similar outcome. Circular dichroism spectroscopy showed that purified, brain-derived αSyn can display more helical content than the recombinant protein, but this result varied. Collectively, our data suggest that purifying αSyn to homogeneity destabilizes native, α-helix-rich multimers that exist in intact and partially purified brain samples. This finding suggests existence of a stabilizing cofactor (e.g., a small lipid) present inside neurons that is lost during final purification.
Missense mutations in and gene multiplications of SNCA, which encodes αSyn, cause autosomal dominant forms of Parkinson’s disease (PD).1−4 Moreover, αSyn is the main component of Lewy bodies and Lewy neurites, insoluble protein aggregates that form the principal cytopathological hallmark of PD and other human synucleinopathies.5 Because of its prominent role in neurodegenerative diseases, the pathological activities of αSyn have been extensively probed, with particular emphasis on the ability of this normally soluble protein to aggregate into amyloid-like fibrils.6−8 However, the physiological form and function of αSyn are still unclear.
αSyn in brain is primarily localized to presynaptic terminals,9,10 and the vast majority of the protein appears cytosolic upon cellular fractionation,9,11,12 suggesting that associations with synaptic membranes are likely transient and/or weak. Mice lacking αSyn display relatively subtle deficits in synaptic function, suggesting that αSyn may be a negative regulator of synaptic vesicle release under conditions of elevated neuronal activity.13−15 Consistent with this idea, mild overexpression of WT αSyn may interfere with synaptic vesicle exocytosis,16,17 although others reported synaptic vesicle phenotypes that may be related to the formation of toxic species.18,19 αSyn may act to modulate membrane remodeling20−22 or perhaps as a chaperone for SNARE complex formation and stability.23 However, a biochemical interaction of αSyn with SNARE proteins remains controversial.11,24−26 Having a source of pure human brain αSyn could allow studies of its interactions with various membrane lipids and proteins, thus elucidating a more clearly defined function for the protein and better differentiating physiological from pathological activities.
Since the early studies characterizing the recombinant protein purified from Escherichia coli, as a “natively unfolded” monomer,27,28 an observation that was recently supported by in-cell NMR on bacteria,29,30 the field has believed that soluble αSyn exists largely in this form physiologically, assuming that the physiological conformational state of the protein is mostly independent of its exact cellular context. As such, in vitro work on the structure–function relationships of αSyn has continued to focus on the unfolded recombinant protein as a normal form. In addition to this unfolded monomeric form and reports of other energetically favorable conformations in vitro,31,32 we recently found that αSyn exists physiologically as an aggregation-resistant, helically folded tetramer, based principally on studying αSyn purified under nondenaturing conditions from fresh human erythrocytes,33 one of the highest αSyn-expressing cell types.34 In follow-up studies using intact cell cross-linking and fluorescence complementation, we have found that abundant cellular tetramers and related low-n multimers of αSyn, as well as those of the aggregation-resistant and not disease-related β-synuclein (βSyn), could be detected in a variety of cells, including rodent primary neurons11,35 and freshly biopsied human brain (unpublished data). A thorough understanding of how neuronal αSyn functions under nonpathological conditions will require in vitro studies of this newly recognized form of the protein after its purification.
While the biophysical and functional properties of monomeric αSyn have been extensively studied, and αSyn isolated from human erythrocytes33 and mouse brain36 have been characterized, almost nothing is known about the properties of αSyn found in the normal human brain. We therefore sought to develop a protocol to fully purify soluble αSyn from human brain, the most relevant tissue source, with the hope that characterizing brain-derived αSyn will aid in studying physiological multimers and enable better comparison to pathological species. Here, we describe methods for the purification of αSyn from frozen human cortex. We show that our principal protocol can yield pure αSyn with greater helical content than recombinant protein, but that the degree of helicity is variable. Cross-linking of the starting material and partially purified samples followed by Western blotting showed that tetramers and related multimers of αSyn are abundant in post-mortem tissue and are readily detectible at intermediate stages of purification; however, the final chromatography step which purifies αSyn to homogeneity can destabilize the native multimers.
Experimental Procedures
Purification of Synucleins from Human Brain
Homogenization and Ultracentrifugation
Frozen cortices with no evidence of αSyn pathology were provided by Dr. M. Frosch (Massachusetts General Hospital/Harvard NeuroDiscovery Center) under an institutional review board (IRB)-approved protocol. Approximately 20 g of white and gray matter from frozen cortical slices were cut into ∼1 cm3 pieces and placed in a glass homogenizer with 3.5 vol of phosphate buffered saline (PBS) with protease inhibitor cocktail (0.5 mg/mL leupeptin, aprotinin, and 0.2 mg/mL pepstatin-A). Homogenization was performed with 24 strokes of a Teflon-coated pestle in an overhead stirrer (Wheaton) at power level 2.4. Total homogenates were centrifuged at 230000g for 50 min at 4 °C to collect the truly soluble protein.
Ammonium Sulfate Precipitation (ASP)
Ammonium sulfate was added to the supernatant of the ultracentrifugation step to a final concentration of 55%, after which samples were incubated at 4 °C for 60 min under nutation and then centrifuged for 20 min at 20000g at 4 °C to pellet precipitated αSyn. At this stage, αSyn-containing pellets were either dried and stored at −80 °C or immediately processed for size exclusion chromatography.
Size Exclusion Chromatography (SEC)
Pellets were resuspended in 5–8 mL of anion exchange buffer A (20 mM HEPES pH 8, 25 mM NaCl, 1 mM EDTA) and injected onto a Superdex 200 XK26/100 gel filtration column (GE Healthcare) that had been equilibrated in anion exchange buffer A. The column was washed with anion exchange buffer A at 1 mL/min, and 1.5 mL fractions were collected. SDS-PAGE/Western blotting (WB) was performed (see below) to identify αSyn-containing fractions.
Anion Exchange Chromatography (AEC)
SEC fractions that contained αSyn but minimal amounts of key contaminating proteins were pooled and loaded onto an equilibrated MonoQ anion exchange column (GE Healthcare) at a rate of 0.5 mL/min. αSyn was eluted using a gradient from 100% anion exchange buffer A to 50% anion exchange buffer A and 50% anion exchange buffer B (20 mM HEPES pH 8, 1000 mM NaCl, 1 mM EDTA). αSyn eluted at approximately 300 mM NaCl. As before, fractions were probed for the presence of αSyn by SDS-PAGE/WB and for purity by SDS-PAGE/Coomassie staining. AEC fractions that contained αSyn, but not βSyn, were used for thiopropyl Sepharose 6b incubation.
Thiopropyl Sepharose 6b (TS6b) Incubation
TS6b resin (GE Healthcare) was prepared by washing dried beads with 200 mL Milli-Q water/0.25 g of dried beads over filter paper. Milli-Q water was added to the hydrated beads to adjust the volume to 1 mL of slurry/0.25 g of dried beads. αSyn-containing AEC fractions were incubated with TS6b resin at a ratio of 1.5:1 overnight at 4 °C with circular rotation. The αSyn-containing supernatant was collected by centrifugation for 5 min at 1500g at 4 °C.
Hydrophobic Interaction Chromatography (HIC)
AEC fractions that contained αSyn (including those with βSyn contamination) were diluted 10-fold in HIC binding buffer A (1.2 M ammonium sulfate, 50 mM phosphate, pH 7.4) and bound to an equilibrated HiPrep Phenyl HP 16/10 column (GE Healthcare) at a rate of 0.5 mL/min. αSyn was eluted using a gradient from 0 to 100% HIC elution buffer B (50 mM phosphate, pH 7.4). αSyn eluted at 44% buffer B (528 mM ammonium sulfate). Fractions were probed for the presence of αSyn by SDS-PAGE/WB and for purity by SDS-PAGE/Coomassie staining. To remove ammonium sulfate prior to downstream analysis, samples underwent buffer exchange using six rounds of dilution and centrifugation in Amicon Ultra 10K filters (EMD Millipore). Amicon filters provided better protein retention than Zeba desalting columns (Thermo Pierce) during this buffer exchange step.
Purifying β-Synuclein
Purification of βSyn was performed identically to αSyn purification except that, when incubating with TS6b, the resin was added to βSyn- and not αSyn-containing fraction(s).
Circular Dichroism (CD) Spectroscopy
Following purification, αSyn was exchanged into 10 mM ammonium acetate using Zeba spin desalting columns (Thermo Fisher), lyophilized, and resuspended in 10 mM ammonium acetate at a concentration of approximately 10 μM. αSyn samples were added to a 1 mm path length quartz cuvette and analyzed using a J-815 CD spectrometer (Jasco). Spectra from at least seven recordings were averaged. A background spectrum of 10 mM ammonium acetate was subtracted from all αSyn spectra. Calculations of helicity were performed using the following formula, as in ref (37)fhelix = ([Θ]222 – [Θ]coil)/([Θ]helix – [Θ]coil) where % helicity = (100)(fhelix). The mean-residue ellipticities at 222 nm for completely helical and unfolded/random coil peptides were obtained from [Θ]helix = −40000(1–2.5/n) + 100T/°C and [Θ]coil = 640–45T/°C. n = 140, the number of amino acids in the αSyn polypeptide, and T = 20 °C
Mass Spectrometry
Gel samples were digested with trypsin using a method described previously.38 Samples were analyzed on an ABI model 4800 time-of-flight (TOF)/TOF matrix assisted laser desorption (MALDI) mass spectrometer (Applied Biosystems, Foster City, CA), a research grade time-of-flight instrument equipped with delayed extraction technology and a reflectron for resolution of up to 20 000 (fwhh) with MSMS capability by way of tandem TOF/TOF technology. Samples previously digested were prepared by mixing 0.5 μL of sample with 0.5 μL of alpha cyano-4-hydroxy-trans-cinnamic acid (10 mg/mL in 70% acetonitrile 0.1%TFA). The sample was rinsed after drying with 0.1%TFA. Intact mass analysis was also performed on the MALDI mass spectrometer but run in linear mode, and spotted using 3,5-dimethoxy-4-hydroxycinnamic acid (10 mg/mL in 70% acetonitrile 0.1%TFA) and calibrated using an external calibration. Data were analyzed using the Mascot algorithm by searching against the updated nonredundant database from NCBI.
Determination of Protein Concentrations
Total protein concentrations were determined by BCA assay (Thermo Scientific) according to the manufacturer’s instructions. αSyn concentrations were determined using an in-house developed sandwich ELISA for total αSyn. 96-well multi-array high bind plates (MSD, Meso Scale Discovery) were coated with the capture antibody 2F12 diluted (6.7 ng/μL) in Tris-buffered saline with 0.1% Tween-20 (TBS-T) in 30 μL vol/well and incubated at 4 °C overnight. Following emptying of the wells, plates were blocked for 1 h at RT in blocking buffer (5% MSD Blocker A; TBS-T). After three washes with TBS-T, samples diluted in TBS-T with 1% MSD Blocker A and 0.5% nonidet P-40 were loaded and incubated at 4 °C overnight. Sulfo-tagged SOY1 mAb (detection Ab) was generated using Sulfo-Tag-NHS-Ester (MSD), diluted in blocking buffer (6.7 ng/μL), added to the plate (30 μL volumes/well) and shaken for 1 h at RT. Following three washes, MSD Reader buffer was added, and the plates were immediately measured using a MSD Sector 2400 imager.
Cross-Linking
For cross-linking of purified or partially purified samples, 1 μL of either dithiobis[succinimidyl propionate] (DSP) or disuccinimidyl glutarate (DSG) dissolved in dimethyl sulfoxide was added to 50 μL of sample to a variety of concentrations noted in the text and figure legends. Typically, and to adjust for changes in total protein concentration, a starting concentration of 0.5 mM DSG/DSP was used for total homogenates, total soluble protein fractions (supernatants from the ultracentrifugation step), and ammonium sulfate-precipitated samples; 0.25 mM was used for SEC and AEC fractions. Upon addition of DSP or DSG, samples were incubated quiescently at 37 °C for 30 min, after which 2.5 μL of 1 M Tris pH 7.6 were added and samples were incubated at room temperature for 15 min under nutation to quench cross-linking reactions.
For intact-cell cross-linking of human brain tissue, samples (frozen, post-mortem human cortical section) were finely minced by two rounds on a McIlwain Tissue Chopper (model MTC/2E, Mickle Laboratory Engineering Co., blade distance set to 100 μm); the sample was turned by 90° for the second round of mincing. The minced brain samples were transferred to 15 mL tubes containing ∼5 mL PBS/PI, and the solution containing the intact brain pieces was resuspended by gentle shaking without homogenization. From this suspension, aliquots were transferred to 1.5 mL tubes, which were spun at 1500g for 5 min at RT. Supernatants were discarded, and pellets (bits of relatively intact tissue) underwent cross-linking, routinely at a ratio of 1000 μL of 1 mM DSG in PBS/PI per 100 mg tissue or else 1000 μL of 2 mM DSP/100 mg tissue. The suspensions were incubated at 37 °C for 30 min while shaking, followed by spinning at 1500g for 5 min at RT. After cross-linking, the supernatant was discarded, and the pellet was resuspended in PBS/PI (in 25–50% of the volume of the cross-linking solution), followed by sonication (Sonic Dismembrator model 300, Fisher Scientific; microtip setting = 40; 2 × 15 s). Spinning at 800g for 5 min at 4 °C yielded the postnuclear supernatant. A second spin at 100000g for 60 min at 4 °C yielded the cytosolic material in the supernatant. Cross-linked samples were stored on ice (for immediate use) or at −20 °C.
SDS-PAGE
Samples were prepared using Milli-Q water and 4× sample buffer containing LDS plus 20% β-mercaptoethanol (βME) (for cross-linked samples), or without βME (for non-cross-linked samples). Samples were electrophoresed on Nu-PAGE 4–12% Bis-Tris gels (Life Technologies) with MES-SDS running buffer.
Western Blotting and Coomassie Staining
Gels were then transferred onto 0.45 μm Immobilon-P PVDF membranes (Millipore) for 60 min at 400 mA constant current at 4 °C in transfer buffer consisting of 25 mM Tris, 192 mM glycine, and 20% methanol. After transfer, membranes were blocked in 5% nonfat milk in PBS with 0.1% v/v Tween-20 (PBS-T) for 30 min at room temperature and then incubated in primary antibody either overnight at 4 °C or for 60 min at room temperature. Membranes were then washed three times for 5 min in PBS-T, incubated with secondary antibody, washed three more times for 5 min in PBS-T, and then developed with ECL Plus or ECL Prime (GE-Amersham) according to the manufacturer’s directions. For Coomassie staining, gels were incubated in Milli-Q water for 10 min and then stained using Gel Code Blue for 45 min at room temperature. Gels were destained by washing with Milli-Q water.
Antibodies
2F12 and SOY1, monoclonal mouse antibodies (mAb) against αSyn were generated by immunizing αSyn −/– mice with αSyn purified from human erythrocytes. Hybridoma cell lines were generated by fusion of mouse splenocytic B lymphocytes with X63-Ag8.653 myeloma cells. Antibodies were generated and purified from hybridoma supernatant by Cell Essentials (Boston, MA). For WB, 2F12 was used at 0.18 μg/mL in 5% milk. Another mouse monoclonal AB, 15G7, was generously provided by the Haass lab,12 and the rabbit polyclonal αSyn antibody C20 was purchased from Santa Cruz. The following commercially available antibodies were also used: mAb EP1537Y to βSyn (Novus Biologicals) and mAb NSE-P1 to γ-enolase (Santa Cruz).
Results
Purification of αSyn from Human Brain
We designed and carried out a five-step protocol for the purification of soluble αSyn from the cerebral cortex of humans with no evidence of αSyn pathology (Figure 1A). Abundant αSyn was detected in cortical white matter in addition to gray matter (Figure 1B), and therefore both were included in our preparations. To minimize the possibility of disrupting intermolecular interactions within native αSyn multimers or inducing their formation, we avoided the use of detergents during our purification. As such, total cortical matter was homogenized in PBS plus protease inhibitors. Though this method does not extract membrane-associated αSyn, we and others have previously shown that the protein is overwhelmingly (≥90%) soluble and localized to the cytosol.9,11,12,36 Using an αSyn-specific, in-house ELISA coupled with total protein assays, we estimated that αSyn comprised ∼0.1% of total cortical homogenate protein and ∼0.3% by weight of protein in the soluble fraction after ultracentrifugation (Figure 1C), in line with published findings.10 PBS homogenates were spun at 240000g to pellet all membrane components and insoluble protein, since we wanted to avoid any possible contribution from αSyn found in incidental Lewy bodies should they exist in this nondiseased tissue. Ammonium sulfate was added to the supernatant to a final concentration of 55% in order to precipitate αSyn quantitatively. The resuspended pellet was applied to a Superdex 200 column for size exclusion chromatography (SEC), and fractions containing αSyn were pooled and further purified using anion exchange chromatography (AEC). The αSyn-containing AEC fractions were collected, and purification was completed using thiopropyl Sepharose 6b (TS6b) resin in batch mode. Since αSyn lacks cysteines, it remained in the unbound supernatant, while the few remaining contaminants bound to this resin and were pelleted by centrifugation.
Figure 1.

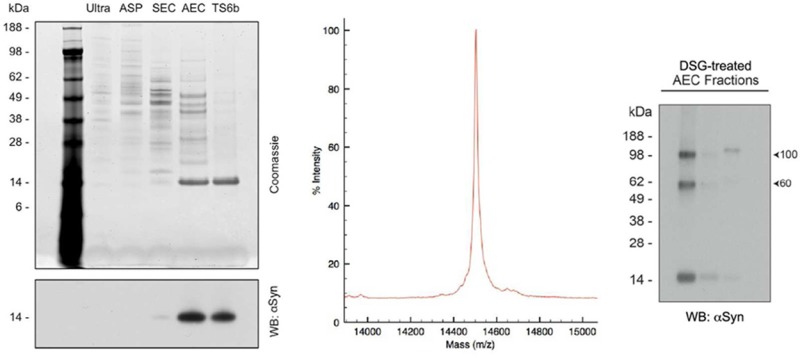
Purification of αSyn from nondiseased, post-mortem human cortex. (A) Schematic of the strategy used to purify αSyn from human cortex. (B) Cortical gray and white matter both contain αSyn. Gray matter (G) and white matter (W) were separated prior to homogenization and processed through the ammonium sulfate purification (ASP) step. Protein normalized samples of gray and white matter from after the ultracentrifugation (ultra.) and ASP steps were run on SDS-PAGE and probed for αSyn. (C) The total protein concentration as determined by BCA assay and αSyn content as measured by ELISA were used to calculate the percentage of αSyn in crude cortical homogenates and the supernatant following ultracentrifugation. Error bars represent the standard deviation from four independent experiments. (D) 400 ng of samples from after each stage of the purification were run on SDS-PAGE and stained with Coomassie blue (upper panel). Thirty nanograms of samples from after each purification step were run on SDS-PAGE and probed for αSyn using the antibody 2F12 (lower panel). All samples were taken after the chromatography step listed at the top of their respective lanes. Ultra = ultracentrifugation, ASP = ammonium sulfate precipitation, SEC = size exclusion chromatography, AEC = anion exchange chromatography, TS6b = thiopropyl Sepharose 6b incubation.
Samples from each stage of the purification were protein-normalized and run on SDS-PAGE (Figure 1D). After the SEC step, Coomassie blue staining of ∼400 ng of total protein revealed a faint 14 kDa band (the approximate molecular weight of the αSyn monomer). This band was greatly enriched following AEC, and became the only remaining band following the final TS6b pull-down of contaminating proteins (Figure 1D, upper panel). Western blotting of 30 ng of protein with the mouse monoclonal αSyn antibody 2F12 matched what was observed with Coomassie blue staining: the 14 kDa αSyn band was strongly enhanced after AEC and was retained in the supernatant after incubation with thiopropyl Sepharose 6b (Figure 1D, lower panel).
Total protein and αSyn content were measured after each stage of purification by a BCA assay and our αSyn-specific sandwich ELISA, respectively. From these results, we calculated the progressive degrees of αSyn enrichment during the purification (Figure 2A). SEC and AEC were particularly effective, with each enriching for αSyn by approximately 1 order of magnitude. The full protocol provided a >3500 fold enrichment of αSyn from the starting total homogenate and yielded 5–10% of the starting cytosolic αSyn. We typically obtained approximately 100 μg of pure αSyn starting from 20 to 25 g wet weight of human cortex (Table 1). After testing various storage conditions on the stability of αSyn in a partially purified state (data not shown), we avoided freezing final samples at −20 °C since each freeze/thaw cycle was associated with protein loss. Instead, we used short-term storage (<5 days) at 4 °C and long-term storage at −80 °C after flash freezing in liquid nitrogen.
Figure 2.
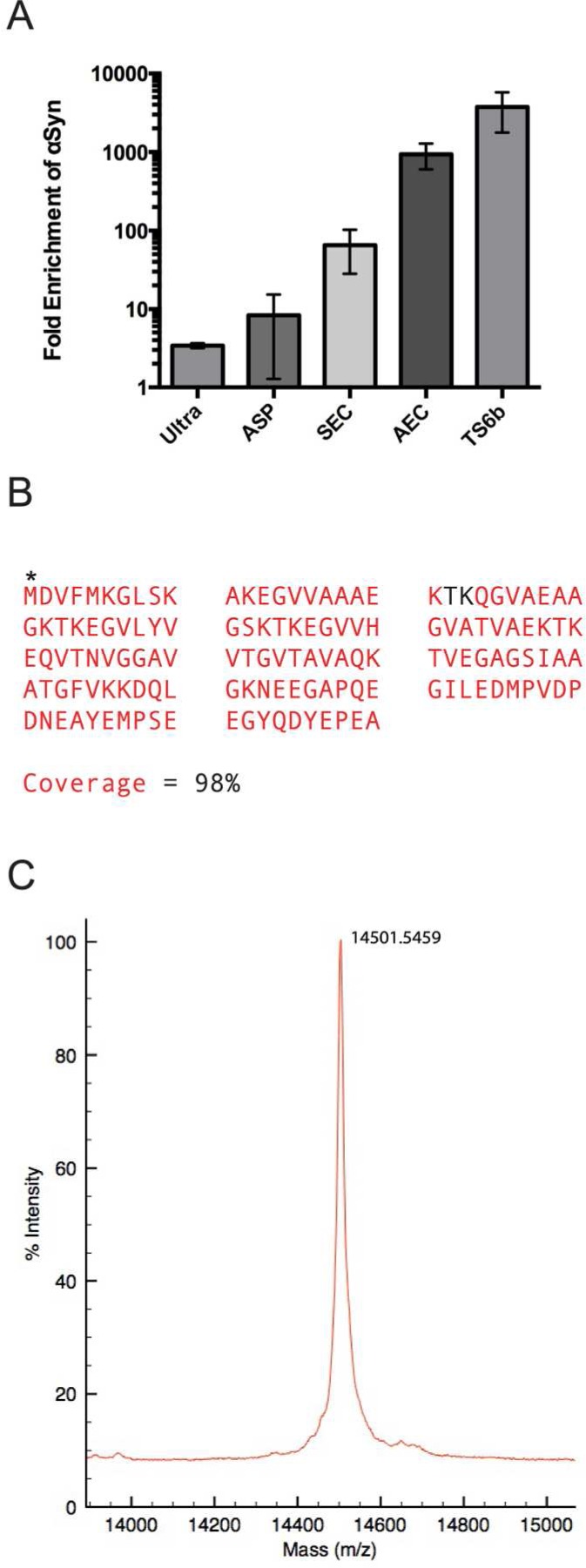
Enrichment and mass spectroscopy of purified αSyn. (A) The fold enrichment of αSyn after each purification step was calculated by dividing the percentage of total protein that is αSyn at each step by the percentage of αSyn in the starting homogenate. (B) Following TS6b incubation, the sample was loaded for SDS-PAGE, and the gel was stopped as soon as the sample entered the gel. The entire sample was excised, trypsin-digested, and analyzed by mass spectroscopy. The αSyn sequence covered by observed peptide fragments is shown in red. Acetylation of the N-terminal methionine was observed (*), but no other post-translational modifications were detected. (C) The mass of the undigested, pure sample was determined by intact mass spectroscopy and is highly consistent with the expected molecular weight of N-acetylated αSyn (14 502 Da).
Table 1. Quantitative Profile of αSyn Purification from Human Braina.
| purification step | total protein (μg/mL) | αSyn (μg/mL) | αSyn yield (μg) |
|---|---|---|---|
| total brain | 20889 (± 3100) | 18 (± 18) | 1432 (± 1484) |
| Ultra Supe | 4637 (± 2478) | 12 (± 6) | 850 (± 440) |
| SEC Start | 7707 (± 8105) | 81 (±46) | 653 (± 364) |
| AEC Start | 337 (± 221) | 7 (± 3) | 191 (± 75) |
| TS6b Start | 111 (± 40) | 61 (± 42) | 131 (± 68) |
| final | 205 (± 158) | 192 (± 128) | 96 (± 64) |
The concentrations of total protein and αSyn were determined at each stage of purification when starting with 20 g wet weight of human cortex. The yield of αSyn was calculated by multiplying the αSyn concentration (determined by an αSyn ELISA) by the volume at each step of the purification. Values represent the mean ± standard deviation from 4 independent experiments.
Purity of the final αSyn sample was confirmed using mass spectrometry. Enzymatic digestion and mass spectroscopy of the final supernatant following thiopropyl Sepharose incubation identified αSyn as the only human protein present in the sample. Analysis of the digested fragments revealed 98% coverage of the αSyn amino acid sequence and the presence of an N-terminal acetylation (Figure 2B, Supplementary Tables S1 and S2), as we had previously observed for αSyn purified from human erythrocytes.33 No phosphorylated residues were detected. Intact mass spectrometry (performed under conditions that do not maintain intermolecular interactions in any multimers) revealed a molecular mass of 14501.5 Da, highly consistent with the expected mass (14 502 Da) of N-acetylated αSyn without additional post-translational modifications (Figure 2C).
Key Contaminants and Their Removal
On many repetitions, the above procedure reproducibly led to pure αSyn. However, four specific proteins occasionally hindered full purification due to their abundance and apparently similar biochemical properties. These contaminating proteins were initially revealed by Coomassie stain: bands at ∼50, 45, 40, and 16 kDa were each excised and analyzed by in-gel digestion and tandem LC-MS/MS, revealing them to be ATP-synthase beta subunit (ATP5B), γ-enolase (NSE), creatine kinase B-type (CKB), and βSyn, respectively. These results were later confirmed by Western blotting with the cognate antibodies (Figure 3). A single final incubation with TS6b resin was usually sufficient to completely purify αSyn (Figure 1D); however, in some preparations, these contaminating proteins were sufficiently abundant that a second incubation with this resin was needed to achieve purity (Figure 3B). In later purification runs, before pooling αSyn-containing SEC fractions for AEC, we routinely probed a range of SEC fractions with specific antibodies that recognize the principal contaminating proteins and one that detects αSyn (Figure 3C). This approach allowed us to take steps to reduce the amounts of these contaminants reaching the final thiopropyl Sepharose step.
Figure 3.

Principal contaminating proteins. (A) WB for γ-enolase (NSE) and αSyn in the AEC fractions shows an overlap in the elution profiles of these two proteins. Start refers to pooled SEC fractions that were used for AEC. (B) Coomassie staining of an αSyn-rich AEC fraction prior to and after 1× and 2× TS6b incubation steps. In some cases, two incubation steps were necessary to remove contaminants such as ATP5b and produce αSyn of >90% purity. (C). Some αSyn-rich AEC fractions also contained abundant βSyn (as confirmed by mass spectroscopy) that was not separated from αSyn by the TS6b incubation step. (D) WB for SNE, βSyn, and αSyn in the SEC fractions shows considerable overlap in the elution profiles of these three proteins; however the peak of αSyn immunoreactivity is shifted relative to these two contaminants.
A protein that was invariably present through the early stages of the purification was βSyn, which copurified with αSyn through the SEC step (Figure 3C). βSyn could usually be separated from αSyn in the AEC step, because it eluted later (i.e., at a slightly higher salt concentration) (Figure 3D), as anticipated from its lower predicted isoelectric point (pI αSyn = 4.67, pI βSyn = 4.41).39 Only αSyn-containing AEC fractions that lacked βSyn were incubated with thiopropyl Sepharose, since βSyn also lacks cysteines and would not be separated from αSyn by this technique (Figure 3D).
Brain-Derived αSyn Contains Variable α-Helical Content
We and others have recently provided evidence that endogenous αSyn in intact cells exists in substantial part as a tetramer and closely related multimers.11,22,33,35,40 Unlike monomeric αSyn, the tetramer has significant α-helical content when purified from erythrocytes or bacteria under nondenaturing conditions.22,33,41−43 We therefore performed circular dichroism (CD) spectroscopy to determine the secondary structure of αSyn purified from human brain. In contrast to recombinant αSyn, which invariably showed the expected random coil secondary structure (with a predictable minimum of ellipticity at 196 nm), the CD spectra of human brain αSyn purified using the above protocol were more variable. In some cases, the spectrum contained significant α-helical content (Figure 4A, red trace), while in others, the protein appeared almost completely unfolded (Figure 4A, blue trace). We quantified the helical content of both purified brain-derived and recombinantly expressed αSyn by comparing their ellipticity minima at 222 nm (a minimum expected for α-helical but not unfolded protein) to those of completely helical and unfolded peptides.37 Recombinant αSyn, as expected, displayed very little helical content, with a mean of 4%. On average, our purified human brain αSyn possessed significantly more helical content, ranging from 7 to 21%, with a mean of ∼12% (Figure 4B). As a point of comparison, lipid vesicle-folded recombinant αSyn, an accepted helical form of αSyn,44 contains ∼43% helicity by these calculations. We occasionally observed a spontaneous increase in α-helical content of purified brain αSyn samples upon storage, which initially were determined to be of random coil conformation. Here, CD reanalysis after 4–5 days of storage of the purified brain αSyn at RT showed a relative loss of unfolded character and gain in helicity (Figure 4C) without any significant protein degradation (data not shown). When the spectra were adjusted for the soluble protein concentration remaining in the sample, they appeared quite similar to those samples that were partially α-helical immediately following purification (compare red trace in Figure 4A to dark green trace in Figure 4C). Together, these data suggest that the human brain contains helical αSyn but that there are factors that can negatively influence its stability over the purification procedure (discussed below).
Figure 4.
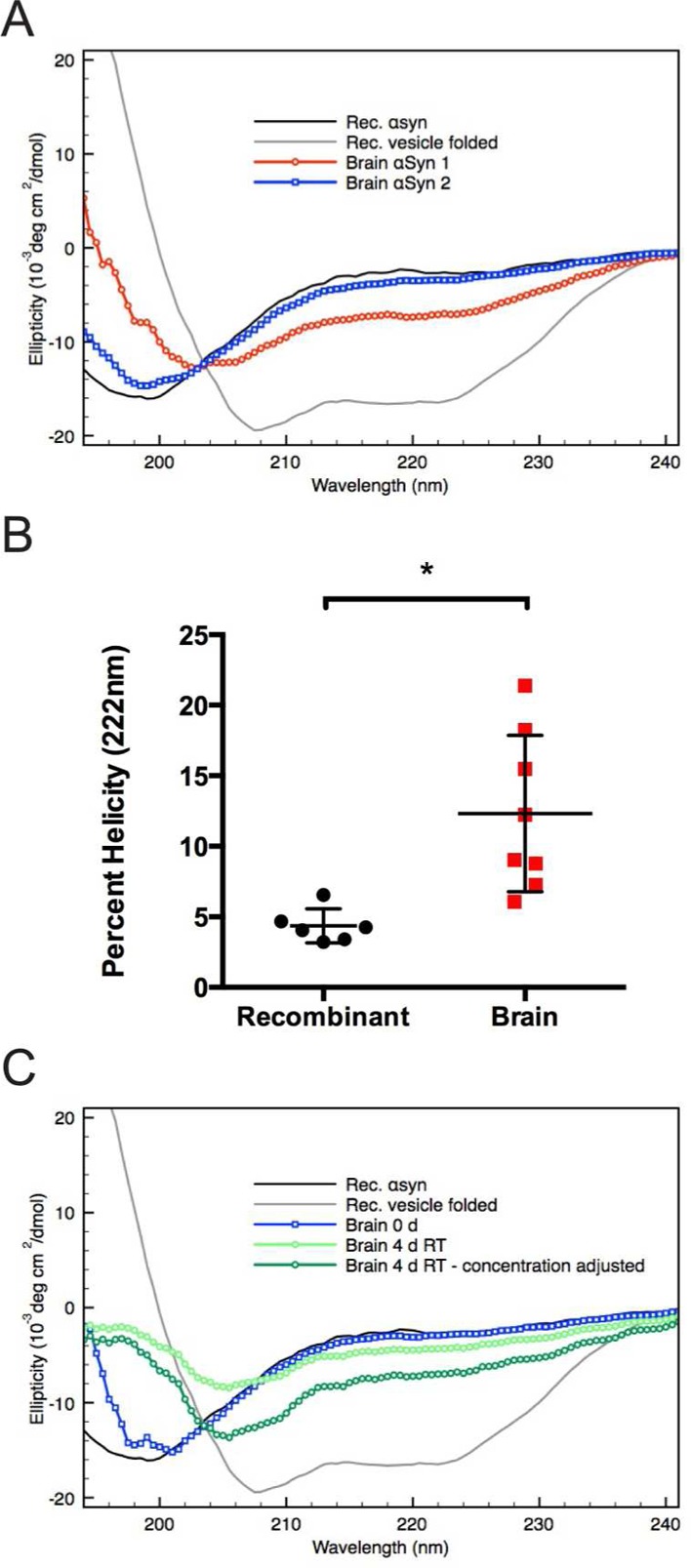
Purified human brain αSyn shows variable helical folding. (A) Circular dichroism (CD) spectroscopy was used to estimate the secondary structure of purified αSyn samples. CD spectra obtained from two samples of αSyn purified from human brain highlighting the variability in helical content. A partially helical sample is shown in red, and a largely unfolded sample is shown in blue. CD spectroscopy of monomeric recombinant αSyn in the absence (unfolded - solid black trace) and presence of POPC/POPS small unilamellar vesicle (α-helical–solid gray trace) are shown for reference. (B) The helicity of αSyn samples purified from human brain are compared to recombinant monomer and are expressed as the percentage of vesicle-folded recombinant αSyn. Relative helicity was determined using the ellipticity value at 222 nm. * = p < 0.01 using an unpaired t-test. Error bars represent the standard deviation of six or eight independent experiments for recombinant and brain-derived αSyn, respectively. (C) CD spectra of human brain αSyn before (blue trace) and after room temperature (RT) incubation (green traces) were compared to unfolded and vesicle-folded recombinant αSyn (black and gray traces, respectively). The raw data obtained after room temperature incubation (light green trace) were scaled to adjust for the reduction in protein concentration following extended room temperature incubation (dark green trace).
Abundant αSyn Multimers Occur in Human Brain
Our previous work indicated that freshly biopsied human brain, primary rodent neurons, human erythrocytes, and a variety of other cell sources contain abundant αSyn tetramers.11,33,35 To determine whether tetramers and related midmolecular weight multimers could similarly be trapped in human cortical cells, we cross-linked small pieces of tissue (to avoid major cell lysis, see Experimental Procedures) from frozen, post-mortem human brain using the homobifunctional, lysine-reactive cross-linker disuccinimidyl glutarate (DSG). Western blotting of the 100000g soluble lysate of cells after DSG cross-linking revealed residual amounts of the 14 kDa monomer and multimers of 60 kDa (probable tetramer, based on all our prior data), and 80 and 100 kDa (probable conformers of the tetramer or slightly larger multimers), all of which have been described in neurons11,45 (Figure 5A). The predominance of the 80 kDa band seen here and also after lysis of primary rodent neurons and other cell types11 (compared to the strong 60 kDa band trapped using in vivo cross-linking11) suggests that some degree of cell lysis has already occurred in the frozen, post-mortem brain tissue. These cross-linking data suggest that soluble multimers constitute a substantial pool of physiological αSyn in normal human neurons.
Figure 5.

Abundant αSyn multimers are detected in intact brain cells and partially purified samples. (A) Cross-linking of intact cells from frozen, post-mortem human cortex using 1 mM DSG revealed abundant midmolecular weight multimers of 60, 80, and 100 kDa. The sample run in the lane marked “R” was cross-linked with 1 mM of the cleavable cross-linker DSP and then reduced (cleaved) by boiling in sample buffer containing βME. This served as a non-cross-linked control. (B) Total brain homogenate, soluble protein (ultra. supernatant), and ammonium sulfate precipitated (ASP) samples were protein normalized and cross-linked with a range of DSG concentrations from 0.5 to 1 mM. A clear 80 kDa band was visible in cross-linked samples from each of these three stages of purification. Residual ammonium sulfate present in the resolubilized ASP samples led to a reduced DSG cross-linking efficiency in those samples. “R” lanes served as non-cross-linked controls. (C) A range of SEC fractions (left panel) and AEC fractions (right panel) were cross-linked with 0.25 mM DSG. Multimeric species, especially the 60 kDa and 100 kDa doublet bands, are predominant in SEC fractions. These multimeric species are also readily detectable following AEC, though the relative abundance of monomer in the main αSyn-containing fractions is higher compared to after SEC.
We have previously demonstrated that physiological αSyn tetramers isolated from erythrocytes and neuroblastoma cells have α-helical content,33 in contrast to the bacterially expressed protein.27,29,30 We therefore hypothesized that brain neurons possess αSyn tetramers and related multimers but that either our purification method is biased toward the monomeric αSyn population or most multimers are destabilized to unfolded monomers at some point during the purification. To address this issue, we cross-linked αSyn-containing fractions at each intermediate stage of purification. Cross-linking of the total brain homogenate or its 230000g soluble supernatant or the resolubilized ammonium sulfate precipitate (ASP) of that supernatant each revealed an abundant ∼80 kDa multimeric band (Figure 5B) that migrates at the same position as the ∼80 kDa multimer trapped by cross-linking of cells immediately after lysis that we have reported11,35 and that was observed above after cross-linking small pieces of brain tissue (Figure 5A). Note that the efficiency of the DSG cross-linking is reduced, as expected, in the ammonium sulfate precipitate due to the abundant residual free amines, which act to quench this lysine-reactive cross-linker. Cross-linking of the material following the SEC step (when little or no ammonium sulfate remains) revealed very small amounts of monomers (14 kDa) and the 60, 80, and 100 kDa multimers (Figure 5c), in accord with our previous observations of cross-linked αSyn from intact normal neurons11 and from minced, post-mortem brain bits (Figure 5A). Cross-linkable multimers were still readily detected in the peak αSyn-containing fractions following AEC, particularly the 60 kDa and 100 kDa species, but relatively more 14 kDa monomer was now present (Figure 5C). The latter result suggests that the AEC step may contribute to some destabilization of endogenous αSyn multimers during our purification (compare left and right sides of blot in Figure 5C).
TS6b Incubation Specifically Destabilizes αSyn Multimers
Cross-linking of multimer-rich fractions before and after TS6b incubation revealed a striking reduction in the ability to trap multimers following this step (Figure 6A, compare lanes 1 and 2). Because there was the expected decrease of total protein with this final purification step, the same DSG concentration we used to trap multimers before the TS6b incubation might have resulted in an “over-crosslinking” of the material after TS6b incubation and thus a reduction in the ability of αSyn to enter the gel. To address this possibility, we tried DSG at lower concentrations (Figure 6A left: lanes 3 and 4) but were still unable to trap more than very small amounts of multimers, compared to the abundant monomer (Figure 6A right: dark exposure). As shown in Table 1, roughly 75% of the αSyn from the start of the TS6b incubation is present in the final sample. Because multimers make up >66% of the total αSyn immunoreactivity, a loss of 25% of the αSyn (even if it were exclusively multimers) would not account for the near-complete loss of DSG-trapped multimers.
Figure 6.

TS6b incubation precludes the detection of multimeric αSyn. (A) A multimer-rich AEC fraction was cross-linked with a range of DSG from 0.50 to 0.25 mM of before and after TS6b incubation. The 60 kDa band seen before TS6b incubation was only faintly visible afterward and the strongly immunoreactive 100 kDa band was completely undetectable (dark exposure). Higher cross-linker concentration did not result in the trapping of multimers, but rather resulted in “over-crosslinking” and a loss of protein, probably due to the exclusion of larger species from entering the gel. (B) AEC fraction containing αSyn and NSE multimers was incubated with TS6b resin for 1 h and then cross-linked with a range of DSG concentrations. Samples were blotted for αSyn (top) and NSE (bottom). Efficient trapping of the native NSE dimer suggests that αSyn multimers are preferentially destabilized. (C) AEC fraction from B was incubated with TS6b resin or dithiopyridine (DTP) for 10 min and then cross-linked with the indicated DSG concentrations. Samples were blotted for αSyn (top) and NSE (bottom). A light and dark exposure are shown for DTP-incubated samples highlighting the specific destabilization of αSyn multimers. (D) AEC fraction containing αSyn multimers was left undiluted (lane 1) or diluted 1:2 (lane 2) or 1:3 (lane 3). Samples were then cross-linked with a range of DSG concentrations such that the cross-linker/protein ratio across the samples remained the same.
We asked whether this inability to detect αSyn multimers after TS6b incubation was due to their depolymerization or to a reduction in general cross-linking efficiency caused by incubation with the TS6b beads. To answer this question, we took advantage of samples that contained some residual NSE (a major contaminant during the purification; see above) after the TS6b incubation. Western blotting for NSE after DSG cross-linking revealed that the residual NSE could still be efficiently cross-linked into its physiological, dimeric form46 (Figure 6B, lower panel). This control suggests that the αSyn multimers were selectively destabilized by the final TS6b step. Potential mechanisms of destabilization include the sharp reduction of the total protein concentration in the purified sample (i.e., lack of molecular crowding) and/or the depletion of a specific αSyn multimer-stabilizing factor by the resin. To address these possibilities, we incubated the αSyn multimer-containing AEC fractions with the closely related chemical, 2,2′-dithiopyridine (DTP) at a concentration equivalent to that of the similar active groups on the TS6b resin. (DTP is the disulfide of 2-thiopyridine, the compound released from TS6b resin upon protein binding.) The addition of DTP to the multimer-containing AEC fraction thus creates an environment similar to that of incubating the AEC fraction with TS6b resin but without actually removing cysteine-containing proteins (such as NSE) from the solution, thereby also avoiding a reduction in protein concentration. As we had observed upon TS6b incubation, incubation with DTP alone sharply reduced our ability to trap multimeric αSyn but not the NSE dimers (Figure 6C). Higher DSG concentrations did not allow recovery of the αSyn multimers (as would have been expected if cross-linking inefficiency were to blame), but instead led to a greater amount of gel-excluded material (Figure 6C: DSG 1 mM lanes). To confirm that protein dilution did not lessen our ability to detect αSyn multimers, we diluted the multimer-containing AEC fractions either 1:2 or 1:3, thereby achieving the range of total protein concentration resulting from the TS6b step (as determined by BCA assay). These diluted AEC samples were still readily cross-linkable by DSG when the ratio of DSG:protein was adjusted accordingly to avoid overcross-linking (Figure 6D), although as shown earlier, this was not the case for samples incubated with TS6b resin (Figure 6A, B). Collectively, these experiments suggest that the αSyn depolymerizing effect of the TS6b resin is due, at least in part, to a chemical interaction induced by its leaving group (DTP). The results indicate that endogenous αSyn multimers are carried through our purification protocol until the final step, at which point contact with TS6b destabilizes multimers and yields monomeric αSyn.
Hydrophobic Interaction Chromatography as an Alternate Final Step
We next explored whether a different final step to obtain pure αSyn after AEC would preserve the ability to cross-link endogenous αSyn multimers. To this end, we employed hydrophobic interaction chromatography (HIC) and found that it afforded a clean and consistent separation of αSyn from βSyn and any other remaining contaminating proteins, as illustrated by the UV chromatogram, the Coomassie staining, and the Western blotting of the HIC fractions (Figure 7A: upper, middle and lower panels, respectively). We then performed CD spectroscopy and cross-linking analysis of these final αSyn fractions after buffer exchange into 10 mM phosphate. CD spectroscopy revealed largely unfolded protein, though some preparations yielded protein with some helical content (Figure 7B). As we observed after the TS6b incubation, cross-linking with DSG after HIC did not yield the familiar pattern of midmolecular weight αSyn multimers plus the monomer that was seen upon cross-linking of intact cells or the partially purified samples through the AEC step. Instead, we observed αSyn monomers together with high molecular weight smearing associated with overcross-linking (due to the higher than optimal DSG/protein ratio) (Figure 7C compare lanes 1 and 2). Even after decreasing the DSG:protein ratio, we were unable to trap midmolecular weight αSyn multimers after this final HIC step (Figure 7C, lanes 3 and 4).
Figure 7.

Purification using HIC as an alternative polishing step also leads to destabilization of αSyn multimers. (A) When used in place of TS6b incubation, HIC results in an effective separation of αSyn away from contaminating proteins as seen by the UV chromatogram obtained during the HIC step (top panel), Coomassie staining (middle panel) and WB of HIC fractions (bottom panel). (B) HIC-purified αSyn appears largely unfolded by CD spectroscopy though some samples did possess greater α-helical content than recombinant αSyn. (C) αSyn multimers present at the start of HIC are destabilized upon full purification. After buffer exchange of the αSyn-containing HIC fraction, cross-linking with a range of DSG concentrations (0.25–0.0625 mM) revealed only monomeric αSyn and high molecular weight smearing.
To determine whether HIC itself was destabilizing to brain-derived αSyn multimers, we employed the protocol previously used to isolated helical multimers from human erythrocytes33 (Figure 8A). As seen by Coomassie staining of αSyn-containing SEC fractions, this procedure did not yield pure αSyn (Figure 8B), highlighting the differences between brain tissue and erythrocytes with regard to the pool of “contaminating” proteins from which αSyn must be separated. However, we were still able to compare the ability of DSG to trap multimeric αSyn before and after HIC. As shown above (Figure 5B), cross-linking following the 230,000 x g soluble supernatant (Ultra) revealed an 80 kDa multimer and 14 kDa monomer (Figure 8C). While cross-linking with DSG immediately after HIC is not possible due to the presence of ammonium sulfate, we were able to efficiently cross-link αSyn to the 80 kDa and 60 kDa positions following SEC, the final chromatography step used here. These results indicate that HIC is not inherently destabilizing to native, brain-derived αSyn multimers. Together, these data suggest that achieving complete purity of brain-derived αSyn leads to a destabilization of native multimers.
Figure 8.
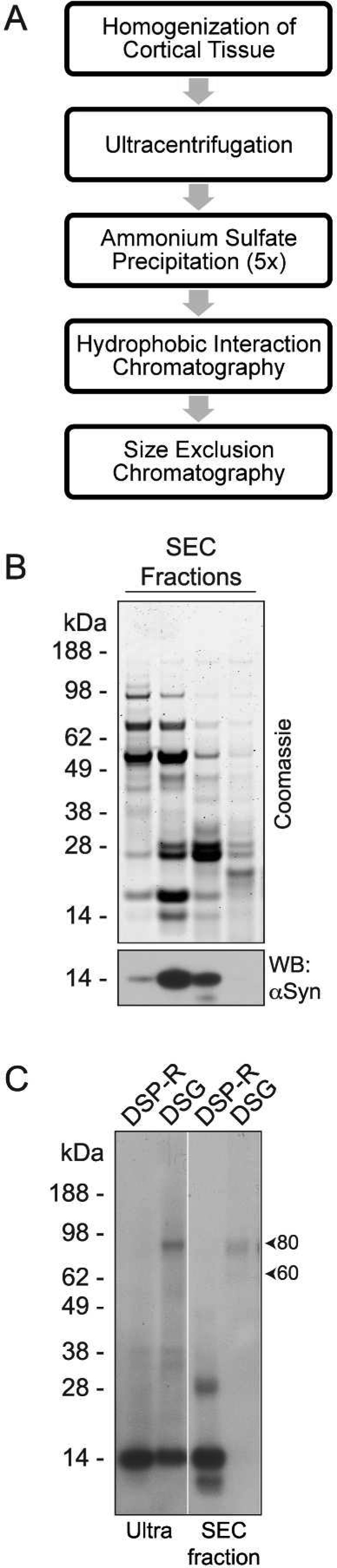
HIC is not inherently destabilizing to brain-derived αSyn. (A) Sequence of chromatography steps used to partially purify αSyn from frozen, post-mortem human cerebral cortex. (B) Coomassie staining (upper panel) and Western blot (lower panel) of SEC fractions illustrating the impurity of αSyn-containing fractions. (C) Western blot of the ultracentrifuge supernatant (Ultra) and SEC fractions cross-linked with 0.5 mM DSG revealed abundant αSyn multimers before (Ultra) and after (SEC fractions) the HIC step. Samples run in the lanes marked “DSP-R” were cross-linked with 0.5 mM DSP and then reduced (cleaved) by boiling in sample buffer containing βME. These served as a non-cross-linked control.
Purification of β-Synuclein
While optimizing our protocol to purify αSyn, we discovered that we could simultaneously purify βsyn. As mentioned above, βSyn copurified with αSyn until the AEC step, at which point βSyn eluted at a higher concentration of NaCl than αSyn, as predicted by its lower isoelectric point (Figure 3D). βSyn-containing AEC fractions occasionally also contained αSyn, but when αSyn was not present, we were able to completely purify βSyn using the TS6B resin (like αSyn, βSyn lacks cysteines and thus can also be separated from remaining contaminating proteins using this step) (Figure 9A). Alternatively, βSyn could be fully purified from fractions containing αSyn using HIC (Figure 9B). Following purification, the identity of βSyn was confirmed by LC-MS/MS (Figure 9C). Cross-linking of βSyn-containing fractions following SEC and AEC demonstrated that human neurons contain βSyn multimers (Figure 9D) that elute from these columns in a pattern similar to that observed for αSyn and have similar migration on Western blots (Figure 5C). This finding is in agreement with the cross-linked βSyn multimers trapped in intact rat primary neurons previously.11 However, cross-linking after either the TS6b (data not shown) or HIC final steps (Figure 9E) yielded only monomers, just as in the case of αSyn (Figure 7C).
Figure 9.

Purification and cross-linking of βSyn. (A) Coomassie stain of AEC fractions illustrating that βSyn elutes later (at a higher salt concentration) than αSyn and is also able to be purified away from contaminating proteins using TS6b incubation provided that no αSyn is present in the AEC fraction. (B) Following AEC, HIC allows for the separation of βSyn from αSyn and other contaminating proteins as seen by Coomassie staining (top panel) and Western blotting (bottom panel) of HIC fractions. (C) Mass spectroscopy was performed on the final purified sample and confirmed the identity of βSyn. * = N-terminal acetylation. The βSyn sequence covered by observed peptide fragments is shown in red. (D) Western blotting of SEC and AEC fractions cross-linked with DSG revealed abundant βSyn multimers in a pattern similar to what was observed with αSyn. (E) Multimeric βSyn present at the start of HIC are unable to be trapped by a range of DSG concentrations (0.25–0.0625 mM) after full purification and buffer exchange.
Discussion
αSyn is an intensely studied protein and is highly abundant in the human nervous system. Anderson and colleagues47 isolated insoluble αSyn from the brains of synucleinopathy patients in their study of disease-associated changes in primary structure, but conformational studies of nonpathologic αSyn isolated from human brain are lacking. This report is the first to describe the purification of physiological human brain αSyn. Moreover, we show that βSyn can be purified simultaneously from the same starting material with only slight modifications. This set of protocols will be useful in further studies of the physiological properties of these two members of the synuclein family.
In recent years, the secondary structure and multimerization state of native, cellular αSyn have been vigorously debated. While the recombinant, unmodified αSyn seems to be mainly unfolded in E. coli,27 even when measured in intact bacteria,29,30E. coli-expressed protein that is N-terminally acetylated, analogous to its state in eukaryotic cells, displays variable helicity and oligomerization states dependent on the exact isolation procedure used.42 The conformation of αSyn in mammalian cells endogenously expressing it is equally under debate. Our lab and others have provided evidence that the protein exists in substantial part in human cells as a helical multimer,11,22,33,48 while other groups have continued to maintain that it exists exclusively49 or in very large part36 as a “natively unfolded” monomer.
We show here that we can purify αSyn from human brain that has significantly higher α-helical content than the unfolded recombinant monomer obtained by bacterial expression but that the degree of helicity is variable among purified samples (Figure 4B). While Fauvet et al.49 used a different purification procedure, making a direct comparison of results difficult, the study of mouse brain performed by Burré and colleagues36 was similar in some ways to our earlier study in erythrocytes and the experiments on human brain presented here. In their recent characterization of αSyn purified from mouse brain, Burré and colleagues36 reported a degree of helical content in their purified sample that is in between what we observed for erythrocyte-isolated tetramer33 and the values obtained for unfolded bacterially expressed monomer. Still they interpreted their data as generally supportive of an unfolded state. Intriguingly, they also observed an increase in α-helical content and a simultaneous reduction in protein concentration of their purified sample upon incubating it at room temperature for 7 days, as shown by a time-dependent reduction in CD amplitude. We likewise observed instances in which purified αSyn initially appeared unfolded by CD spectroscopy but developed a partially helical spectrum after 4-day room temperature incubation. This phenomenon could be interpreted as (a) an initial induction of helicity in the purified sample during its incubation; (b) the refolding of protein that had become unfolded at some point in the purification; or (c) an apparent gain of helical CD signal due to an increase in the relative abundance (i.e., enrichment) of folded αSyn in the sample. On the basis of our data and the result reported,36 we believe the third interpretation is the most likely. We previously observed that, unlike the unfolded monomer, helical αSyn tetramers and related physiological multimers are resistant to aggregation.33 One would therefore predict that in a mixed sample containing both unfolded and α-helical αSyn and under storage conditions that allow for aggregation, the helical αSyn would remain soluble while unfolded αSyn would aggregate and become insoluble. Since insoluble protein does not contribute to a CD spectrum, the sample will appear to have undergone a random coil-to-α-helical transition when instead a larger percentage of the CD-measurable αSyn is now helical. In line with this interpretation, we observed a reduction over time in the amplitude of the CD signal concurrent with an apparent increase in helicity; the former change suggesting that there is less total soluble protein in the sample. Our data, as well as those of Burré et al.,32 are consistent with the hypothesis that cerebral tissue normally contains a pool of helical αSyn that is aggregation resistant. Furthermore, our findings reinforce the idea that novel therapeutics designed to stabilize α-helical conformations of αSyn may reduce the relative abundance of aggregation-prone monomers, thereby interfering with cytopathological processes in PD and related synucleinopathies.
Different structural states of αSyn (including unfolded monomer and helical multimers) may have distinct functional roles within the cell. It has been proposed that helical multimers may represent a storage mechanism for regulating the concentration of lipid-binding monomer.22,32 A recent publication, however, suggests a direct function for physiological multimers in synaptic vesicle clustering.48 It is also possible that the unstructured nature of the αSyn monomer confers upon it a flexibility that could allow it to perform a variety of functions.50 In fact, this appears to be a hallmark of proteins that have been proposed to be “intrinsically disordered”, as their conformation is usually dependent on binding partners and cellular context.50 Therefore, we do not believe that αSyn has only one functional form, as there is likely a dynamic equilibrium among physiological conformations. Accordingly, additional studies of both the monomeric and multimeric forms of αSyn (and, importantly, factors (e.g., lipids) and cellular events that influence their interconversion) will enhance our understanding of the function of this pleomorphic protein.
Our purified human brain αSyn did not consistently contain as much α-helical content as αSyn isolated from human erythrocytes.33 One possible explanation for this could be related to the tissue from which the respective samples originated. The source material dictates the nature and array of contaminating proteins, which, in turn, determine what chromatographic techniques are best for isolating αSyn. We optimized the protocol presented here to remove contaminants that are highly expressed in brain but may not be abundant in erythrocytes and other cell types, and this led to an increased number of steps and therefore handling time necessary to achieve full purity. The sequence and combination of chromatography techniques we used for the full purification from brain appears, in the end, to be detrimental to the preservation of the native αSyn multimers that were detectable by cross-linking at all intermediate steps during the purification. In fact, until the final chromatography step (TS6b incubation or HIC), we observed abundant multimers by DSG cross-linking that comigrated with the αSyn multimers trapped by DSG when cross-linking intact neurons (Figure 5). The variation in helical content of our final purified samples can likely be attributed to differences in the retention of multimeric forms of αSyn as one approaches full purity at the last step of purification and/or the coincident removal of an unknown tetramer-stabilizing cofactor. It has proven difficult to pinpoint a particular purification step as the one responsible for destabilization. For example, HIC appears to be less harmful when used earlier in the purification scheme (Figure 8) suggesting that the total number of purification steps needed, and not a particular step per se, is detrimental to multimeric stability. Alternative explanations for the difference in the recovery of α-helical tetramers when purifying from erythrocytes vs brain tissue are that the former were fresh and the latter was frozen (i.e., underwent one or more freeze–thaw cycles) or that αSyn in brain tissue can more readily convert between helical multimer and unfolded monomer.
We have previously shown that Western blotting for αSyn after in vivo cross-linking of neurons11 and fresh brain tissue (unpublished data) yields a major 60 kDa tetramer and minor 80 and 100 kDa multimers. Here, we observed a greater relative abundance of the 80 kDa band relative to the 60 kDa upon cross-linking of minced tissue bits from frozen, post-mortem human cortex. Moreover, the relative amounts of these three species shift reproducibly during certain steps of the purification (e.g., cell lysis and SEC), suggesting a dynamic relationship between these species. This is reminiscent of our previous finding that cell lysis at high protein concentrations enhances the ability to trap the normally lysis-sensitive 60 kDa αSyn species (see Figure 5D from Dettmer et al.,11). We are as yet unable to explain this consistent interconversion of αSyn multimers, though we speculate that changes in the sample environment induced by these steps may allow for the preferential stabilization of certain conformers of the αSyn tetramer that are trapped by DSG. This interpretation is supported by intact mass spectrometry of purified, cross-linked αSyn species, which suggests that these multimeric bands represent conformers of the tetramer rather than multimers of distinct sizes (i.e., tetramers, hexamers, octamers) (unpublished data).
Though αSyn has been reported to interact with other proteins within cells, we have no evidence at this time that partially (or fully) purified αSyn exists in a heteromultimer with other proteins. The cross-linked multimers of αSyn that we detected upon DSG treatment of partially purified brain samples migrate at the same positions as those that we observe upon cross-linking of intact cells, and we have systematically ruled out the possibility that any published αSyn-interacting proteins exist in heteromultimers with αSyn in those cross-linked cell samples.11 Further, we have analyzed the ∼60 kDa putative tetrameric band cross-linked in intact cells by mass spectrometry and found that it contains only αSyn (unpublished data). Interactions of αSyn with other proteins in cells may be too weak/transient to be trapped quantitatively with DSG, but it is conceivable that the use of different cross-linking reagents could reveal other interactors. Overall, we hypothesize that αSyn occurs in substantial part in the brain as an intraneuronal, α-helically folded homotetramer and related homomultimers that are in a complex equilibrium with unfolded monomers.
Taken together, our CD data (Figure 4) and the cross-linking findings at intermediate steps in the purification (Figure 5) support the existence of helical αSyn multimers in normal human brain. They also highlight the difficulty of purifying these labile species and the need to avoid any conditions that destabilize native αSyn−αSyn interactions. The apparent ease with which cross-linkable multimers can be destabilized during full purification helps provide an explanation for why some groups have had difficulty obtaining α-helical material even from erythrocytes49 while others were successful.22 Our brain purification protocol should be useful in screening for agents (small molecules; lipids) that can serve to stabilize native αSyn or allow refolding of purified, brain-derived αSyn into the tetramers present in intact, living neurons.
Acknowledgments
We thank M. Frosch, K Fitch, and I. Costantino at Massachusetts General Hospital for providing tissue samples, J. Lee at the Dana Farber Cancer Institute Molecular Biology Core facility for assistance with mass spectrometry, and A. Newman and other members of the Selkoe laboratory for helpful comments and suggestions.
Glossary
Abbreviations
- AEC
anion exchange chromatography
- αSyn
α-synuclein
- ASP
ammonium sulfate precipitate
- ATP5b
ATP-synthase beta subunit
- βME
beta-mercaptoethanol
- βSyn
β-synuclein
- CD
circular dichroism
- CKB
creatine kinase B-type
- DSG
disuccinimidyl glutarate
- DSP
dithiobis[succinimidyl propionate]
- DTP
dithiopyridine
- HIC
hydrophobic interaction chromatography
- NSE
γ-enolase
- PD
Parkinson’s disease
- SEC
size exclusion chromatography
- TS6b
thiopropyl Sepharose 6b
Supporting Information Available
Raw data from mass spectrometry of purified, trypsin-digested samples are included as Supplemental Tables S1 and S2. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by NIH Grant 1 RO1 NS083845 (to D.J.S.).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Polymeropoulos M. H.; Lavedan C.; Leroy E.; Ide S. E.; Dehejia A.; Dutra A.; Pike B.; Root H.; Rubenstein J.; Boyer R.; Stenroos E. S.; Chandrasekharappa S.; Athanassiadou A.; Papapetropoulos T.; Johnson W. G.; Lazzarini A. M.; Duvoisin R. C.; Di Iorio G.; Golbe L. I.; Nussbaum R. L. (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin M.-C.; Kachergus J.; Roumier C.; Mouroux V.; Douay X.; Lincoln S.; Levecque C.; Larvor L.; Andrieux J.; Hulihan M.; Waucquier N.; Defebvre L.; Amouyel P.; Farrer M.; Destée A. (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169. [DOI] [PubMed] [Google Scholar]
- Kara E.; Lewis P. A.; Ling H.; Proukakis C.; Houlden H.; Hardy J. (2013) α-Synuclein mutations cluster around a putative protein loop. Neurosci. Lett. 546, 67–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton A. B.; Farrer M.; Johnson J.; Singleton A.; Hague S.; Kachergus J.; Hulihan M.; Peuralinna T.; Dutra A.; Nussbaum R.; Lincoln S.; Crawley A.; Hanson M.; Maraganore D.; Adler C.; Cookson M. R.; Muenter M.; Baptista M.; Miller D.; Blancato J.; Hardy J.; Gwinn-Hardy K. (2003) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841. [DOI] [PubMed] [Google Scholar]
- Spillantini M. G.; Schmidt M. L.; Lee V. M.; Trojanowski J. Q.; Jakes R.; Goedert M. (1997) Alpha-synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Breydo L.; Wu J. W.; Uversky V. N. (2012) A-synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 1822, 261–285. [DOI] [PubMed] [Google Scholar]
- Cookson M. R.; van der Brug M. (2008) Cell systems and the toxic mechanism(s) of alpha-synuclein. Exp. Neurol. 209, 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles M. J.; Lansbury P. T. (2003) Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson’s disease. Biochemistry 42, 7871–7878. [DOI] [PubMed] [Google Scholar]
- George J. M.; Jin H.; Woods W. S.; Clayton D. F. (1995) Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 15, 361–372. [DOI] [PubMed] [Google Scholar]
- Iwai A.; Masliah E.; Yoshimoto M.; Ge N.; Flanagan L.; de Silva H. A.; Kittel A.; Saitoh T. (1995) The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 14, 467–475. [DOI] [PubMed] [Google Scholar]
- Dettmer U.; Newman A. J.; Luth E. S.; Bartels T.; Selkoe D. (2013) In vivo cross-linking reveals principally oligomeric forms of α-synuclein and β-synuclein in neurons and non-neural cells. J. Biol. Chem. 288, 6371–6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle P. J.; Neumann M.; Ozmen L.; Muller V.; Jacobsen H.; Schindzielorz A.; Okochi M.; Leimer U.; van Der Putten H.; Probst A.; Kremmer E.; Kretzschmar H. A.; Haass C. (2000) Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J. Neurosci. 20, 6365–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abeliovich A.; Schmitz Y.; Fariñas I.; Choi-Lundberg D.; Ho W. H.; Castillo P. E.; Shinsky N.; Verdugo J. M.; Armanini M.; Ryan A.; Hynes M.; Phillips H.; Sulzer D.; Rosenthal A. (2000) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252. [DOI] [PubMed] [Google Scholar]
- Yavich L.; Tanila H.; Vepsäläinen S.; Jäkälä P. (2004) Role of alpha-synuclein in presynaptic dopamine recruitment. J. Neurosci. 24, 11165–11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavich L.; Jäkälä P.; Tanila H. (2006) Abnormal compartmentalization of norepinephrine in mouse dentate gyrus in alpha-synuclein knockout and A30P transgenic mice. J. Neurochem. 99, 724–732. [DOI] [PubMed] [Google Scholar]
- Larsen K. E.; Schmitz Y.; Troyer M. D.; Mosharov E.; Dietrich P.; Quazi A. Z.; Savalle M.; Nemani V.; Chaudhry F. A.; Edwards R. H.; Stefanis L.; Sulzer D. (2006) Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 26, 11915–11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani V. M.; Lu W.; Berge V.; Nakamura K.; Onoa B.; Lee M. K.; Chaudhry F. A.; Nicoll R. A.; Edwards R. H. (2010) Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boassa D.; Berlanga M. L.; Yang M. A.; Terada M.; Hu J.; Bushong E. A.; Hwang M.; Masliah E.; George J. M.; Ellisman M. H. (2013) Mapping the subcellular distribution of α-synuclein in neurons using genetically encoded probes for correlated light and electron microscopy: implications for Parkinson’s disease pathogenesis. J. Neurosci. 33, 2605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D. A.; Tabarean I.; Tang Y.; Cartier A.; Masliah E.; Roy S. (2010) A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J. Neurosci. 30, 8083–8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp F.; Exner N.; Lutz A. K.; Wender N.; Hegermann J.; Brunner B.; Nuscher B.; Bartels T.; Giese A.; Beyer K.; Eimer S.; Winklhofer K. F.; Haass C. (2010) Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 29, 3571–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkey J.; Isas J. M.; Mizuno N.; Jensen M. B.; Bhatia V. K.; Jao C. C.; Petrlova J.; Voss J. C.; Stamou D. G.; Steven A. C.; Langen R. (2010) Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J. Biol. Chem. 285, 32486–32493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal C. H.; Chandra S. S. (2013) Monomeric synucleins generate membrane curvature. J. Biol. Chem. 288, 1829–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J.; Sharma M.; Tsetsenis T.; Buchman V.; Etherton M. R.; Südhof T. C. (2010) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt D. C.; Rhoades E. (2013) α-Synuclein Can Inhibit SNARE-Mediated Vesicle Fusion through Direct Interactions with Lipid Bilayers. Biochemistry 52, 2385–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S.; Gallardo G.; Fernández-Chacón R.; Schlüter O. M.; Südhof T. C. (2005) Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396. [DOI] [PubMed] [Google Scholar]
- Diao J.; Burré J.; Vivona S.; Cipriano D. J.; Sharma M.; Kyoung M.; Südhof T. C.; Brunger A. T. (2013) Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. eLife 2, e00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb P. H.; Zhen W.; Poon A. W.; Conway K. A.; Lansbury P. T. (1996) NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35, 13709–13715. [DOI] [PubMed] [Google Scholar]
- Kim J. (1997) Evidence that the precursor protein of non-A beta component of Alzheimer’s disease amyloid (NACP) has an extended structure primarily composed of random-coil. Mol. Cells 7, 78–83. [PubMed] [Google Scholar]
- Binolfi A.; Theillet F. X.; Selenko P. (2012) Bacterial in-cell NMR of human α-synuclein: a disordered monomer by nature?. Biochem. Soc. Trans. 40, 950–954. [DOI] [PubMed] [Google Scholar]
- Waudby C. A.; Camilloni C.; Fitzpatrick A. W. P.; Cabrita L. D.; Dobson C. M.; Vendruscolo M.; Christodoulou J. (2013) In-cell NMR characterization of the secondary structure populations of a disordered conformation of α-synuclein within E. coli cells. PLoS One 8, e72286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman O.; Fisher C. K.; Stultz C. M. (2011) Explaining the structural plasticity of α-synuclein. J. Am. Chem. Soc. 133, 19536–19546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurry T.; Ullman O.; Fisher C. K.; Perovic I.; Pochapsky T.; Stultz C. M. (2013) The dynamic structure of α-synuclein multimers. J. Am. Chem. Soc. 135, 3865–3872. [DOI] [PubMed] [Google Scholar]
- Bartels T.; Choi J. G.; Selkoe D. J. (2011) α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour R.; Kling K.; Anderson J. P.; Banducci K.; Cole T.; Diep L.; Fox M.; Goldstein J. M.; Soriano F.; Seubert P.; Chilcote T. J. (2008) Red blood cells are the major source of alpha-synuclein in blood. Neurodegener. Dis. 5, 55–59. [DOI] [PubMed] [Google Scholar]
- Newman A. J.; Selkoe D.; Dettmer U. (2013) A new method for quantitative immunoblotting of endogenous α-synuclein. PLoS One 8, e81314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J.; Vivona S.; Diao J.; Sharma M.; Brunger A. T.; Südhof T. C. (2013) Properties of native brain α-synuclein. Nature 498, E4–6–discussion E6–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholtz J. M.; Qian H.; York E. J.; Stewart J. M.; Baldwin R. L. (1991) Parameters of helix-coil transition theory for alanine-based peptides of varying chain lengths in water. Biopolymers 31, 1463–1470. [DOI] [PubMed] [Google Scholar]
- Shevchenko A.; Wilm M.; Vorm O.; Mann M. (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858. [DOI] [PubMed] [Google Scholar]
- (2014) Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 42, D191–D198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klucken J.; Outeiro T. F.; Nguyen P.; McLean P. J.; Hyman B. T. (2006) Detection of novel intracellular alpha-synuclein oligomeric species by fluorescence lifetime imaging. FASEB J. 20, 2050–2057. [DOI] [PubMed] [Google Scholar]
- Wang W.; Perovic I.; Chittuluru J.; Kaganovich A.; Nguyen L. T. T.; Liao J.; Auclair J. R.; Johnson D.; Landeru A.; Simorellis A. K.; Ju S.; Cookson M. R.; Asturias F. J.; Agar J. N.; Webb B. N.; Kang C.; Ringe D.; Petsko G. A.; Pochapsky T. C.; Hoang Q. Q. (2011) A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. U. S. A. 108, 17797–17802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trexler A. J.; Rhoades E. (2012) N-Terminal acetylation is critical for forming α-helical oligomer of α-synuclein. Protein Sci. 21, 601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels T.; Kim N. C.; Luth E. S.; Selkoe D. J. (2014) N-Alpha-acetylation of α-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS One 9, e103727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson W. S.; Jonas A.; Clayton D. F.; George J. M. (1998) Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449. [DOI] [PubMed] [Google Scholar]
- Gould N.; Mor D.; Lightfoot R.; Malkus K.; Giasson B.; Ischiropoulos H. (2014) Evidence of Native α-Synuclein Conformers in the Human Brain. J. Biol. Chem. 14, 7929–7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai G.; Brewer J. M.; Lovelace L. L.; Aoki T.; Minor W.; Lebioda L. (2004) Expression, purification and the 1.8 angstroms resolution crystal structure of human neuron specific enolase. J. Mol. Biol. 341, 1015–1021. [DOI] [PubMed] [Google Scholar]
- Anderson J. P.; Walker D. E.; Goldstein J. M.; de Laat R.; Banducci K.; Caccavello R. J.; Barbour R.; Huang J.; Kling K.; Lee M.; Diep L.; Keim P. S.; Shen X.; Chataway T.; Schlossmacher M. G.; Seubert P.; Schenk D.; Sinha S.; Gai W. P.; Chilcote T. J. (2006) Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. [DOI] [PubMed] [Google Scholar]
- Wang L.; Das U.; Scott D. A.; Tang Y.; McLean P. J.; Roy S. (2014) α-Synuclein Multimers Cluster Synaptic Vesicles and Attenuate Recycling.. Curr. Biol. 24, 2319–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauvet B.; Mbefo M. K.; Fares M.-B.; Desobry C.; Michael S.; Ardah M. T.; Tsika E.; Coune P.; Prudent M.; Lion N.; Eliezer D.; Moore D. J.; Schneider B.; Aebischer P.; El-Agnaf O. M.; Masliah E.; Lashuel H. A. (2012) α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 287, 15345–15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N.; Dunker A. K. (2010) Understanding protein non-folding. Biochim. Biophys. Acta 1804, 1231–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.