

Abstract
The circadian clock is a global regulatory system that interfaces with most other regulatory systems and pathways in mammalian organisms. Investigations of the circadian clock–DNA damage response connections have revealed that nucleotide excision repair, DNA damage checkpoints, and apoptosis are appreciably influenced by the clock. Although several epidemiological studies in humans and a limited number of genetic studies in mouse model systems have indicated that clock disruption may predispose mammals to cancer, well-controlled genetic studies in mice have not supported the commonly held view that circadian clock disruption is a cancer risk factor. In fact, in the appropriate genetic background, clock disruption may instead aid in cancer regression by promoting intrinsic and extrinsic apoptosis. Finally, the clock may affect the efficacy of cancer treatment (chronochemotherapy) by modulating the pharmacokinetics and pharmacodynamics of chemotherapeutic drugs as well as the activity of the DNA repair enzymes that repair the DNA damage caused by anticancer drugs.
The circadian clock controls the daily rhythmicity of the biochemistry, physiology, and behavior of organisms. In mammals, it is comprised mainly of a core transcription–translation feedback loop (TTFL) that is cell autonomous and self-sustained. The core clock consists of CLOCK-BMAL1 transcriptional activators and cryptochrome (CRY) and period (PER) transcriptional repressors.1−4 The primary TTFL is stabilized by secondary, consolidating loops. The effect of the circadian clock on the DNA damage response and the related questions of circadian clock–cancer connections may be considered under two broad categories:5−7 regulation of DNA damage responses by the clock and circadian rhythmicity as a variable in carcinogenesis and in cancer treatment.
I. Circadian Clock and the DNA Damage Response
It has been reported that the three major DNA damage response networks, DNA repair, DNA damage checkpoints, and apoptosis, are affected by the circadian clock.7
1. Circadian Clock Regulation of DNA Repair
The effect of the circadian rhythm on the cell cycle in epidermal and gastrointestinal tissues was well-documented long before the discovery of the clock genes and the development of the current molecular model.1,2 Although it has been known that the cell cycle phase varies by the time of day in proliferating tissues such as epidermis and intestine,8,9 only recently has clock control of the cell cycle in these tissues been demonstrated in mouse genetic models.10,11 In mice, DNA replication/S phase peaks in the morning and mitosis peaks in the evening.8 In parallel with these findings, it was reported that certain genotoxic chemicals such as N-methyl-N-nitrosourea12 and cyclophosphamide13,14 were more toxic in terms of tumor incidence and mortality when they were administered in the early morning compared to delivery in the evening. However, there is no evidence that this differential effect of toxicity was due to a differential effect on damage induction or repair because similar preferential mortality has been observed with other treatments that do not induce DNA damage, such as injections of massive doses of bacterial cultures or of lipopolysaccharides.15 Circadian clock regulation of DNA repair was discovered more recently and is further explained below.
There are five major pathways of DNA repair:16 direct repair, base excision repair, nucleotide excision repair, mismatch repair, and recombination/cross-link repair (Table 1). Currently available evidence indicates that of all these pathways, only nucleotide excision repair is directly controlled by the circadian clock.10,17−19 However, the possibility that the other repair pathways exhibit low-amplitude circadian rhythmicity has not been eliminated. In addition, because mismatch repair is coupled to replication,20 which is controlled by the clock,10,11 it is indirectly affected by the circadian clock in replicating tissues. Similarly, the mode of double-strand break/cross-link repair is affected by the clock because the homologous recombination mode of double-strand break repair is restricted to the S and G2 phases of the cell cycle, whereas double-strand break repair by nonhomologous end joining occurs throughout the cell cycle.21
Table 1. DNA Repair Mechanismsa.
| DNA repair mechanisms | types of DNA damage repaired |
|---|---|
| direct repair | pyrimidine dimers, O6-methylguanines |
| base excision repair | oxidized bases, alkylated bases |
| nucleotide excision repair | pyrimidine dimers, oxidized bases, cisplatin (GG) diadducts |
| mismatch repair | base mismatches, insertions, deletions |
| recombination/cross-link repair | double-strand breaks and interstrand cross-links |
These are nearly universal mechanisms. However, mammalian organisms do not have photolyase that directly repairs pyrimidine dimers. Instead, repair of pyrimidine dimers is by nucleotide excision repair, which is controlled by cryptochrome, the evolutionary cousin of photolyase.
In humans and mice, nucleotide excision repair (excision repair) is the sole repair mechanism for eliminating bulky DNA base lesions produced by ultraviolet (UV) light [cyclobutane pyrimidine dimers and (6–4)photoproducts], chemical carcinogens such as benozo[a]pyrene and 2-acetylaminofluorene, and anticancer drugs such as cis-diamminedichloroplatinum(II) (cisplatin).22 Humans with mutations in excision repair genes suffer from xeroderma pigmentosum (XP), a disease characterized by a 5000-fold increase in skin cancer incidence because of an inability to repair UV-induced DNA damage.23 The reaction mechanism of excision repair is remarkably similar from Escherichia coli to humans, although the proteins carrying out the repair reaction, in contrast to all other repair systems, are not evolutionarily related (Figure 1). Human excision repair is carried out by the concerted action of six core repair factors comprising 16–20 polypeptides: RPA, XPA, XPC, TFIIH, XPG, and XPF-ERCC1. Damage is recognized by RPA, XPA, and XPC, and the enzyme specificity is ensured by the kinetic proofreading function of TFIIH, which hydrolyzes ATP along several steps of the damage recognition pathway to discern damaged DNA (Figure 2). Then, at the damage site, RPA, XPA, and TFIIH form a stable complex. This complex recruits the XPG and XPF-ERCC1 nucleases, which incise the damaged strand at the 6th ± 3 and 20th ± 5 phosphodiester bonds 3′ and 5′ to the lesion, respectively. The excised 24–32-nucleotide damage-containing oligomer (canonical 30-mer) is released in a complex with TFIIH24,25 and degraded by intracellular nucleases. The resulting gap is filled by DNA polymerases and ligated. Nearly all of the proteins involved in the basic damage recognition/excision reaction are rate-limiting, and therefore, circadian control of any of the proteins involved in the dual incision could potentially confer circadian rhythmicity to nucleotide excision repair.7
Figure 1.
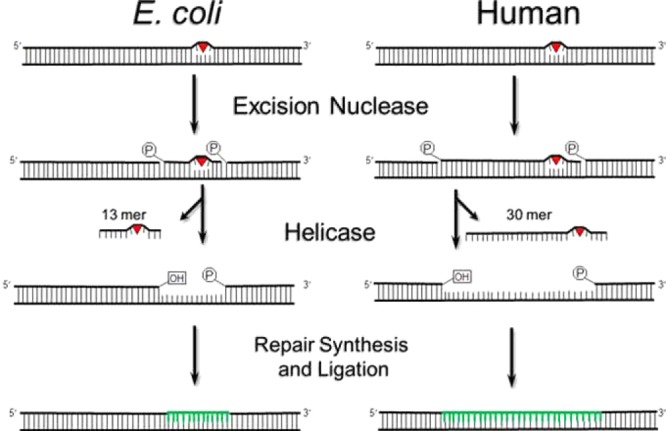
Nucleotide excision repair. An excision nuclease makes dual incisions 5′ and 3′ to the DNA damage to generate a 12–13- or 24–32-nucleotide damage-containing oligonucleotide in E. coli or humans, respectively. The excised fragment is released by helicases, and the gap is filled and ligated by DNA polymerases. Adapted from ref (104).
Figure 2.
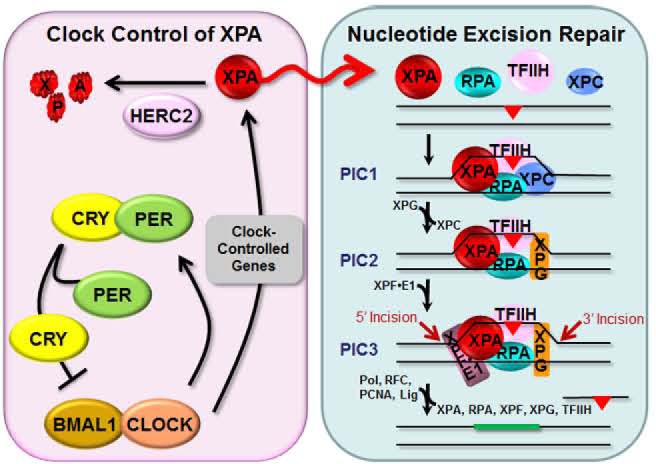
Model for the circadian regulation of XPA and excision repair by the clock and the ubiquitin–proteasome system. The transcription factors CLOCK and BMAL1 make a heterodimer that binds to E-boxes (CACGTG) in the promoters and activates transcription of the core clock genes Per and Cry, as well as clock-controlled genes, including Xpa. The CRY and PER proteins dimerize and, after a time delay, enter the nucleus where CRY dissociates from PER and inhibits CLOCK-BMAL1-activated transcription, thus generating an oscillatory pattern of gene expression. Robust protein oscillation is achieved by the short half-lives of CRY and PER proteins because of proteolysis by the ubiquitin–proteasome system.1,2 HERC2 is the ubiquitin ligase that targets XPA for relatively rapid degradation.18 Excision of DNA damage is accomplished by coordinated functions of six core repair factors. Damage is recognized by RPA, XPA, and XPC, followed by recruitment of TFIIH by XPC and XPA. The DNA near the damage site is unwound by the helicase activity of the XPD subunit of TFIIH to form stable preincision complex 1 (PIC1) that recruits XPG, and XPC is displaced from the complex to form PIC2. XPF-ERCC1 is then recruited to form PIC3. Within PIC3, XPG makes the 3′ incision 6 ± 3 phosphodiester bonds 3′ and XPF makes the 5′ incision 20 ± 5 phosphodiester bonds 5′ to the damage. The excised 24–32-nucleotide oligomer carrying the damage is released, and the resulting gap is filled by DNA polymerases and ligated. The XPA protein plays an essential role in damage recognition and is a rate-limiting factor that is regulated by the clock and by the HERC2 ubiquitin ligase. A consequence of the daily oscillation of XPA (red sinusoidal arrow) is that excision repair activity exhibits a daily rhythm that increases during the late afternoon and evening and decreases during the early morning in mouse.
When nuclear extracts from mouse brain or liver were tested over a circadian cycle, it was found that the excision activity, as measured by the removal of (6–4)photoproduct or cisplatin-d(GpTpG) diadducts, exhibited circadian rhythmicity, with a zenith at ∼5 p.m. (ZT10) and a nadir around 5 a.m. (ZT22)17,18 (Figure 3). When the expression patterns of the nucleotide excision repair proteins were analyzed, it was found that only the level of XPA exhibited circadian rhythmicity.17 Further analyses revealed that Xpa transcription exhibits high-amplitude oscillation in all tissues that were tested (brain, liver, and skin) except testis,18 which is known to lack a circadian clock.26Xpa transcription, XPA protein level, and excision activity all exhibit amplitudes of 5–10, and all are in phase with one another. As is the case with other proteins with high-amplitude rhythmicity, the XPA protein also has a relatively short half-life (t1/2 ∼ 3 h).18 The protein is ubiquitinated by HERC2 E3 ligase and degraded by the ubiquitin–proteosome system (UPS), and downregulation of HERC2 by siRNA leads to the stabilization of XPA and to a constitutively high level of the protein.18 Similarly, in Cry1/2–/– mouse tissues, XPA levels are at a constitutively high level. However, the increased XPA level, whether it is a result of constitutive transcription or reduced ubiquitination and proteolytic degradation, does not increase, or only marginally increases, the nucleotide excision repair activity because under these conditions other excision repair proteins become rate-limiting.
Figure 3.
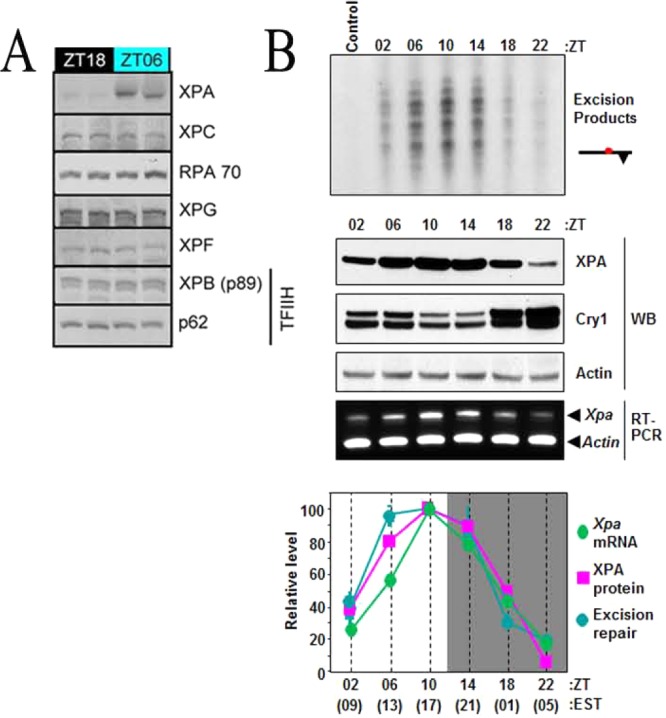
Effect of the circadian clock on day and night expression patterns of excision repair proteins and excision activity. (A) Nucleotide excision repair proteins were analyzed by immunoblotting in duplicate C57BL/6 mouse brain samples harvested at ZT(zeitgeber time)18 (midnight) and ZT06 (noon).17 XPA is highly expressed during the day and downregulated at night. The other excision repair proteins do not exhibit circadian variation between ZT18 and ZT06. (B) Circadian rhythm of XPA transcript, protein, and nucleotide excision repair in C57BL/6 mouse liver.18 (Top) Excision repair. The liver extracts were tested for repair activity. The control lane contained DNA substrate but no extract. The excision products [indicated as a bar with a circle (isotope-labeled) and a triangle (Pt adduct)] in the range of 24–32 nucleotides were detected by autoradiography. (Middle) Transcription and protein expression. WB indicates Western blot for XPA and Cry1. Note the antiphase relation between the Cry1 repressor and the clock-controlled XPA protein. RT indicates analysis of XPA transcript by quantitative RT-PCR. (Bottom) Quantitative analysis. Values of Xpa transcription, XPA protein level, and excision repair activity over a circadian cycle are expressed relative to the maximum of each variable (ZT = 0 is light on, and ZT = 12 is light off; EST is Eastern Standard Time). The removal of both (6–4)photoproduct and cisplatin-d(GpTpG) diadducts exhibits circadian rhythmicity under conditions of both ZT and CT (circadian time).
In tissue culture, individual mammalian cells maintain their circadian rhythmicity.27 However, because of stochastic perturbations the cells are out of phase with one another, and as a consequence the culture as a whole does not exhibit circadian rhythm. The circadian phase of the entire culture can be synchronized by extracellular signals that activate essentially any of the major intracellular signaling pathways (“a bewildering variety of signaling pathways”27), including MAP kinase, nuclear receptors, and DNA damage response pathways.27−29 Activation of these pathways by such treatments (serum shock and dexamethasone) synchronizes the phases of the cells in culture, and the culture as a whole becomes a circadian system. It should be noted, however, that although circadian synchronization in tissue culture has been quite useful in studying the basic mechanisms of the molecular clock,27,28 this method is of limited use in studying most of the clock-controlled genes and pathways because in tissue culture the clock is supplanted by homeostatic control mechanisms.30 As a consequence, even though excision repair exhibits high-amplitude circadian rhythmicity at the animal and organ level, its rhythmicity in circadian synchronized tissue culture is either nonexistent or below the resolution limit of repair assays, which is true for other circadian-controlled outputs in cell-based systems.30
Finally, with respect to evolutionary considerations regarding the circadian control of nucleotide excision repair, we note that one of the most commonly held views in circadian biology is the “escape from light” hypothesis according to which circadian rhythmicity evolved to minimize exposure to UV light-induced DNA damage in primitive organisms by diel vertical movement to and from the surface of the ocean.16,31,32 We also note that photolyase, which repairs UV-induced DNA damage, uses blue light as a cosubstrate and that while mammals do not have photolyase,33 its close evolutionary relative cryptochrome regulates excision repair of UV-induced DNA damage. Thus, photolyase and cryptochrome may have evolved from a common ancestor into an enzyme that repairs UV damage and into a clock protein, which though it does not repair UV damage, does regulate the excision repair system that repairs UV damage in addition to many other types of DNA damage.
2. DNA Damage Checkpoints
Cell cycle checkpoints regulate the progression of the cell cycle,34 and DNA damage checkpoints are biochemical pathways or networks that delay cell cycle progression in response to DNA damage.35 DNA damage checkpoints are damage-amplified forms of cell cycle checkpoints. The DNA damage checkpoints promote cell survival by preventing cells from entering the S phase while the DNA is damaged (G1/S checkpoint), by preventing the firing of late replication origins while the early replicons are blocked by DNA damage (intra-S phase checkpoint), by blocking cells from entering mitosis while the S phase is ongoing albeit at a slow rate because of DNA damage (S-M checkpoint), and by preventing cells from entering into mitosis while DNA carries damage in the G2 phase (G2/M checkpoint).22
Although coupling of the cell cycle to the circadian clock has been reported in organisms ranging from unicellular organisms36−38 to mammals27 by various mechanisms and to confer an evolutionary advantage, the significance of this coupling in mammals is unclear at present. In mammals, it has been reported that the clock controls the cell cycle by several mechanisms, including G1/S regulation by p21,39 p20,40 and NONO,41 and G2/M regulation by Wee1.42 The significance of these findings, as well as the converse findings that the cell cycle controls the circadian cycle in proliferating tissues or tissue culture,28,43,44 to the DNA damage checkpoint response is unclear. In addition, circadian/cell cycle coupling may not be that strong. A study with immortalized fibroblasts revealed that the two become uncoupled at higher temperatures; with an increase in temperature, the cell cycle is accelerated in accordance with the rules governing the rate of chemical reactions as a function of temperature, but the circadian period remained constant, which has been known to occur in a bona fide circadian system (temperature compensation).45
In mammalian organisms, two main DNA damage checkpoint pathways and/or systems have been defined on the basis of the PIKK (phosphoinositol 3-kinase-related kinase) family member damage sensor that initiates the signal transduction cascade (Figure 4). These two pathways are governed by the ATM (Ataxia Telangiectasia-Mutated) and ATR (ATM and Rad3-related) protein kinases.22 Although there is some overlap and crosstalk between the two pathways, as a general rule, DNA damage by ionizing radiation (IR) and other agents that induce double-strand breaks activates the ATM pathway while DNA damage by UV and UV-mimetic agents that produce bulky base adducts activates the ATR pathway. Both pathways encompass approximately 10 core proteins, but both are also affected by many other proteins that are involved in cell growth and differentiation.22 It would be expected that such pathways with pervasive effects on cellular homeostasis would form an interface with the circadian clock molecular clockwork, which is another global regulatory system with wide-ranging effects on cellular physiology.7 Indeed, interfacing of both the ATR– and ATM–DNA damage checkpoints with the clock has been reported.
Figure 4.
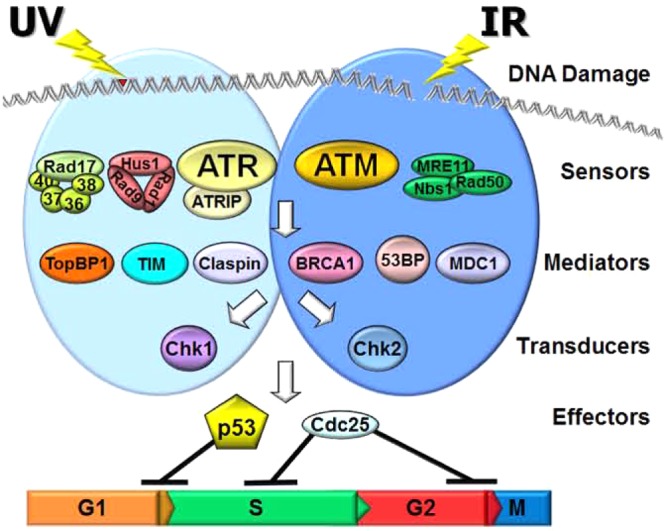
Model for the mammalian DNA damage checkpoint pathways and/or systems. Two main DNA damage checkpoint pathways and/or systems have been defined on the basis of the PIKK family member damage sensor (ATR or ATM) that initiates the signal transduction cascade. DNA damage by ionizing radiation (IR) and other agents that induce double-strand breaks activates the ATM pathway, while DNA damage by UV and UV-mimetic agents that produce bulky base adducts activates the ATR pathway, although there is overlap and crosstalk between the two pathways.
a. Coupling of the Circadian Clock with the ATR–Chk1 DNA Damage Signaling System
The first evidence that the circadian clock interfaces with the DNA damage checkpoints came from the analysis of Timeless (TIM) protein in human cells.46 There are two TIM paralogs in Drosophila, Tim1 (the classic Tim gene) and Tim2 (the subsequently discovered timeout gene).47,48 dTIM1 is a core clock protein in Drosophila with no other known functions. The mammalian Tim gene is more homologous to dTim2 and plays an essential role in growth and differentiation at the organismal level49 and replication fork stability at the cellular level,50,51 and as a consequence, TIM knockout is embryonic lethal in the mouse.52 Nevertheless, cumulative evidence from a variety of experimental approaches indicates that the human (and mouse) TIM is a bona fide clock gene, as well, albeit with additional essential functions required for viability.52−54
The clue about the essential function for TIM came from studies of its yeast orthologs.55,56 These are Swi1 in Schizosaccharomyces pombe and Tof1 in Saccharomyces cerevisiae. Moreover, it was shown that both Swi1 and Tof1 function as heterodimers (Swi1–Swi3 and Tof1–Csm3, respectively) and play important roles in stabilizing replication forks and in S phase (replication) checkpoint activation. Similarly, most human TIM is in the form of a TIM–TIPIN (Timeless interacting protein) complex.57 Thus, the two lines of research, circadian studies in mammalian cells, implicating TIM in the circadian clock, and replication checkpoint studies in yeasts, implicating it in replication fork stability, suggested the possibility that the TIM–TIPIN complex may participate in both circadian and cell cycle (checkpoint) regulation in mammalian cells.
Experimental analyses supported this expectation. It was found that human TIM associates both with CRY2 on the circadian side and with ATR–ATRIP damage sensor and Chk1 signal transducer kinases on the checkpoint side of the cellular signaling network.46 At the functional level, the downregulation of TIM by siRNA caused a significant reduction in PER2 protein levels and disruption of circadian rhythmicity in the suprachiasmatic nucleus (SCN) (clock effect)53 and a significant reduction in the level of phosphorylation of Chk1 protein under conditions of replication inhibition (Figure 5A), which caused both intra-S (replication fork collapse) and S-M (premature mitosis) checkpoint defects (cell cycle effect).46 These findings led to the conclusion that TIM, either alone or in the form of the TIM–TIPIN complex, is both a clock and a checkpoint protein by being a direct participant in both processes (Figure 5B). This was named parallel coupling of the circadian and cell cycles to indicate the participation of the same protein in both systems, as opposed to serial coupling whereby a clock-regulated protein or a cell cycle-regulated protein influences the cell cycle or circadian clock phase and amplitude, respectively. In further support of the clock–DNA damage checkpoint connection, it was reported that downregulation of CRY1 in mouse NIH3T3 cells by siRNA abolished ATR-mediated Chk1 phosphorylation after DNA damage;58 however, a different study with mouse skin fibroblasts found that CRY1 downregulation had no effect on ATR activation.59
Figure 5.
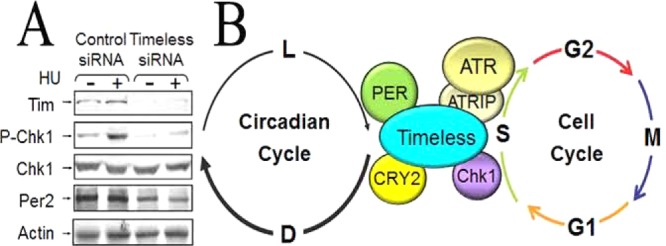
Coupling the circadian clock to the cell cycle/checkpoint response. (A) Timeless (TIM) is required for Chk1 activation. HeLa cells were transfected with control or TIM siRNA twice over a period of 3 days. Seventy-two hours after the initial transfection, cells were treated with 10 mM hydroxyurea (HU) for 1.5 h or left untreated. Two hundred micrograms of cell lysates was immunoblotted with anti-Tim (α-Timeless), anti-P-Chk1 (Ser345), anti-Chk1, anti-Per2, and anti-actin antibodies.46 (B) Model for parallel coupling of the circadian clock and the cell cycle/checkpoint response. The core circadian clock protein CRY2, in conjunction with TIM, participates in the ATR–Chk1 signaling pathway in response to UV and UV-mimetic agents.
Finally, we note that in parallel with studies of human TIM, work on dTIM2 (timeout) has shown that this protein plays a role in both circadian clock and checkpoint functions.60,61
b. Coupling of the Circadian Clock with the ATM–Chk2 DNA Damage Signaling System
Findings similar to those reported for the ATR–Chk1 system were reported for the ATM–Chk2 signaling pathway.6,62 It was reported that PER1, in addition to its role as a clock protein, also participates in ATM → Chk2 signaling by directly interacting with both ATM and Chk2. It was found that downregulation of PER1 interfered with IR-induced phosphorylation of Chk2 by ATM and reduced the level of apoptosis by agents that induce double-strand breaks.62 In further support of this function of PER1 as a pro-apoptotic protein, it was reported that PER1 is downregulated in a number of cancers, including lung and breast cancers.62
Against this background, a systematic study of the clock effect on the DNA damage response in tissue culture yielded unexpected results,59 which suggests that caution must be exercised in relying on data from tissue culture experiments regarding the clock–checkpoint–apoptosis–repair connection. First, it was found that mutations in clock genes in either the positive arm (CLOCK and BMAL1) or the negative arm (CRYs and PERs) in mouse cell lines had no effect on the two major DNA repair pathways tested, nucleotide excision repair and recombination/cross-link repair. These results indicate that if these repair systems are circadian-controlled at the organismic level, this control mechanism is lost or overridden by homeostatic mechanisms in tissue culture. This was not surprising because a systematic analysis of circadian-controlled genes revealed that even though in a given organ 1000–3000 genes are transcriptionally clock-controlled,1,2,30,63 in tissue culture of most commonly used mammalian cell lines, transcription of only 10–20 genes, mainly those of the core clock genes, was controlled by the TTFL of the circadian clock.30 Second, when attempts were made to compare the effects of clock gene mutations on checkpoint and apoptosis using immortalized cell lines, it was realized that these cell lines isolated in different laboratories have different amounts of checkpoint and apoptosis proteins (both apoptosis proteases and their targets), which makes comparison between wild-type and mutant cell lines impossible. Thus, it was concluded that the effect of the clock on checkpoint and apoptosis could be reliably compared only in the same cell strain in which the particular clock protein is downregulated by siRNA. To this end, primary mouse skin fibroblasts (MSFs) were studied. In MSFs, downregulation of CLOCK, BMAL1, CRY1, CRY2, PER1, or PER2 had no effect on Chk1 or Chk2 phosphorylation after DNA damage.59
Because of a previous report indicating that CLOCK knockdown rendered cells sensitive to IR and mitomycin C,64 the MSFs were also analyzed for sensitivity to these DNA-damaging agents, and there was no indication that eliminating any of the clock proteins had any effect on the sensitivity to these DNA-damaging agents.59 Third, when apoptosis was analyzed in MSFs in which clock gene expression was downregulated by siRNA, no effect on apoptosis could be detected as probed by PARP and caspase-3 cleavage following UV irradiation.59 Finally, in contrast to the study that reported that certain human cancer cell lines had reduced levels of Per1 transcription and as a consequence were resistant to IR-induced apoptosis and clonogenic killing by IR, and that they could be made sensitive by PER1 overexpression,62 no such effect could be detected in a follow-up report.59 Similarly, in contrast to the reports linking PER1 levels to IR sensitivity, when the NCI-H460 human lung cancer cell line and the HCT-116 human colorectal cancer cell line were transfected with a PER1 expression vector and then tested for IR-induced apoptosis and clonogenic killing there was no effect (Figure 6A). There was also no effect on either apoptosis or clonogenic survival in response to IR treatment, relative to control cells or the same cell lines in which PER1 was downregulated by siRNA (Figure 6B).59 In agreement with these latter results, it has been reported that in human gingival and pancreatic cancer cell lines, CA9-22 and MTA PCa-2, respectively, and in the hepatocellular carcinoma cell line HepG2, PER1 actually acts as an anti-apoptotic protein because downregulation of PER1 by siRNA in these cell lines reduced the level of spontaneous and cisplatin-induced apoptosis and had an only marginal effect on clonogenic survival.65−67
Figure 6.

Neither PER1 overexpression nor downregulation sensitizes human lung cancer cells to apoptosis or affects Chk2 phosphorylation following ionizing radiation (IR). (A) Overexpression of PER1. Protein levels were analyzed by immunoblotting 48 h after irradiation with IR with the indicated antibodies from NCI-H460 cells following transfection with either empty vector (pcDNA3) or a mPer1-overexpressing vector for 72 h. T-PARP and C-PARP stand for total PARP and cleaved PARP, respectively. (B) Downregulation of PER1. Protein levels from NCI-H460 cells transfected with either negative control (nontarget, NT) or Per1 siRNAs were analyzed by immunoblotting 48 h after irradiation with 0, 10, or 30 Gy of IR with the indicated antibodies.59
To recapitulate, quantitative analyses of the DNA damage response with either immortalized cell lines or primary fibroblasts, with or without any of the core clock proteins, do not provide convincing evidence that in these systems the core clock affects the DNA damage response. The only exception to this generalization is TIM. However, TIM is an essential cell cycle gene in addition to its role in the circadian cycle, and therefore, the effects of its downregulation or mutation on DNA damage checkpoints are more likely the reflection of its cell cycle function.
3. Circadian Clock and Apoptosis
Although apoptosis is one of the pathways activated by DNA damage, it is a more general cell death program that can be activated by a variety of stimuli,68 and hence, the clock–apoptosis connection deserves a consideration separate from the general topic of DNA damage responses. Apoptosis is the most common form of programmed cell death and has been classified into two categories: intrinsic apoptosis, which is initiated by DNA-damaging agents, and extrinsic apoptosis, which is initiated by cytokines, including death ligands such as tumor necrosis factor α (TNFα).68 Recently, the clock has been connected to both intrinsic and extrinsic apoptosis (Figure 7).
Figure 7.
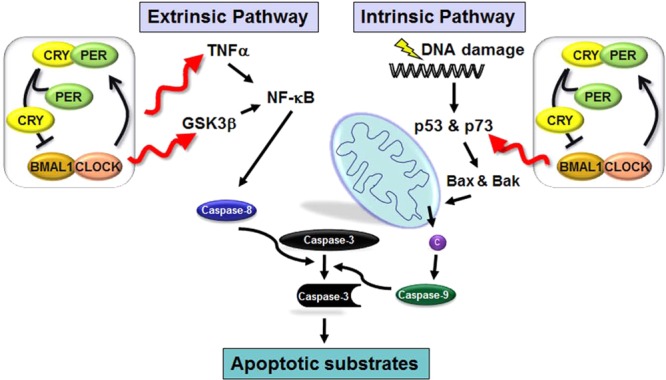
Model for effects of the clock on apoptosis. Circadian regulation of the extrinsic pathway occurs via regulation of the synthesis of TNFα and the phosphorylation of GSK3β, both of which regulate NF-kB signal transduction to caspase 8, which in turn activates executioner caspases, including caspase 3. In the intrinsic pathway, DNA damage causes the upregulation of p73 (in a circadian-regulated manner), which activates transcription of Bax and Bak causing the release of cytochrome c (c) from the mitochondria, apoptosome assembly, and the eventual cleavage and activation of transducer caspase 9, which in turn cleaves and activates executioner caspase 3.
a. Clock and Intrinsic Apoptosis
In the intrinsic (or mitochondrial) pathway, cellular damage, including DNA damage and cellular stress caused by unfolded proteins, leads to an increased level of expression of pro-apoptotic members (Bax and Bak) of the Bcl-2 family. The p53 tumor suppressor plays an important role in the DNA damage-initiated intrinsic apoptosis pathway.68 In fact, it appears that p53 acts as a tumor suppressor to a large extent by inducing apoptosis in oncogenically transformed cells by upregulating the expression of Bax and Bak in response to genotoxic stress.69 p53 is a member of a family of proteins comprised of p53, p63, and p73.69 Among these three proteins, p53 is the main tumor suppressor and does so by promoting the apoptosis of damaged or oncogenically transformed and hence stressed cells; p63 and p73 function primarily in growth and differentiation with a secondary role as pro-apoptotic proteins, with p73 as the stronger tumor suppressor of the two.70 As we will detail below, it was found that oncogenically transformed p53–/–Cry1/2–/– cells were more sensitive to genotoxic agents than p53–/– cells,71 which suggested that the absence of CRYs activated a p53-independent apoptosis pathway that is not apparent in p53+/+ cells because of the strong pro-apoptotic effect of p53.
Detailed analyses of the enhanced apoptosis in p53–/– cells by CRY mutation led to the discovery that p73 is upregulated in these cells after DNA damage.72 Like p53 protein levels, p73 protein levels also increase after DNA damage. However, in contrast to p53, whose upregulation occurs mainly through post-translational modifications, DNA damage-induced upregulation of p73 occurs mainly by transcriptional induction. The p73 promoter contains five binding sites for Egr1, three binding sites for E2F1, and one binding site for C-EBPα transcription factors. Egr1 and E2F1 are transcriptional activators, whereas C-EBPα functions as a repressor. Egr1 is controlled by the circadian clock in addition to other ill-defined mechanisms related to growth control.72 Thus, with respect to circadian regulation, Egr1, which contains an E-box in its promoter, is a first-order clock-controlled gene (CCG) and p73 is a second-order CCG (Figure 8A). In most cell types in the absence of DNA damage, the level of expression of p73 is rather low. DNA damage affects all three transcription factors in a way to enhance p73 transcriptional activation. DNA damage by UV or UV-mimetic agents causes C-EBPα phosphorylation, its release from the p73 promoter, and exclusion from the nucleus, facilitating p73 transcription. Egr1 is also activated by UV and the UV-mimetic agent oxaliplatin; therefore, p73 induction by these DNA-damaging agents has a strong circadian component, and Cry mutation sensitizes p53–/– cells to oxaliplatin73 (Figure 8B). In summary, in cells with wild-type p53, the circadian clock does not appreciably affect intrinsic apoptosis, because of the dominant effect of p53 in promoting apoptosis in response to DNA damage. However, in p53 mutant cells (50% of cancers have p53 mutations) for intrinsic apoptosis initiated by UV-mimetic agents, including the widely used anticancer drug oxaliplatin, the clock affects apoptosis through a gene (p73) that becomes a second-order clock-controlled gene only after DNA damage.
Figure 8.
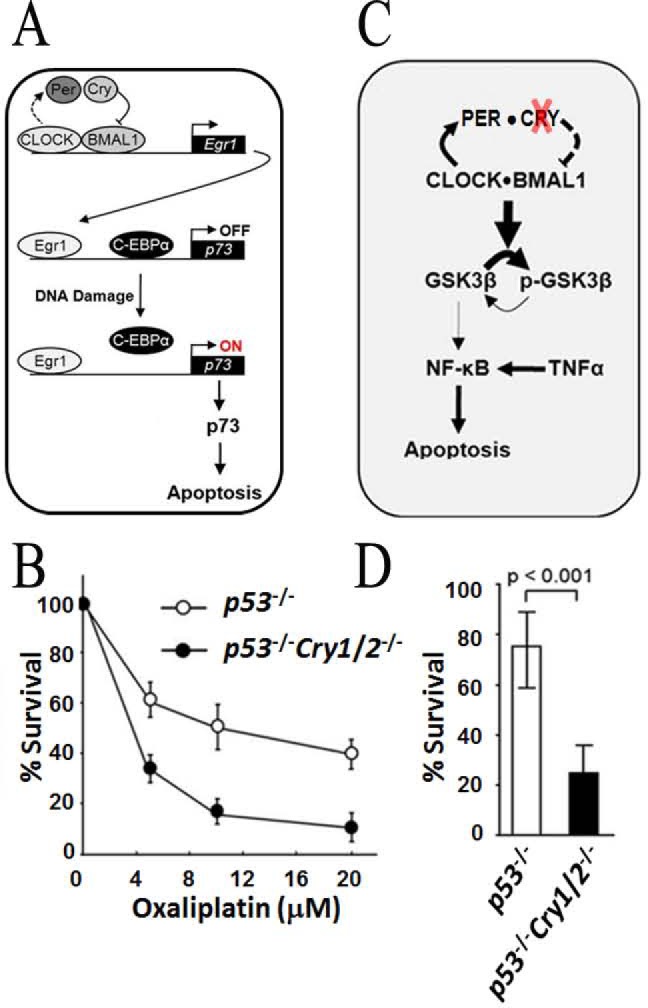
Circadian clock and apoptosis. (A) Model for targeting the clock to improve the efficacy of oxaliplatin treatment of p53-null tumors. CRY regulates Egr1 expression, which transactivates p73. In Cry mutants, Egr1 is upregulated. Following oxaliplatin treatment, there is a decreased level of binding of C-EBPα and an increased level of binding of Egr1 to the p73 promoter, causing massive production of p73 and enhanced apoptosis.72 (B) Effect of cryptochrome mutation on clonogenic cell death by oxaliplatin. Cells of the indicated genotypes were treated with the indicated doses of oxaliplatin and incubated for 9–10 days, and then colonies were stained with 5% methylene blue and counted to obtain the UV survival curves.73 (C) Model for the regulation of cytokine-initiated NF-κB anti-apoptotic function through the clock-controlled GSK3β activity in p53-deficient tumor cells and the consequence of cryptochrome inactivation.75 In the absence of CRY, GSK3β becomes hyperphosphorylated and unable to phosphorylate NF-κB, leading to weakened anti-apoptotic function of NF-κB. The absence of CRY also causes overexpression of TNFα, which contributes to enhanced extrinsic apoptosis. (D) Clonogenic cell survival assay showing the viable cells remaining after treatment with TNFα (50 ng/mL) for 48 h as a percentage of viable untreated cells.75
b. Clock and Extrinsic Apoptosis
The finding that mice of the p53–/–Cry1/2–/– genotype exhibit delayed onset of cancer relative to p53–/– mice71 suggested that the absence of CRY may amplify the extrinsic pathway of apoptosis that eliminates cancerous cells by upregulation or amplification of cytokines that promote extrinsic apoptosis. It was found that clock disruption through CRY mutation promotes extrinsic apoptosis by two mechanisms (Figure 8C). First, in Cry mutant mice, levels of inflammatory and pro-apoptotic cytokines such as TNFα are elevated.74 Second, in p53 mutant cells with a functional clock, these cytokines activate the transcription of anti-apoptotic genes in a manner gated by the circadian clock through GSK3β phosphorylation.74 In contrast, the Cry-null mutation in p53-mutated cells leads to hyperphosphorylation and inactivation of GSK3β and hence its failure to phosphorylate and activate the anti-apoptotic activity of NF-κB, making cells more sensitive to TNFα-induced apoptosis (Figure 8D).75 Thus, CRY mutation promotes extrinsic apoptosis by two different mechanisms, by leading to overproduction of pro-apoptotic cytokines such as TNFα and by leading to GSK3β hyperphosphorylation, which in turn results in its failure to phosphorylate NF-κB to perform its anti-apoptotic function. An additional level of regulation may also be mediated by the direct interaction between CLOCK and the p65 subunit of NF-κB.76 Thus, the combination of an elevated level of TNFα with reduced NF-κB anti-apoptotic activity because of its reduced level of phosphorylation leads to enhanced apoptosis of malignant cells that are known to be more susceptible to both intrinsic and extrinsic apoptosis as a general rule.
Finally, with regard to circadian control of DNA damage responses, we should also note that, conversely, evidence exists that DNA damage also affects the clock by resetting the phase.29,77 However, as activation of any of the major intracellular signaling pathways causes phase shifting, the physiological relevance of these observations remains to be established.27
II. Circadian Clock and Cancer
The circadian clock–cancer connection may be analyzed as three separate entities, including circadian control of carcinogenesis by the time of the day, circadian clock disruption as carcinogenic or as a modality in cancer treatment, and the circadian rhythm as a guide to administer anticancer treatment for improvement of the therapeutic index.
1. Circadian Control of Carcinogenesis
It was recently shown that the capacity for excision repair in mouse skin oscillates with a circadian rhythm that is correlated with rhythmic XPA protein oscillation.10 In addition to excision repair oscillation, using Cry1/2–/– mice it was shown that DNA replication is circadian in nature in proliferating tissues such as the skin and intestine, and excision repair is antiphase to that of DNA replication. Both excision repair and accurate replication are critical for maintenance of genomic stability, and presumably also for melanoma and non-melanoma skin cancer prevention. In fact, mice exposed to UV radiation in the early morning when excision repair is at its minimum and replication is at its maximum are more prone to squamous cell carcinoma with 5-fold more invasive carcinomas than mice exposed to UV radiation in the evening when excision repair is at its maximum and replication is at its minimum (Figure 9). These studies establish a rationale for chrono-photobiological response and suggest that timing or modulating the circadian clock mechanism may reduce the risk of exposure to sunlight and other sources of radiation.
Figure 9.
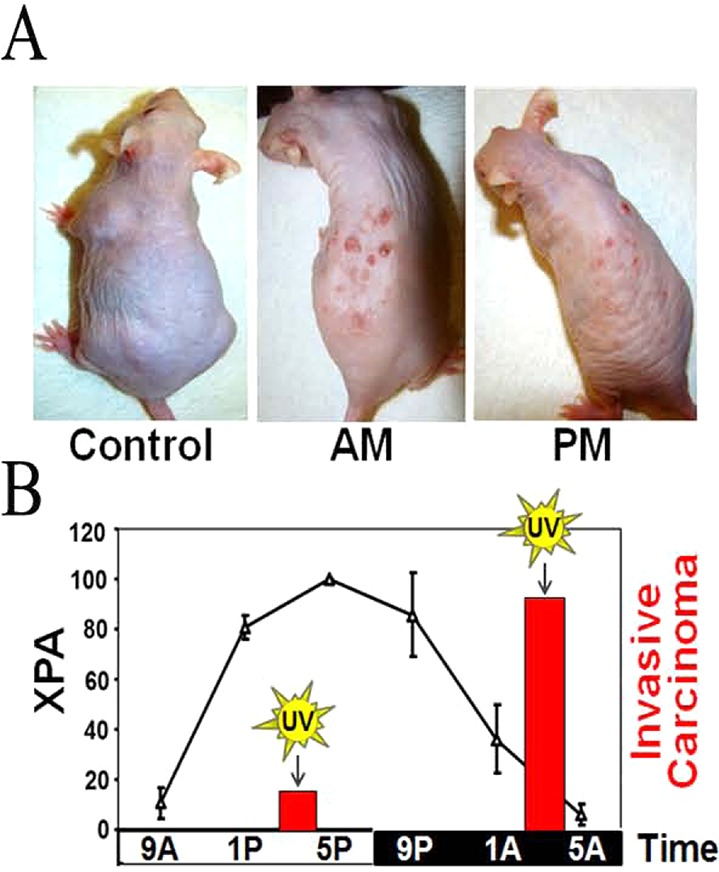
Effect of the time of day of exposure to UVB on skin carcinogenesis in SKH-1 hairless mice. The control was not irradiated; the AM group was irradiated at 4 a.m. (XPA minimum), and the PM group was irradiated at 4 p.m. (XPA maximum). (A) Physical appearance of mice from the three experimental groups 23 weeks after the initiation of irradiation (353 J/m2 of UVB). (B) Effect of the circadian clock on repair and cancer. XPA protein levels in epidermis (line plot) over the entire cycle and invasive cancer caused by UV at 4 a.m. and 4 p.m. (bar graph) are shown.10
2. Circadian Clock Disruption as a Carcinogenic Factor
Because the circadian clock directly or indirectly affects all metabolic processes, signaling pathways, and intracellular systems and/or networks,1−3,78,79 it would be expected that clock disruption would have an effect on cancer development and progression.80 With this general premise as a starting point, a number of studies have been conducted to demonstrate that clock disruption increases cancer risk and promotes cancer progression. These studies fall into three general categories: epidemiologic analysis in human populations, xenograft growth in mice with a surgically or environmentally disrupted clock, and analysis of the incidence of spontaneous or induced cancer in mice in which the clock has been permanently disrupted by gene targeting technologies. These studies will be briefly summarized below, but it must be stated at the outset that the evidence considered in its totality suggests that any link between clock disruption and cancer is either anecdotal or so weak that it might be considered nonexistent.
a. Epidemiological Studies
Several studies have reported that individuals with occupational clock disruption have an increased risk of cancer.81,82 Thus, it has been reported that night shift or rotating shift workers in health and food industries and flight attendants working on intercontinental routes have increased risk of breast and colorectal cancers, and correlations were made between the length of rotating shift work and cancer incidence.81,82 In fact, on the basis of a ruling by the WHO-IARC that “shift work that involves circadian disruption is probably carcinogenic to humans”, night shift workers with breast cancer were awarded compensation in Denmark.83 More recently, an American Medical Association policy statement has recognized clock disruption by light at night as a cancer risk based on consideration of meta-analyses of several epidemiologic studies.84 While these studies are important from a public health perspective, they do not establish causality, nor do they provide a mechanistic insight into clock disruption and cancer. In fact, it has been argued that night shift and rotating shift work and cancer are two independent outcomes of certain personality traits.85 In support of this latter argument, it was reported that in a large study of breast cancer incidence in Chinese women, the incidence of breast cancer in night shift and rotating shift workers was actually lower than the breast cancer incidence in women with conventional working schedules.86 More recently, the validity of epidemiological studies linking clock disruption to increased cancer risk has been criticized on methodological grounds.87,88 Hence, when all relevant factors are taken into account, the epidemiological data fall short of establishing an association between circadian clock disruption and an elevated risk of cancer. Studies with animal models and cell biological experiments have therefore been conducted to provide a more direct approach to the question of a circadian disruption–cancer connection.
b. Animal Models
Two types of animal studies have been done to investigate the effect of circadian clock disruption on carcinogenesis.
i. Surgical Clock Disruption
In this approach, the master circadian clock, the SCN, is surgically destroyed, tumors (or cancer cell lines) are inserted into mice subcutaneously, and tumor growth is monitored relative to xenografts in mice with a normal SCN. It was reported that both pancreatic carcinoma and Glasgow osteosarcoma xenografts grew at a faster rate in SCN-lesioned animals.89 However, as the clock was disrupted only in the SCN of the host mice, it is not known whether the implanted tumors had an intrinsic clock or whether the tumor clock was influenced by the host circadian clock. In a related study, when Clock gene expression was downregulated in Lewis lung carcinoma (LLC1) cells by siRNA and then inoculated subcutaneously into mice, the resulting tumor grew at a rate slower than that of the control, suggesting that in this experimental system disruption of the clock actually interfered with tumor growth.90 However, it could be argued that clock disruption by CLOCK mutation or downregulation stops the circadian clock at around midnight (when CLOCK-BMAL1 activity is at its minimum), and hence, stopping the clock by other means and at other times of the biological time may differentially affect tumor growth. This qualification notwithstanding, on the basis of this evidence alone the generalization that clock disruption by whatever means promotes tumorigenesis cannot be made.
ii. Genetic Disruption of the Circadian Clock
In this method, one (or both paralogs when they exist) of the clock genes is knocked out genetically, and in these genetically engineered mice, the incidence of spontaneous and DNA damage-induced cancers is monitored. In the first such experiment, it was reported that Per2 mutant mice were predisposed to spontaneous and IR-induced cancers.29 This was ascribed to elevated c-Myc expression and reduced p53 levels, which together were presumed to promote cell proliferation and inhibit apoptosis. However, subsequent studies failed to support this conclusion.80 Because CRY and PER constitute the negative arm of the circadian TTFL, it was reasoned that CRY-null mice would exhibit the same phenotype vis-à-vis spontaneous and IR-induced carcinogenesis as Per2 mutant mice. However, it was found that Cry1/2–/– mice were indistinguishable from wild-type mice.91 While this could be ascribed to PER or CRY having unique functions in addition to their functions as repressors of CLOCK-BMAL1, a recent study in which wild-type, Per1–/–, and Per2–/– mice in a C57/BL6 background were compared showed no significant difference among the three strains with respect to spontaneous and IR-induced cancer incidence92 (Figure 10), raising serious doubts about the validity of the report that Per2 is a tumor suppressor. Similarly, neither CLOCK nor BMAL1 null mutant mice have elevated incidence of cancers, although they exhibit some early aging phenotypes.93 Thus, considered in its totality, the available data lead to the conclusion that circadian clock disruption by genetic mutations does not predispose mice to cancer. Other reports claiming not-permanent clock disruption but repeated phase shifts might be carcinogenic await independent verification.94 Clearly, the circadian clock disruption–cancer connection is a very important public health issue, and further well-controlled epidemiologic and animal model studies are needed to provide a consensus about this subject, upon which public health policy decisions can be made.
Figure 10.
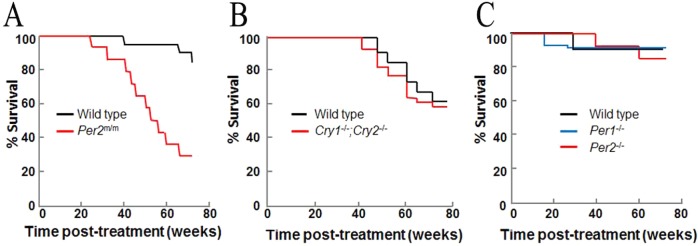
Kaplan–Meier plots of death from cancer from three different studies of mice with clock gene mutations. Eight-week-old mice of the indicated genotypes were exposed to 4 Gy of IR at ZT10 and observed for 90 weeks. Data for tumorigenesis and mortality are replotted from (A) ref (29) (B) ref (91), and (C) ref (92).
3. Circadian Clock and Cancer Treatment
Chronotherapy is the timing of drug delivery with the appropriate phase of the circadian rhythm to achieve optimal efficacy.95 Chonochemotherapy is the administration of chemotherapeutic drugs at the appropriate phases of the circadian clock to obtain the best therapeutic index.79,95 Although the application of chronotherapy to certain pathologic conditions such as asthma, hypertension, and cardiovascular disease has led to significant improvements in developing drug regimens for these diseases,96,97 the application of chronotherapy to cancer treatment has not. This is due in large part to the empirical design of the treatment regimens and to the anecdotal nature of the reports on the efficacy of chronochemotherapy. With recent advances in the mechanistic basis of the molecular clock, attempts are being made to develop mechanism-based chronochemotherapy regimens. Two such approaches, one based on the circadian clock control of excision repair and one based on the effect of circadian clock disruption on apoptosis, will be summarized below.
a. Clock Control of DNA Excision Repair and Chemotherapy
As noted above, the essential nucleotide excision repair protein XPA is a first-order clock-controlled protein, and as a consequence, the rate of nucleotide excision repair exhibits high-amplitude circadian rhythmicity.17 Because excision repair is the sole mechanism for removing the predominant DNA adducts induced by the anticancer drug cisplatin (and its second-generation derivatives oxaliplatin and carboplatin), Pt-d(GpG) and Pt-d(GpXpG), the repair rates of these adducts exhibit circadian rhythmicity in brain, liver, kidney, skin, and all other tissues tested except testis.10,17−19 Thus, in tumors arising in tissues with circadian rhythmicity, provided the tumor maintains rhythmicity in phase with the normal tissue, administration of cisplatin when excision repair is in the descending phase is expected to improve the therapeutic index by administering a less toxic dose. Conversely, if the tumor is rhythmic but out of phase with the normal tissue, the therapeutic index may be improved by drug delivery when repair is ascending in normal tissue. Similarly, if the cancerous tissue lacks rhythmicity, delivery of the drug in the ascending phase of the excision repair rate in the normal tissue would be expected to improve the therapeutic index.
Application of these findings to the development of practical chronochemotherapy regimens faces some technical problems that must be overcome to be of clinical value. First, accurate information regarding cisplatin pharmacokinetics and pharmacodynamics is needed for various tissues, including the tumor. Second, a noninvasive method for determining if the tumor has rhythmicity and, if it does, the phase of the rhythmicity relative to the normal tissue must be known. Finally, the timing of the other drugs that are used in combination chemotherapy, such as doxorubicin, which induces double-strand breaks, must be considered. There is no evidence that enzymes involved in double-strand break repair are controlled by the circadian clock. However, the mode of double-strand break repair is affected by the cell cycle (which is affected by the clock), with repair by homologous recombination dominating in the S phase and nonhomologous end joining in G1 and G2 phases.21 Similarly, cisplatin induces interstrand cross-links that are repaired by interstrand cross-link repair pathways22 that are also cell cycle-dependent, and their contribution to cisplatin lethality needs to be quantified. In summary, while the finding that excision repair is controlled by the circadian clock has provided the opportunity for a rational chronochemotherapy regimen, other factors will have to be incorporated into the overall approach in a clinical setting.
b. Clock Control of Apoptosis and Cancer Therapy
Preferential killing of cancer cells relative to normal cells is the predominant rationale in all therapeutic approaches. A major mechanism of drug-induced cell killing is programmed cell death (apoptosis). Analyses of the effect of clock disruption on the cellular response to DNA damage led to the finding that the circadian clock affects apoptosis in certain genetic backgrounds and to the conclusion that this effect must be considered in chronotherapy regimens.
The initial finding that the clock affects apoptosis came from investigations into the effect of clock disruption on carcinogenesis. To understand the effect of CRY mutation on carcinogenesis, the Cry1/2–/– mutations were combined with a p53-null mutation following a commonly used strategy to uncover the carcinogenicity of weakly penetrant tumorigenic genes.71 The p53–/– mice, as expected, developed lymphoma and lymphosarcomas and had an average life span of 5.5 months. Contrary to expectation, the p53–/–Cry1/2–/– mice had a reduced age-adjusted incidence of cancer and lived 1.5-fold longer than the p53–/– mice (Figure 11A). This finding led to the suggestion that in the p53–/–Cry1/2–/– background, cells become more sensitive to DNA-damaging agents, and therefore, the two cell lines after transformation with ras(V12G)/rasT24 were examined for their DNA repair, DNA damage checkpoint, and apoptosis activities, all of which are known to affect cell survival. It was found that the triple knockout had nucleotide excision repair and checkpoint responses indistinguishable from those in p53 mutant cells.71 However, the triple mutant exhibited increased sensitivity to apoptosis, which was detailed in the earlier section on the effect of the circadian clock on the cellular response to DNA damage. These findings are briefly recapitulated here to emphasize the potential of this increased sensitivity to apoptosis as a variable in cancer therapy. It should be noted, however, that in contrast to the effect of Cry mutation on increasing the survival of p53-null mice, no such effect was seen in another mouse cancer model system. Ink4a–/–;ras(V12G) tumor suppressor/oncogene mutant mice are used as a model for UV-induced melanomas, and these mice develop malignant melanomas with 100% frequency in light-exposed areas of the animal under standard mouse facility lighting conditions.98 The combination of Cry mutation with the ink4a–/–;ras(V12G) mutation did not affect the melanoma incidence (Figure 11B), and hence, it was concluded that in this genetic background, Cry mutation has no moderating effect on cancer incidence or progression.
Figure 11.
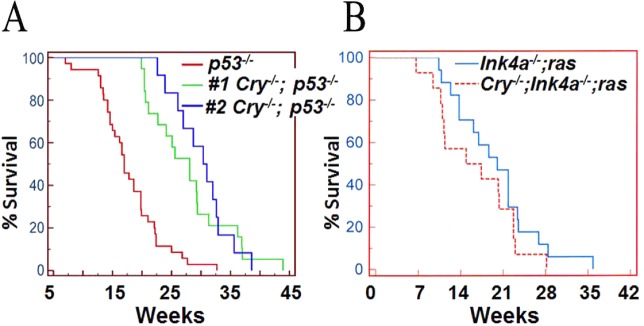
Effect of CRY mutation on cancer incidence and mortality in mouse strains with a predisposition to cancer. Kaplan–Meier plots of death from cancer are shown. (A) p53–/– (red) and p53–/–;Cry1/2–/– (green and blue) survival probabilities. Data shown by the green line have been published,71 and the unpublished data shown by the blue line were obtained by a different member of the lab in a blind experimental design. (B) Tumor-free survival of ink4a–/–;ras(V12G) (blue) and ink4a–/–;ras(V12G);Cry1/2–/– (red) mice. The activated HRAS gene is carried on the Y chromosome, and the experiment was conducted in male mice maintained under standard conditions of 12 h light–12 h dark cycles and monitored regularly for the appearance of melanomas. There is no statistical significance between the two survival curves (p = 0.2), and hence, it is concluded that in this genetic background Cry mutation has no mitigating effect on cancer incidence or progression.
i. CRY Inactivation as a Modality in Chemotherapy (intrinsic apoptosis)
To determine if the apoptosis enhanced by CRY mutation could be used in chemotherapy, xenograft experiments were performed.72 NOD/SCID mice were injected in different flanks with ras-transformed p53–/– or p53–/–;Cry1/2–/– cells, and tumor growth was followed (Figure 12A). Tumors of both cell types grew at the same rate, and when they reached a certain size, they were treated with oxaliplatin (Figure 12B,D). It was found that oxaliplatin had only marginal effects on p53–/– tumors but caused massive apoptosis and tumor regression in p53–/–;Cry1/2–/– tumors. Mechanistic analysis of this phenomenon revealed that in the absence of p53, p73 (of the p53, p63, p73 family) becomes the major driver of apoptosis and p73 is highly upregulated after DNA damage as a second-order clock-controlled gene. The p53 gene is mutated in ∼50% of cancers,99 and therefore, these findings along with the recent report of identification of small molecule CRY inhibitors100 would suggest that CRY inhibition might be applicable to cancer chemotherapy.
Figure 12.

Cry mutation increases the efficacy of cancer treatment by chemotherapy and immunotherapy. (A) Diagram of the NOD/SCID mouse bearing a p53–/– tumor xenograft on the left flank (one asterisk) and a p53–/–;Cry1/2–/– tumor xenograft on the right flank (two asterisks). (B and C) Effect of oxaliplatin and TNFα on tumor growth (metabolism). (B) In the chemotherapy experiment, following xenograft growth to 0.1 cm3 the mouse was treated with oxaliplatin. (C) In the case of the biotherapy experiment, the transformed cells were treated with TNFα before injection. Tumor metabolism was measured either 38 days (oxaliplatin) or 40 days (TNFα) after xenograft injection. Colored panels show micro PET scans, and charts at the right show quantification of the [18F]FDG signal as the ratio of the percent injected dose (%ID) within the circled area to the %ID in the background region. (D) Plot of tumor growth with red arrows indicating days of oxaliplatin injection following the xenograft injection on day 0.72 (E) Plot of tumor growth following injection of TNFα-treated transformed cells.
ii. CRY Inactivation as a Modality in Biotherapy (extrinsic apoptosis)
The finding that Cry1/2–/–;p53–/– mice developed tumors later than the p53–/– mice71 suggested that transformed cells of the Cry1/2–/–;p53–/– genotype were more sensitive to being eliminated by cytotoxic cytokines. Indeed, it was found that Cry1/2–/–;p53–/– cells were more sensitive to TNFα-induced extrinsic apoptosis than p53–/– cells.75 To determine if this selective sensitivity could be used as a therapeutic approach, p53–/–;ras(p24T) and Cry1/2–/–;p53–/–;ras(p24T) xenografts were treated with TNFα. It was found that the absence of CRY made tumors very sensitive to TNFα-induced apoptosis, and as a consequence, TNFα stopped the growth of the CRY mutant but not the control tumors (Figure 12C,E). Although TNFα is rather toxic and its systemic administration for cancer treatment is not practical, this finding may provide an explanation for slower tumor progression in Cry mutant p53–/– mice: TNFα is itself controlled by the clock,74 and in the absence of CRY, this cytokine would be overproduced and act on cells already sensitized to TNFα-induced apoptosis because of the reduced anti-apoptotic effect of NF-κB as a result of CRY mutation.
III. Perspective
The molecular circadian clock is integrated into all major transcriptional and signal transduction networks,2,101,102 and therefore, it is expected that the clock would influence and be influenced by essentially all major intracellular communication systems and affect physiological and pathophysiological events. Indeed, it is well-established that the clock affects the daily rhythmicity of physiological functions, such as sleep and mental and physical performance, and the time of occurrence of certain pathophysiological events, such as allergic reactions, including bronchial asthma and cardiovascular and cerebrovascular incidents.3,79,96,97
Generation of mouse strains with mutations in the core clock genes has made it possible to directly test the contribution of the molecular clock to the normal functioning at cellular and organismal levels. These studies have yielded some expected and some unexpected and rather surprising results. Thus, while Bmal1 and, to a lesser extent, Clock mutant mice exhibit premature aging phenotypes, and most clock gene mutations including Bmal1, Clock, Per, and Cry mutations have been reported to predispose mice to metabolic syndrome,101 it also appears that Clock, Cry, and Per mutant mice have essentially normal growth properties and normal life spans.80
With regard to coupling of the molecular clock to cell cycle and DNA damage checkpoints, while the coupling does exist, its contribution to normal cell physiology is limited and not essential. This is either because the coupling of the clock to these systems is weak or nonresonant with noncircadian pathways or because disruption of the molecular clock activates compensatory homeostatic adjustments in other molecular pathways and/or networks that mitigate the effect of clock disruption on cellular and organismic biology.
From the perspective of clock disruption as a causative factor in cancer, and circadian rhythmicity as a modality in cancer therapy, the following generalizations might be made. First, causality between clock disruption and cancer has not been convincingly demonstrated. It remains to be seen whether future research with more specific experimental designs will uncover such a connection. In contrast, it appears that disruption of the circadian clock in appropriate genetic backgrounds may actually interfere with cancer growth.71 Finally, it is clear that the nucleotide excision repair rate has a strong circadian component17−19 and that this feature may be used in developing more efficient chemotherapy regimens. In addition to excision repair, it has been reported that the gating of the cell cycle by the clock imposes a circadian pattern of hair follicle loss by ionizing radiation.103 Thus, it is conceivable that the efficiencies of both chemotherapy and radiation therapy alone or in combination (combination therapy) may be improved by appropriate circadian timing of delivery or by modulating the core circadian system.
Glossary
Abbreviations
- TTFL
transcription–translation feedback loop
- CRY
cryptochrome
- PER
period
- UV
ultraviolet
- cisplatin
cis-diamminedichloroplatinum(II)
- UPS
ubiquitin–proteasome system
- PIKK
phosphainositol 3-kinase-related kinase
- ATM
Ataxia Telangiectasia-Mutated
- ATR
ATM and Rad3-related
- TIM
Timeless
- TIPIN
Timeless interacting protein
- SCN
suprachiasmatic nucleus
- IR
ionizing radiation
- MSFs
mouse skin fibroblasts
- CCG
clock-controlled gene
- TNF
tumor necrosis factor
- ZT
zeitgeber time
- CT
circadian time
- HU
hydroxyurea.
Author Present Address
† J.H.L.: Department of Food and Biotechnology, Korea University, Sejong, Republic of Korea.
Author Present Address
‡ N.O.: Department of Molecular Biology and Genetics, Gebze Institute of Technology, 41400 Kocaeli, Turkey.
This work was supported by National Institutes of Health Grants GM31082 and GM32833 and by the National Science Council and Academia Sinica, Taiwan, ROC.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Partch C. L.; Green C. B.; Takahashi J. S. (2014) Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 24, 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahar S.; Sassone-Corsi P. (2009) Metabolism and cancer: The circadian clock connection. Nat. Rev. Cancer 9, 886–896. [DOI] [PubMed] [Google Scholar]
- Hastings M. H.; Reddy A. B.; Maywood E. S. (2003) A clockwork web: Circadian timing in brain and periphery, in health and disease. Nat. Rev. Neurosci. 4, 649–661. [DOI] [PubMed] [Google Scholar]
- Reppert S. M.; Weaver D. R. (2002) Coordination of circadian timing in mammals. Nature 418, 935–941. [DOI] [PubMed] [Google Scholar]
- Antoch M. P.; Kondratov R. V. (2013) Pharmacological modulators of the circadian clock as potential therapeutic drugs: Focus on genotoxic/anticancer therapy. Handb. Exp. Pharmacol. 289–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratov R. V.; Antoch M. P. (2007) Circadian proteins in the regulation of cell cycle and genotoxic stress responses. Trends Cell Biol. 17, 311–317. [DOI] [PubMed] [Google Scholar]
- Sancar A.; Lindsey-Boltz L. A.; Kang T. H.; Reardon J. T.; Lee J. H.; Ozturk N. (2010) Circadian clock control of the cellular response to DNA damage. FEBS Lett. 584, 2618–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown W. R. (1991) A review and mathematical analysis of circadian rhythms in cell proliferation in mouse, rat, and human epidermis. J. Invest. Dermatol. 97, 273–280. [DOI] [PubMed] [Google Scholar]
- Bjarnason G. A.; Jordan R. C.; Wood P. A.; Li Q.; Lincoln D. W.; Sothern R. B.; Hrushesky W. J.; Ben-David Y. (2001) Circadian expression of clock genes in human oral mucosa and skin: Association with specific cell-cycle phases. Am. J. Pathol. 158, 1793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaddameedhi S.; Selby C. P.; Kaufmann W. K.; Smart R. C.; Sancar A. (2011) Control of skin cancer by the circadian rhythm. Proc. Natl. Acad. Sci. U.S.A. 108, 18790–18795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyfman M.; Kumar V.; Liu Q.; Ruiz R.; Gordon W.; Espitia F.; Cam E.; Millar S. E.; Smyth P.; Ihler A.; Takahashi J. S.; Andersen B. (2012) Brain and muscle Arnt-like protein-1 (BMAL1) controls circadian cell proliferation and susceptibility to UVB-induced DNA damage in the epidermis. Proc. Natl. Acad. Sci. U.S.A. 109, 11758–11763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beland F. A.; Dooley K. L.; Sheldon W. G.; Delongchamp R. R. (1988) Circadian variation in the induction of intestinal tumors by N-methyl-N-nitrosourea in male C57BL/6N mice. J. Natl. Cancer Inst. 80, 325–330. [DOI] [PubMed] [Google Scholar]
- Gorbacheva V. Y.; Kondratov R. V.; Zhang R.; Cherukuri S.; Gudkov A. V.; Takahashi J. S.; Antoch M. P. (2005) Circadian sensitivity to the chemotherapeutic agent cyclophosphamide depends on the functional status of the CLOCK/BMAL1 transactivation complex. Proc. Natl. Acad. Sci. U.S.A. 102, 3407–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoch M. P.; Kondratov R. V.; Takahashi J. S. (2005) Circadian clock genes as modulators of sensitivity to genotoxic stress. Cell Cycle 4, 901–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberg F. (1960) Temporal coordination of physiologic function. Cold Spring Harbor Symp. Quant. Biol. 25, 289–310. [DOI] [PubMed] [Google Scholar]
- Sancar A. (2004) Regulation of the mammalian circadian clock by cryptochrome. J. Biol. Chem. 279, 34079–34082. [DOI] [PubMed] [Google Scholar]
- Kang T. H.; Reardon J. T.; Kemp M.; Sancar A. (2009) Circadian oscillation of nucleotide excision repair in mammalian brain. Proc. Natl. Acad. Sci. U.S.A. 106, 2864–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T. H.; Lindsey-Boltz L. A.; Reardon J. T.; Sancar A. (2010) Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 107, 4890–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T. H.; Reardon J. T.; Sancar A. (2011) Regulation of nucleotide excision repair activity by transcriptional and post-transcriptional control of the XPA protein. Nucleic Acids Res. 39, 3176–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrich P. (2006) Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 281, 30305–30309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Filippo J.; Sung P.; Klein H. (2008) Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 77, 229–257. [DOI] [PubMed] [Google Scholar]
- Sancar A.; Lindsey-Boltz L. A.; Unsal-Kacmaz K.; Linn S. (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85. [DOI] [PubMed] [Google Scholar]
- Kraemer K. H.; Lee M. M.; Scotto J. (1984) DNA repair protects against cutaneous and internal neoplasia: Evidence from xeroderma pigmentosum. Carcinogenesis 5, 511–514. [DOI] [PubMed] [Google Scholar]
- Kemp M. G.; Reardon J. T.; Lindsey-Boltz L. A.; Sancar A. (2012) Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J. Biol. Chem. 287, 22889–22899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.; Choi J. H.; Gaddameedhi S.; Kemp M. G.; Reardon J. T.; Sancar A. (2013) Nucleotide excision repair in human cells: Fate of the excised oligonucleotide carrying DNA damage in vivo. J. Biol. Chem. 288, 20918–20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto Y.; Sancar A. (1999) Circadian regulation of cryptochrome genes in the mouse. Brain Res. 71, 238–243. [DOI] [PubMed] [Google Scholar]
- Nagoshi E.; Brown S. A.; Dibner C.; Kornmann B.; Schibler U. (2005) Circadian gene expression in cultured cells. Methods Enzymol. 393, 543–557. [DOI] [PubMed] [Google Scholar]
- Balsalobre A.; Damiola F.; Schibler U. (1998) A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 93, 929–937. [DOI] [PubMed] [Google Scholar]
- Fu L.; Pelicano H.; Liu J.; Huang P.; Lee C. (2002) The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 111, 41–50. [DOI] [PubMed] [Google Scholar]
- Hughes M. E.; DiTacchio L.; Hayes K. R.; Vollmers C.; Pulivarthy S.; Baggs J. E.; Panda S.; Hogenesch J. B. (2009) Harmonics of circadian gene transcription in mammals. PLoS Genet. 5, e1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring W.; Rosbash M. (2003) The coevolution of blue-light photoreception and circadian rhythms. J. Mol. Evol. 57(Suppl. 1), S286–S289. [DOI] [PubMed] [Google Scholar]
- Pittendrigh C. S. (1993) Temporal organization: Reflections of a Darwinian clock-watcher. Annu. Rev. Physiol. 55, 16–54. [DOI] [PubMed] [Google Scholar]
- Sancar A. (2003) Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem. Rev. 103, 2203–2237. [DOI] [PubMed] [Google Scholar]
- Hartwell L. H.; Weinert T. A. (1989) Checkpoints: Controls that ensure the order of cell cycle events. Science 246, 629–634. [DOI] [PubMed] [Google Scholar]
- Paulovich A. G.; Hartwell L. H. (1995) A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell 82, 841–847. [DOI] [PubMed] [Google Scholar]
- Johnson C. H. (2010) Circadian clocks and cell division: What’s the pacemaker?. Cell Cycle 9, 3864–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T.; Binder B.; Johnson C. H. (1996) Circadian gating of cell division in cyanobacteria growing with average doubling times of less than 24 h. Proc. Natl. Acad. Sci. U.S.A. 93, 10183–10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido S. S.; Johnson C. H. (2000) Daily and circadian variation in survival from ultraviolet radiation in Chlamydomonas reinhardtii. Photochem. Photobiol. 71, 758–765. [DOI] [PubMed] [Google Scholar]
- Grechez-Cassiau A.; Rayet B.; Guillaumond F.; Teboul M.; Delaunay F. (2008) The circadian clock component BMAL1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J. Biol. Chem. 283, 4535–4542. [DOI] [PubMed] [Google Scholar]
- Laranjeiro R.; Tamai T. K.; Peyric E.; Krusche P.; Ott S.; Whitmore D. (2013) Cyclin-dependent kinase inhibitor p20 controls circadian cell-cycle timing. Proc. Natl. Acad. Sci. U.S.A. 110, 6835–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalska E.; Ripperger J. A.; Hoegger D. C.; Bruegger P.; Buch T.; Birchler T.; Mueller A.; Albrecht U.; Contaldo C.; Brown S. A. (2013) NONO couples the circadian clock to the cell cycle. Proc. Natl. Acad. Sci. U.S.A. 110, 1592–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo T.; Yamaguchi S.; Mitsui S.; Emi A.; Shimoda F.; Okamura H. (2003) Control mechanism of the circadian clock for timing of cell division in vivo. Science 302, 255–259. [DOI] [PubMed] [Google Scholar]
- Nagoshi E.; Saini C.; Bauer C.; Laroche T.; Naef F.; Schibler U. (2004) Circadian gene expression in individual fibroblasts: Cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell 119, 693–705. [DOI] [PubMed] [Google Scholar]
- Feillet C.; Krusche P.; Tamanini F.; Janssens R. C.; Downey M. J.; Martin P.; Teboul M.; Saito S.; Levi F. A.; Bretschneider T.; van der Horst G. T.; Delaunay F.; Rand D. A. (2014) Phase locking and multiple oscillating attractors for the coupled mammalian clock and cell cycle. Proc. Natl. Acad. Sci. U.S.A. 111, 9828–9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeom M.; Pendergast J. S.; Ohmiya Y.; Yamazaki S. (2010) Circadian-independent cell mitosis in immortalized fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 107, 9665–9670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsal-Kacmaz K.; Mullen T. E.; Kaufmann W. K.; Sancar A. (2005) Coupling of human circadian and cell cycles by the timeless protein. Mol. Cell. Biol. 25, 3109–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotter A. L.; Manganaro T.; Weaver D. R.; Kolakowski L. F. Jr.; Possidente B.; Sriram S.; MacLaughlin D. T.; Reppert S. M. (2000) A time-less function for mouse timeless. Nat. Neurosci. 3, 755–756. [DOI] [PubMed] [Google Scholar]
- Benna C.; Scannapieco P.; Piccin A.; Sandrelli F.; Zordan M.; Rosato E.; Kyriacou C. P.; Valle G.; Costa R. (2000) A second timeless gene in Drosophila shares greater sequence similarity with mammalian tim. Curr. Biol. 10, R512–R513. [DOI] [PubMed] [Google Scholar]
- Xiao J.; Li C.; Zhu N. L.; Borok Z.; Minoo P. (2003) Timeless in lung morphogenesis. Dev. Dyn. 228, 82–94. [DOI] [PubMed] [Google Scholar]
- Unsal-Kacmaz K.; Chastain P. D.; Qu P. P.; Minoo P.; Cordeiro-Stone M.; Sancar A.; Kaufmann W. K. (2007) The human Tim/Tipin complex coordinates an intra-S checkpoint response to UV that slows replication fork displacement. Mol. Cell. Biol. 27, 3131–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotter A. L.; Suppa C.; Emanuel B. S. (2007) Mammalian TIMELESS and Tipin are evolutionarily conserved replication fork-associated factors. J. Mol. Biol. 366, 36–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylka M. J.; Shearman L. P.; Levine J. D.; Jin X.; Weaver D. R.; Reppert S. M. (1998) Molecular analysis of mammalian timeless. Neuron 21, 1115–1122. [DOI] [PubMed] [Google Scholar]
- Barnes J. W.; Tischkau S. A.; Barnes J. A.; Mitchell J. W.; Burgoon P. W.; Hickok J. R.; Gillette M. U. (2003) Requirement of mammalian Timeless for circadian rhythmicity. Science 302, 439–442. [DOI] [PubMed] [Google Scholar]
- Engelen E.; Janssens R. C.; Yagita K.; Smits V. A.; van der Horst G. T.; Tamanini F. (2013) Mammalian TIMELESS is involved in period determination and DNA damage-dependent phase advancing of the circadian clock. PLoS One 8, e56623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi E.; Noguchi C.; McDonald W. H.; Yates J. R. III; Russell P. (2004) Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol. Cell. Biol. 24, 8342–8355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty B. K.; Bairwa N. K.; Bastia D. (2006) The Tof1p-Csm3p protein complex counteracts the Rrm3p helicase to control replication termination of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 103, 897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotter A. L. (2003) Tipin, a novel timeless-interacting protein, is developmentally co-expressed with timeless and disrupts its self-association. J. Mol. Biol. 331, 167–176. [DOI] [PubMed] [Google Scholar]
- Kang T. H.; Leem S. H. (2014) Modulation of ATR-mediated DNA damage checkpoint response by cryptochrome 1. Nucleic Acids Res. 42, 4427–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaddameedhi S.; Reardon J. T.; Ye R.; Ozturk N.; Sancar A. (2012) Effect of circadian clock mutations on DNA damage response in mammalian cells. Cell Cycle 11, 3481–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan R. C.; Chan A.; Jeon M.; Wu T. F.; Pasqualone D.; Rougvie A. E.; Meyer B. J. (2003) Chromosome cohesion is regulated by a clock gene paralogue TIM-1. Nature 423, 1002–1009. [DOI] [PubMed] [Google Scholar]
- Benna C.; Bonaccorsi S.; Wulbeck C.; Helfrich-Forster C.; Gatti M.; Kyriacou C. P.; Costa R.; Sandrelli F. (2010) Drosophila timeless2 is required for chromosome stability and circadian photoreception. Curr. Biol. 20, 346–352. [DOI] [PubMed] [Google Scholar]
- Gery S.; Komatsu N.; Baldjyan L.; Yu A.; Koo D.; Koeffler H. P. (2006) The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol. Cell 22, 375–382. [DOI] [PubMed] [Google Scholar]
- Oishi K.; Miyazaki K.; Kadota K.; Kikuno R.; Nagase T.; Atsumi G.; Ohkura N.; Azama T.; Mesaki M.; Yukimasa S.; Kobayashi H.; Iitaka C.; Umehara T.; Horikoshi M.; Kudo T.; Shimizu Y.; Yano M.; Monden M.; Machida K.; Matsuda J.; Horie S.; Todo T.; Ishida N. (2003) Genome-wide expression analysis of mouse liver reveals CLOCK-regulated circadian output genes. J. Biol. Chem. 278, 41519–41527. [DOI] [PubMed] [Google Scholar]
- Cotta-Ramusino C.; McDonald E. R. III; Hurov K.; Sowa M. E.; Harper J. W.; Elledge S. J. (2011) A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science 332, 1313–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato F.; Wu Y.; Bhawal U. K.; Liu Y.; Imaizumi T.; Morohashi S.; Kato Y.; Kijima H. (2011) PERIOD1 (PER1) has anti-apoptotic effects, and PER3 has pro-apoptotic effects during cisplatin (CDDP) treatment in human gingival cancer CA9-22 cells. Eur. J. Cancer 47, 1747–1758. [DOI] [PubMed] [Google Scholar]
- Sato F.; Nagata C.; Liu Y.; Suzuki T.; Kondo J.; Morohashi S.; Imaizumi T.; Kato Y.; Kijima H. (2009) PERIOD1 is an anti-apoptotic factor in human pancreatic and hepatic cancer cells. J. Biochem. 146, 833–838. [DOI] [PubMed] [Google Scholar]
- Geusz M. E.; Blakely K. T.; Hiler D. J.; Jamasbi R. J. (2010) Elevated mPer1 gene expression in tumor stroma imaged through bioluminescence. Int. J. Cancer 126, 620–630. [DOI] [PubMed] [Google Scholar]
- Hotchkiss R. S.; Strasser A.; McDunn J. E.; Swanson P. E. (2009) Cell death. N. Engl. J. Med. 361, 1570–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiewe T. (2007) The p53 family in differentiation and tumorigenesis. Nat. Rev. Cancer 7, 165–168. [DOI] [PubMed] [Google Scholar]
- Yu J.; Baron V.; Mercola D.; Mustelin T.; Adamson E. D. (2007) A network of p73, p53 and Egr1 is required for efficient apoptosis in tumor cells. Cell Death Differ. 14, 436–446. [DOI] [PubMed] [Google Scholar]
- Ozturk N.; Lee J. H.; Gaddameedhi S.; Sancar A. (2009) Loss of cryptochrome reduces cancer risk in p53 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 106, 2841–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H.; Sancar A. (2011) Circadian clock disruption improves the efficacy of chemotherapy through p73-mediated apoptosis. Proc. Natl. Acad. Sci. U.S.A. 108, 10668–10672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H.; Gaddameedhi S.; Ozturk N.; Ye R.; Sancar A. (2013) DNA damage-specific control of cell death by cryptochrome in p53-mutant ras-transformed cells. Cancer Res. 73, 785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiramoto A.; Yamane T.; Tsumiyama K.; Yoshida K.; Komai K.; Yamada H.; Yamazaki F.; Doi M.; Okamura H.; Shiozawa S. (2010) Mammalian Clock Gene Cryptochrome Regulates Arthritis via Proinflammatory Cytokine TNF-α. J. Immunol. 184, 1560–1565. [DOI] [PubMed] [Google Scholar]
- Lee J. H.; Sancar A. (2011) Regulation of apoptosis by the circadian clock through NF-κB signaling. Proc. Natl. Acad. Sci. U.S.A. 108, 12036–12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spengler M. L.; Kuropatwinski K. K.; Comas M.; Gasparian A. V.; Fedtsova N.; Gleiberman A. S.; Gitlin I. I.; Artemicheva N. M.; Deluca K. A.; Gudkov A. V.; Antoch M. P. (2012) Core circadian protein CLOCK is a positive regulator of NF-κB-mediated transcription. Proc. Natl. Acad. Sci. U.S.A. 109, E2457–E2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oklejewicz M.; Destici E.; Tamanini F.; Hut R. A.; Janssens R.; van der Horst G. T. (2008) Phase resetting of the mammalian circadian clock by DNA damage. Curr. Biol. 18, 286–291. [DOI] [PubMed] [Google Scholar]
- Baggs J. E.; Price T. S.; DiTacchio L.; Panda S.; Fitzgerald G. A.; Hogenesch J. B. (2009) Network features of the mammalian circadian clock. PLoS Biol. 7, e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi F.; Schibler U. (2007) Circadian rhythms: Mechanisms and therapeutic implications. Annu. Rev. Pharmacol. Toxicol. 47, 593–628. [DOI] [PubMed] [Google Scholar]
- Yu E. A.; Weaver D. R. (2011) Disrupting the circadian clock: Gene-specific effects on aging, cancer, and other phenotypes. Aging 3, 479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schernhammer E. S.; Kroenke C. H.; Laden F.; Hankinson S. E. (2006) Night work and risk of breast cancer. Epidemiology 17, 108–111. [DOI] [PubMed] [Google Scholar]
- Stevens R. G. (2009) Light-at-night, circadian disruption and breast cancer: Assessment of existing evidence. Int. J. Epidemiol. 38, 963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise J. (2009) Danish night shift workers with breast cancer awarded compensation. BMJ [Br. Med. J.] 338, b1152. [DOI] [PubMed] [Google Scholar]
- Stevens R. G.; Brainard G. C.; Blask D. E.; Lockley S. W.; Motta M. E. (2013) Adverse health effects of nighttime lighting: Comments on American Medical Association policy statement. Am. J. Prev. Med. 45, 343–346. [DOI] [PubMed] [Google Scholar]
- Kantermann T.; Roenneberg T. (2009) Is light-at-night a health risk factor or a health risk predictor?. Chronobiol. Int. 26, 1069–1074. [DOI] [PubMed] [Google Scholar]
- Pronk A.; Ji B. T.; Shu X. O.; Xue S.; Yang G.; Li H. L.; Rothman N.; Gao Y. T.; Zheng W.; Chow W. H. (2010) Night-shift work and breast cancer risk in a cohort of Chinese women. Am. J. Epidemiol. 171, 953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ijaz S.; Verbeek J.; Seidler A.; Lindbohm M. L.; Ojajarvi A.; Orsini N.; Costa G.; Neuvonen K. (2013) Night-shift work and breast cancer: A systematic review and meta-analysis. Scand. J. Work, Environ. Health 39, 431–447. [DOI] [PubMed] [Google Scholar]
- Stevens R. G.; Hansen J.; Schernhammer E. S.; Davis S. (2013) Response to Ijaz S, et al. “Night-shift work and breast cancer: A systematic review and meta-analysis”. Scand. J. Work, Environ. Health 39, 631–632. [DOI] [PubMed] [Google Scholar]
- Filipski E.; King V. M.; Li X.; Granda T. G.; Mormont M. C.; Liu X.; Claustrat B.; Hastings M. H.; Levi F. (2002) Host circadian clock as a control point in tumor progression. J. Natl. Cancer Inst. 94, 690–697. [DOI] [PubMed] [Google Scholar]
- Antoch M. P.; Gorbacheva V. Y.; Vykhovanets O.; Toshkov I. A.; Kondratov R. V.; Kondratova A. A.; Lee C.; Nikitin A. Y. (2008) Disruption of the circadian clock due to the Clock mutation has discrete effects on aging and carcinogenesis. Cell Cycle 7, 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauger M. A.; Sancar A. (2005) Cryptochrome, circadian cycle, cell cycle checkpoints, and cancer. Cancer Res. 65, 6828–6834. [DOI] [PubMed] [Google Scholar]
- Antoch M. P.; Toshkov I.; Kuropatwinski K. K.; Jackson M. (2013) Deficiency in PER proteins has no effect on the rate of spontaneous and radiation-induced carcinogenesis. Cell Cycle 12, 3673–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khapre R. V.; Kondratova A. A.; Patel S.; Dubrovsky Y.; Wrobel M.; Antoch M. P.; Kondratov R. V. (2014) BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging 6, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopalle H. M.; Partch C. L. (2014) An imPERfect link to cancer?. Cell Cycle 13, 507. [DOI] [PubMed] [Google Scholar]
- Hrushesky W. J. (1985) Circadian timing of cancer chemotherapy. Science 228, 73–75. [DOI] [PubMed] [Google Scholar]
- Paschos G. K.; Baggs J. E.; Hogenesch J. B.; FitzGerald G. A. (2010) The role of clock genes in pharmacology. Annu. Rev. Pharmacol. Toxicol. 50, 187–214. [DOI] [PubMed] [Google Scholar]
- Dallmann R.; Brown S. A.; Gachon F. (2014) Chronopharmacology: New insights and therapeutic implications. Annu. Rev. Pharmacol. Toxicol. 54, 339–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless N. E.; Kannan K.; Xu J.; Bosenberg M. W.; Chin L. (2003) Both products of the mouse Ink4a/Arf locus suppress melanoma formation in vivo. Oncogene 22, 5055–5059. [DOI] [PubMed] [Google Scholar]
- Attardi L. D.; Jacks T. (1999) The role of p53 in tumour suppression: Lessons from mouse models. Cell. Mol. Life Sci. 55, 48–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S. K.; Jang J.; Chung S.; Yun H.; Kim N. J.; Jung J. W.; Son G. H.; Suh Y. G.; Kim K. (2014) Identification and validation of cryptochrome inhibitors that modulate the molecular circadian clock. ACS Chem. Biol. 9, 703–710. [DOI] [PubMed] [Google Scholar]
- Green C. B.; Takahashi J. S.; Bass J. (2008) The meter of metabolism. Cell 134, 728–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan S. D.; Lamia K. A. (2013) AMPK at the crossroads of circadian clocks and metabolism. Mol. Cell. Endocrinol. 366, 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plikus M. V.; Vollmers C.; de la Cruz D.; Chaix A.; Ramos R.; Panda S.; Chuong C. M. (2013) Local circadian clock gates cell cycle progression of transient amplifying cells during regenerative hair cycling. Proc. Natl. Acad. Sci. U.S.A. 110, E2106–E2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. C.; Svoboda D. L.; Reardon J. T.; Sancar A. (1992) Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5′ and the 6th phosphodiester bond 3′ to the photodimer. Proc. Natl. Acad. Sci. U.S.A. 89, 3664–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]