

Abstract
Electrochemical DNA-based (E-DNA) sensors are utilized to detect a variety of targets including complementary DNA, small molecules, and proteins. These sensors typically employ surface-bound single-stranded oligonucleotides that are modified with a redox-active molecule on the distal 3′ terminus. Target-induced flexibility changes of the DNA probe alter the efficiency of electron transfer between the redox active methylene blue and the electrode surface, allowing for quantitative detection of target concentration. While numerous studies have utilized the specific and sensitive abilities of E-DNA sensors to quantify target concentration, no studies to date have demonstrated the ability of this class of collision-based sensors to elucidate biochemical-binding mechanisms such as cooperativity. In this study, we demonstrate that E-DNA sensors fabricated with various lengths of surface-bound oligodeoxythymidylate [(dT)n] sensing probes are able to quantitatively distinguish between cooperative and noncooperative binding of a single-stranded DNA-binding protein. Specifically, we demonstrate that oligo(dT) E-DNA sensors are able to quantitatively detect nM levels (50 nM–4 μM) of gene 32 protein (g32p). Furthermore, the sensors exhibit signal that is able to distinguish between the cooperative binding of the full-length g32p and the noncooperative binding of the core domain (*III) fragment to single-stranded DNA. Finally, we demonstrate that this binding is both probe-length- and ionic-strength-dependent. This study illustrates a new quantitative property of this powerful class of biosensor and represents a rapid and simple methodology for understanding protein–DNA binding mechanisms.
Introduction
Electrochemical-based sensors that utilize nucleic acids as recognition elements enable the detection of a wide range of biologically relevant target analytes.1−17 These sensors typically comprise electrode-appended short-chain nucleic acids that are modified at the distal end with a redox-active reporter molecule.18 Using this strategy, several reports demonstrate the detection of complementary DNA targets,2,6,9,10,15 single- and double-stranded DNA binding proteins,16 and triplex-forming oligonucleotides.8 The use of specific, target-binding sequences, e.g. aptamers, enables an even broader range of target detection including proteins7,12,13 and small molecules.1,3−5,11,12,17
The signaling mechanism of electrochemical, DNA-based (E-DNA) sensors affords their excellent sensitivity, specificity, and selectivity. Signaling relies on target-binding-induced changes in the flexibility and/or conformation of the electrode-appended nucleic acid probe.19−21 These changes alter the efficiency with which the covalently linked redox marker can be oxidized or reduced at the interrogating electrode surface. The voltammetric peak current associated with the reduction and oxidation of the redox marker is used to quantify the amount of target analyte present. To date, E-DNA sensors have achieved detection down to pM levels of DNA,2 pM levels of protein,7 and low μM levels of small molecule targets.1,11 Additionally, the nature of the nucleic acid probe and target interaction (e.g., DNA–DNA, DNA-binding protein–DNA, and target–aptamer binding interactions) allows for specific analyte detection. Finally, by combining the specific recognition abilities of the DNA probes and the selectivity of the electrochemical measurement, E-DNA sensors have been shown to function when employed in complex environments.3,5,7,13,17,22
E-DNA sensors have emerged as a promising biosensor device in the detection and quantification of target analytes; however, they have seen little use in elucidating information about the biomolecular interactions occurring upon analyte binding. Furthermore, while several reports have described the utility of alternative DNA-modified electrode platforms for studying DNA–protein binding interactions,23−28 to the best of the authors’ knowledge, this is the first report of E-DNA sensors used for discerning cooperative DNA-protein binding. Here, we demonstrate the utility of the E-DNA sensing platform toward the investigation of cooperative and noncooperative binding of proteins. Using a simple E-DNA sensor, we show that this class of sensor is able to distinguish between cooperative and noncooperative binding by monitoring the interactions between oligodeoxythymidylate [(dT)n] DNA probes of various lengths and the single-strand DNA binding protein bacteriophage T4 gene 32 protein (g32p).29,30 Using the full-length protein, which binds cooperatively to single-strand DNA,29 and a truncated core domain (*III)30 fragment that binds noncooperatively to DNA,29−31 the oligo(dT) E-DNA sensors are able to quantify target concentrations down to nM levels and are able to qualitatively and quantitatively distinguish between the two binding interactions. This new ability adds to the growing analytical capabilities of electrochemical, DNA-based sensors and may represent a relatively rapid and simple methodology in investigating DNA–binding protein interactions.
Materials and Methods
Chemicals
6-Mercapto-1-hexanol, tris(2-carboxyethyl)phosphine hydrochloride, tris base, sodium chloride, magnesium chloride, tris-hydrocholoric acid, ethylenediaminetetraacetic acid, glycerol, β-mercaptoethanol, and hexaammineruthenium(III) chloride (98%) were all purchased from Sigma-Aldrich and were used as received. Tris buffer (100 mM NaCl, 20 mM Tris, 5 mM MgCl2, pH = 7.4) was used in the preparation of the sensors, while protein storage buffer (20 mM Tris-HCl, pH 8.1, 1 mM EDTA, 10% glycerol, 1 mM β-mercaptoethanol, 20 mM NaCl) was used in the electrochemical measurements unless otherwise specified. The (dT)n DNA probes (HPLC-purified, Biosearch Technologies, Inc., Novato, CA), modified with thiol on the 5′ terminus and redox active methylene blue on the 3′ end, were diluted with 1X Tris-EDTA buffer (pH 8.1) and were used as is without further purification. Full-length bacteriophage T4 gene 32 protein (g32p) and the core domain (*III) fragment were isolated, purified, and characterized using previously described methods.29,32−34 Stock solutions of purified g32p (20 μM) and *III (60 μM) protein were used without further purification. The concentrations of the intact and core domain proteins were determined spectrophotometrically (ε280 = 3.7 × 104 M–1 cm–1).29,35 Bovine thrombin (Akron Biotech) was generously provided by Dr. Minjoung Kyoung (UMBC) and used as received. The concentration of the stock thrombin solution (20 μM) was determined using its absorbance at 280 nm (ε280 = 1.95 × 103 M–1 cm–1).
Sensor Fabrication
Electrochemical DNA sensors (oligo(dT) E-DNA) were fabricated on 2 mm polycrystalline gold rod electrodes (CH Instruments, Austin, TX) using a previously described method.18 Briefly, the electrodes were mechanically polished in a microcloth using a small amount of monocrystalline diamond suspension in water (Buehler, Lake Bluff, IL) and were sonicated in ultrapure water for 5 min. The electrodes were then rinsed with ultrapure water, followed by polishing in a micropolish containing aluminum oxide in water (Buehler, Lake Bluff, IL). Finally, the electrodes were thoroughly rinsed and sonicated in ultrapure water for 5 min. Electrochemical cleaning of the polished electrodes was done by first scanning in 0.5 M NaOH to reduce and remove any sulfur molecules linked to the gold surface, followed by a series of oxidation–reduction scans in 0.5 M H2SO4 to remove any organic contaminants while forming and reducing the gold oxide layer. The chloride etching step is then performed in 0.1 M H2SO4/0.01 M KCl, followed by scanning in 0.05 M H2SO4 to completely reduce the gold oxide monolayer on the surface, and allows for an estimation of the electrode area.
Sensor surface modification was performed using the “insertion” method recently described by Josephs and Ye.36 Briefly, the clean electrodes were thoroughly rinsed with ultrapure water and incubated in 3 mM 6-mercapto-1-hexanol (in Tris Buffer) for 1 h to allow for the gold–thiol monolayer to form at the electrode surface. While the electrodes were being incubated in mercaptohexanol, the oligodeoxythymidylate (e.g., (dT)7, (dT)14, and (dT)21) DNA probes were simultaneously incubated in 10 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) for 1 h to reduce any disulfide linkages that may be present in the DNA probe solution arising from the synthesis of the thiol-modified DNA probes. Following incubation, the electrodes were immersed in the solution containing the reduced oligo(dT) DNA probes (diluted in Tris buffer to 500 nM, the concentration of which was determined spectrophotometrically at 260 nm) for 1 h, allowing the DNA to anchor itself onto the surface of the electrodes by “inserting” into the “defect” sites of the mercaptohexanol monolayer.36 Finally, the sensors were thoroughly washed with ultrapure water and stored in protein storage buffer prior to titration.
Electrochemical Measurements and Data Analysis
All titrations were performed using square wave voltammetry in a three-electrode setup (Ag/AgCl reference electrode, platinum wire counter electrode) on a CH Instruments 620D Electrochemical Workstation (CH Instruments, Austin, TX), employing a step potential of 0.001 V, an amplitude of 0.025 V, and a frequency of 500 Hz. All titrations were carried out in a 3 mL glass cell using the protein storage buffer unless otherwise stated. Data analysis was carried out utilizing the voltammetric peak currents recorded for the reversible oxidation–reduction of methylene blue, as a result of target-induced conformational changes within the olido(dT) DNA probes. The varying voltammeric responses were expressed as % signal changes ± SD and were plotted against the concentration of target. All titrations and sensor measurements were performed using at least three fabricated sensors. The error bars reported represent the standard deviation of measurements made on each sensor and, thus, represent sensor-to-sensor variability. Surface coverage measurements were performed using the well-established chronocoulometric method of electrostatically associated hexaamine ruthenium described by Steel et al.37 Briefly, probe surface density was quantified by calculating the number of cationic redox molecules that were electrostatically trapped at the anionic DNA backbone, which is directly proportional to the amount of phosphate molecules (hence, DNA) on the gold electrode surface using a 500 μM hexaammineruthenium(III) chloride solution in 10 mM Tris, pH = 7.3. The values were expressed as probe surface density ± SD in units of molecules/cm2 and were plotted for each electrode, as detailed in the Supporting Information (Figure S1).
Results and Discussion
To demonstrate the ability of the E-DNA sensor platform to monitor cooperative binding, we utilized a representative, well-characterized DNA-binding protein–bacteriophage T4-coded gene 32 protein (g32p). This protein is involved in DNA replication, recombination, and repair.30,34,38 The 33.5 kDa protein comprises 301 amino acid residues that are classified into three domains: the N-domain (residues 1–21), the core domain (residues 22–253), and the C-domain (residues 254–301).30,34 The full-length g32p has a (calculated) isoelectric point (pI) of 4.8; however, the isoelectric point of the protein by itself does not generally relate to its DNA-binding activity, or the electrostatic contribution to this activity. Critical is the charge of the binding surface, which typically represents only a small part of the protein’s exterior. Although the pI of the full-length protein includes the contribution of the acidic C-domain (pI = 3.6), even the core domain (*III), the single-stranded nucleic acid binding domain, is moderately acidic (pI = 5.4). If one examines the surface of *III, there are only two places where one finds a strongly positive electrostatic potential: a narrow cleft within which ssDNA binds and a section adjacent to the cleft.34 As such, full-length gene 32 protein binds single-strand nucleic acids, occluding ∼7 nucleotide residues per g32p monomer (n)31,35 and does so cooperatively, as a result of interactions between the N-terminal domain of one protein and the core domain on the adjacent protein.29−31 Cooperativity is dependent on the presence of the N-terminal domain, as the truncate with only the C-domain removed binds cooperatively, whereas removal of the N-domain destroys cooperativity.31 Deletion of both the N- and C-termini regions creates a core domain (*III) fragment (lacking cooperativity) with a binding site size (n) reduced by about one to two nucleotide residues (n = ∼5.5) relative to the full-length g32p.39 Because the interactions between g32p and single-strand DNA (ssDNA) are well characterized, the full-length g32p and the *III cleavage product represent good model proteins to study cooperative and noncooperative binding using the E-DNA sensing platform.
As model E-DNA sensors, we fabricated sensors using oligo(dT) DNA strands of various lengths ((dT)7, (dT)14, and (dT)21; Scheme 1). These DNA probe strands are expected to be free of any secondary structure and should be relatively flexible. The E-DNA sensors were fabricated using the “insertion” method recently described by Josephs and Ye,36 as opposed to the “backfill” method commonly employed in fabricating this type of sensor.18 Josephs and Ye demonstrate that the “insertion” method leads to a more uniform distribution of DNA molecules on the sensor surface in contrast to DNA aggregates or islands. Characterization of the packing density of surfaces prepared with (dT)21 probes using both methods demonstrates that the “insertion” method consistently and reproducibly yields lower DNA packing density (Supporting Information Figure S1). Specifically, with a constant DNA concentration (500 nM) during the fabrication process, we find that the “backfill” method yields packing densities of 9.72 × 1012 ± 5.70 × 1011 molecules/cm2 while the “insertion” method yields surfaces with 2.11 × 1012 ± 1.36 × 1011 molecules/cm2. Moreover, we find that sensors fabricated using the “insertion” method yield much more reproducible results with lower apparent binding affinities (KDapp) when compared to those fabricated using the “backfill” method as indicated by the sensor-to-sensor reproducibility (Supporting Information Figure S2).
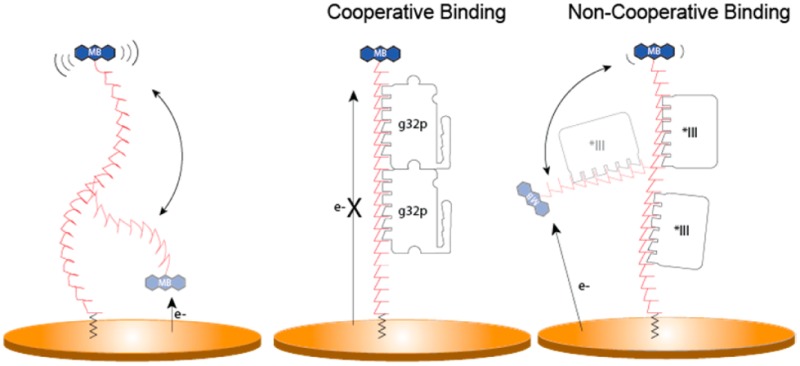
Scheme 1. E-DNA Sensors with Oligodeoxythymidylate [(dT)n] DNA Probes Exhibit a Decrease in the Current Signal When a Target g32 Protein Is Bound.
Oligo(dT) E-DNA sensors comprise a sensing electrode modified with single-stranded, unstructured oligo(dT), [(dT)n], DNA strands of various lengths (7, 14, and 21 nucleotides) modified at their distal ends with a redox-active methylene blue. Protein binding to the oligo(dT) E-DNA sensor causes a change in the flexibility of the (dT)n probe; thus, the efficiency with which electrons can be transferred results in a decrease in the measured current (signal-off).
Electrochemical DNA Sensors Specifically and Quantitatively Respond to g32p
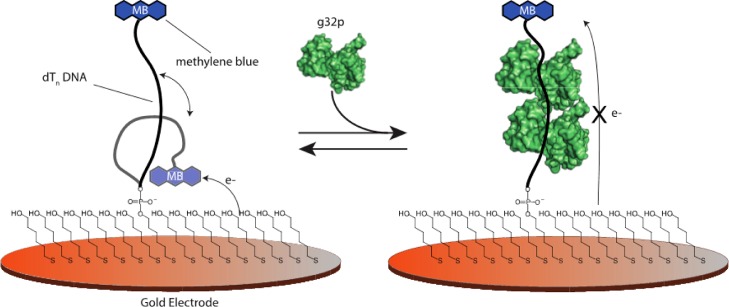
E-DNA sensors fabricated with varying lengths of oligo(dT) DNA respond specifically to full-length g32p binding in a “signal-off” manner. To test the detection abilities of the E-DNA sensor, we fabricated sensors using a (dT)21 DNA probe sequence. The (dT)21 probes were modified with a thiol at the 5′-terminus and with a redox-active methylene blue at the 3′-terminal for electrochemical signaling (Scheme 1). In the absence of a g32p target, the redox-labeled (dT)21 DNA probe is relatively flexible, allowing for efficient electron transfer between the methylene blue and the gold electrode surface. This is confirmed via the observation of a relatively high voltammetric peak current for the reversible reduction of methylene blue as characterized using square wave voltammetry (Figure 1). Conversely, when a saturating amount of full-length g32p is added (4 μM, determined vide infra), binding of g32p to the single-strand DNA causes a readily measurable decrease in the peak current after a ∼30 min equilibration time (Figure 1). The bound ∼33.5 kDa protein decreases the flexibility and, thus, the collision rate, of the (dT)21 DNA probe, which in turn reduces the efficiency of electron transfer between methylene blue and the interrogating gold electrode surface. Because target binding results in a decrease in the measured signal, this sensor is classified as a “signal-off” sensor.15
Figure 1.

Oligo(dT) E-DNA sensor specifically responds to the presence of gene 32 protein (g32p) in solution. E-DNA sensors modified with (dT)21 probe are employed to detect the presence of g32p. Upon binding of g32p, a decrease in the voltammetric peak current is observed, which is readily measured using square wave voltammetry.
The observed sensor signal change is a result of specific single-strand DNA–protein interactions. To test the specificity of the single-strand oligo(dT) E-DNA sensor signaling to g32p, we employed a control sensor comprising double-stranded DNA (dsDNA) as the surface probe. We achieved this sensor architecture by adding an oligodeoxyadenylate [(dA)21] DNA complement to our prepared (dT)21 E-DNA sensors. As expected, the addition of saturating levels of (dA)21 DNA (500 nM) causes a decrease in the observed peak current (Figure 2, top) as a result of the transition from flexible ssDNA to the relatively rigid (but still dynamic) dsDNA.20,40 This observation confirms that the surface probes are dsDNA. The sensors were then immersed in a fresh buffer solution containing 4 μM g32p and exhibited no appreciable changes in the voltammetric peak current after 30 min of incubation time (Figure 2, top). The lack of binding to dsDNA demonstrates that the observed sensor response is a result of specific single-strand g32p-DNA binding interactions as opposed to nonspecific interactions with the sensor surface. To provide further evidence that signaling is specific to g32p interacting with the surface-bound ssDNA, we performed a control experiment by challenging our sensors with a similarly sized protein. Specifically, we challenged our sensors against the 37-kDa protein bovine thrombin (pI = 7.05–7.1), which possesses two positively charged binding sites.41,42 Sensors fabricated with (dT)21 incubated with 4 μM thrombin for 30 min exhibited no change in voltammetric peak current (Figure 2, bottom), further confirming the specificity of our modified surface to g32p binding.
Figure 2.

(dT)21 E-DNA sensor does not show any significant nonspecific interactions with the gold electrode surface, double-strand DNA (dsDNA), and non-DNA-binding protein. (Top) A double-strand E-DNA sensor architecture is achieved by adding the complement, (dA)21, and used to test the selectivity and specificity of the single-strand E-DNA sensor. No significant changes in the current signals are observed, which indicates that the sensors are specific to single strand DNA binding. (Bottom) To further test specificity, E-DNA sensors modified with (dT)21 DNA probes are tested against a similarly sized protein biomolecule, thrombin (∼37 kDa), using the same experimental conditions. Again, no significant changes in the current signals are obtained, which strongly demonstrates a highly specific DNA-binding protein binding sensor response.
To quantify g32p concentrations present in solution, we analyzed the voltammetric peak currents at various concentrations by calculating percent signal changes. Herein, percent signal change is calculated as the difference in the peak currents (itarget – io) normalized with respect to the peak current without target (io). In low salt conditions (20 mM NaCl; Figure 3), we find that the (dT)21 E-DNA sensor responds quantitatively to g32p concentrations saturating at 4 μM protein with ∼ –34% signal change. It is difficult to obtain intrinsic binding affinities from E-DNA sensors since the apparent extent of binding is dependent on the sensitivity and signaling of the nucleic acid architecture utilized.11,43 With this caveat, we estimate an apparent dissociation constant (KDapp) that is useful for comparing relative affinities using a Langmuir isotherm. The Langmuir isotherm used to fit this data is given by eq 1:
| 1 |
where S and Smax are signal and signal at saturation, respectively, and [L] is the free ligand concentration ([g32p]) in solution. KDapp is the apparent, or observed, dissociation constant (M). This expression is derived with the assumption that each binding site represents an independent, noninteracting binding site (which is only true for surfaces with (dT)7 probes) and that g32p binding does not appreciably alter the concentration of free g32p in solution. A full derivation of the expression can be found in the Supporting Information. Thus, under the low salt conditions utilized, the apparent dissociation constant for g32p binding to a (dT)21-based sensor is ∼100 nM (Figure 3).
Figure 3.

E-DNA sensors quantitatively detect varying concentrations of gene 32 protein. Sensors modified with (dT)21 probe are used to detect increasing concentrations of g32p. As g32p binds to the (dT)21 E-DNA sensor, a decrease in the voltammetric peak current, expressed as percent signal change, is observed at low salt conditions (20 mM NaCl). Titration at low ionic conditions leads to tight binding of g32p to the DNA-based electrochemical sensor, with an observed binding affinity (KDapp) estimated to be about ∼100 nM.
g32p Binding and Consequently Sensor Signaling is Dependent on Both Ionic Strength and DNA Probe Length
In order to determine the conditions at which g32p-DNA binding will be optimal for detecting cooperative binding, we varied the ionic strength of the buffer solution and the length of the nucleotide probe, both of which have been shown to affect g32p binding to single-strand nucleic acids.29,31 Specifically, the cooperative intrinsic binding affinity of the full-length protein to single-stranded nucleic acids capable of binding two or more proteins (>8 nucleotide residues in length) exhibits NaCl-dependent binding through the relationship ∂ log K/∂ log[NaCl] ≅ −7, where K is the effective cooperative binding constant.31
At low ionic strength, where g32p binding is dominated by electrostatic interactions and is tighter than at higher salt conditions, the observed percent signal changes were independent of probe length (Figure 4, top), although the calculated current densities for these sensors do show a quantitative dependence on the DNA probe length as expected (Supporting Information Figure S3). Sensors fabricated with (dT)7, (dT)14, and (dT)21 all exhibit similar binding curves, within error of each other when tested in 20 mM NaCl solution. Sensor signaling saturates at ∼4 μM with an average signal change of −37 ± 2%. While the line in Figure 4 (top) is to guide the reader’s eye, the fit to the (dT)7 sensor, which is capable of binding only one protein,31 is approximated using a Langmuir isotherm fit as described above. Again, the assumption is that each probe represents a single, noninteracting binding site, which is reasonable given that the probe can only accommodate one protein. As such, we calculated an apparent dissociation constant (KDapp) of approximately 100 nM for g32p binding to a (dT)7 E-DNA sensor under low ionic strength conditions.
Figure 4.

Protein binding and consequently sensor signaling are dependent on both ionic strength and DNA probe length. (Top) Oligo(dT) E-DNA sensors modified with 7, 14, and 21 thymidylate residues are used to evaluate the ionic-strength- and probe-length- dependence of g32p binding. Under low salt conditions, the binding curves for all probe lengths are not significantly different from each other, resulting from electrostatic DNA–protein binding interactions (KDapp = ∼100 nM). (Bottom) Under high salt conditions, the binding curves show dependence on DNA probe length because more binding sites per probe exist (2 and 3 for (dT)14 and (dT)21 probe, respectively). A decrease in the apparent affinity is calculated and expected with the onset of cooperative binding. Lines are drawn to guide the reader’s eye.
At high ionic strength, the oligo(dT) E-DNA sensors exhibited probe-length-dependent signaling, where the observed percent signal change is greater with increasing DNA length (Figure 4, bottom). Again, sensor signaling saturates at about 4 μM, with a signal change of −16 ± 6%, −36 ± 4%, and −49 ± 4% for the (dT)7, (dT)14, and (dT)21 E-DNA sensors, respectively. Using the Langmuir isotherm fit, we calculated a KDapp of 198 nM for g32p binding to a (dT)7 E-DNA sensor at high ionic strength, which is only slightly higher than the KDapp at low ionic conditions (∼100 nM). Consistent with our observation of only a modest two-fold change in apparent dissociation constants for (dT)7 sensors in low and high ionic strength, Von Hippel and coworkers demonstrated that the overall binding affinity of g32p to ssDNA capable of binding one protein (short oligonucleotides n < 8) exhibits no ionic strength dependence. Conversely, oligonucleotides that are capable of binding multiple proteins exhibit an overall salt-dependent binding affinity equal to (Kint · ω), where Kint is the affinity for an isolated site on the nucleic acid and ω is the cooperativity parameter.44−46 The salt dependence of binding is related to Kint, which increases with increasing salt concentrations while the cooperativity (ω) is salt-independent. Furthermore, DNA probes with multiple bound proteins will likely exhibit a larger decrease in the measured current, thus increasing the magnitude of the signal change. Consistent with this, the sensors modified with the (dT)21 DNA probe exhibited the greatest percent signal change (−49%), while the shortest (dT)7 DNA probe had the smallest change (−16%), both at saturating concentrations of g32p (4 μM; Figure 4, bottom).
E-DNA Sensors Distinguish between Cooperative and Non-Cooperative Binding
Sensors fabricated with (dT)n DNA probes exhibit distinctly different binding curves for the cooperatively binding full-length g32p and its noncooperatively binding *III fragment. Specifically, we find that the effect of cooperativity is amplified at longer probe lengths (Figure 5). We investigated the sensor response using various lengths of DNA probes (e.g., (dT)7, (dT)14, (dT)21, and (dT)80), which interacted with both the full-length protein and the *III core domain. As noted above, the cooperative property of g32p requires the presence of the N-terminal domain; thus, the *III core domain, lacking both the N- and C-domains, is not expected to display cooperative binding.39 Previous reports have shown that oligo- and polynucleotides bind both full-length g32p and *III protein,29,39 which correlate with the specific responses we obtained using our oligo(dT) E-DNA sensors (Figure 5). The binding curves for the *III protein (Figure 5, gray curves) are similar to each other, regardless of the DNA probe length, displaying a maximum of ∼ –24% signal change at saturation. In contrast, the binding curves for the full-length g32p (Figure 5, black curves) exhibit a pronounced dependence on DNA probe length, showing an increase in sensor signaling (from ∼ –16% to −49% maximum signal change at saturation) as the probe length is increased.
Figure 5.

Oligo(dT) E-DNA sensors distinguish between cooperative binding of full-length g32p and noncooperative binding of the core domain (*III) fragment. Sensors modified with (dT)7, (dT)14, (dT)21, and (dT)80 DNA probes are utilized to differentiate between cooperative and noncooperative binding, with the full-length g32p and truncated core domain (*III) protein used as targets. At high ionic strength, the full-length g32p cooperatively binds single-strand DNA in a probe-length-dependent manner (black lines), while the *III protein binds tightly and noncooperatively to DNA (gray lines). As seen in the binding curves, the full-length g32p generally exhibits weaker DNA–protein binding interactions relative to *III protein, which is characteristic of cooperativity between protein molecules. Except in the plots showing binding to (dT)7, which is fit to the Langmuir expression, lines are drawn to guide the reader’s eye.
Of note, we observe a larger signal change (−26%) when the *III fragment binds to the (dT)7 probe in comparison to binding of the full protein ( ∼ –16%) to the same probe. Since the binding site for the full-length protein is ∼7 nucleotides per g32p monomer, the poor binding ability that we observed may be due to deleterious steric interactions of the N- and C-domains with the electrode surface. With these portions of the protein missing on the *III fragment and the slightly smaller binding site (n = ∼5.5 nucleotide residues per g32p monomer), the *III protein is able to approach the sensor surface more effectively, and thus binds with greater affinity.
These results strongly suggest that the difference in signaling for oligo(dT) E-DNA sensors binding the full-length protein versus the *III fragment is a result of the cooperative binding of the full-length protein to DNA, as opposed to the *III fragment, which lacks the component involved in cooperative binding (N-domain). The increased variability in the maximum signal changes observed for the full-length protein is a consequence of cooperative binding of g32p to the DNA probes. Cooperative binding is dependent on the presence of the N-domain. As a result, the full-length g32p is able to bind to the growing length of the DNA strand, while undergoing protein–protein interactions that lead to cooperative protein binding to ssDNA (Figure 5) and thus results in greater signal suppression.
Conclusion
In this report, we have developed a simple electrochemical-DNA sensor modified with varying lengths of oligo(dT) probes for the quantitative detection of gene 32 protein and have monitored cooperative and noncooperative binding of the protein to ssDNA. We have successfully demonstrated the quantitative sensing ability of the oligo(dT) E-DNA sensors with high sensitivity, specificity, and reproducibility. Specifically, we have created E-DNA sensors that were sensitive to nanomolar concentrations of g32p and were highly specific since they did not show any significant nonspecific interactions with either the gold electrode surface or dsDNA. Using these sensors, we were able to characterize some of the binding properties of the full-length g32p by quantitatively evaluating the protein’s dependence on ionic strength and DNA probe length. Lastly, the E-DNA sensors were able to quantitatively and qualitatively distinguish between the cooperative binding of full-length g32p and the noncooperative binding of truncated *III protein to single-strand nucleic acids, in agreement with reports in the literature.
We have demonstrated the quantitative ability of the E-DNA sensing platform to characterize DNA–protein binding interactions and its adaptation to determine the presence or absence of cooperativity. This new ability adds to the growing toolbox enabled by the use of electrochemical, DNA-based sensors and may represent a relatively rapid and simple way for understanding DNA-binding protein interactions.
Acknowledgments
This work was supported by the start-up fund (R.J.W.), the NIH/NIGMS T32GM066706 (K. Radtke; F.C.M.) CBI grant at the Department of Chemistry and Biochemistry, University of Maryland Baltimore County (UMBC), a grant from the UMBC Designated Research Initiative Fund, and the National Institutes of Health (GM52049, R.L.K.).
Supporting Information Available
A derivation of the Langmuir isotherm used in this manuscript, comparison of probe packing densities and protein titrations for sensing monolayers formed using the “backfill” and “insertion” method, titration curves plotted in terms of absolute current densities can all be found in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Baker B. R.; Lai R. Y.; Wood M. S.; Doctor E. H.; Heeger A. J.; Plaxco K. W. An electronic, aptamer-based small-molecule sensor for the rapid, label-free detection of cocaine in adulterated samples and biological fluids. J. Am. Chem. Soc. 2006, 128, 3138–3139. [DOI] [PubMed] [Google Scholar]
- Fan C.; Plaxco K. W.; Heeger A. J. Electrochemical interrogation of conformational changes as a reagentless method for the sequence-specific detection of DNA. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 9134–9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferapontova E. E.; Gothelf K. V. Effect of serum on an RNA aptamer-based electrochemical sensor for theophylline. Langmuir 2009, 25, 4279–4283. [DOI] [PubMed] [Google Scholar]
- Ferapontova E. E.; Gothelf K. V. Optimization of the Electrochemical RNA–Aptamer Based Biosensor for Theophylline by Using a Methylene Blue Redox Label. Electroanalysis 2009, 21, 1261–1266. [Google Scholar]
- Ferapontova E. E.; Olsen E. M.; Gothelf K. V. An RNA aptamer-based electrochemical biosensor for detection of theophylline in serum. J. Am. Chem. Soc. 2008, 130, 4256–4258. [DOI] [PubMed] [Google Scholar]
- Lai R. Y.; Lagally E. T.; Lee S.-H.; Soh H.; Plaxco K. W.; Heeger A. J. Rapid, sequence-specific detection of unpurified PCR amplicons via a reusable, electrochemical sensor. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 4017–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai R. Y.; Plaxco K. W.; Heeger A. J. Aptamer-based electrochemical detection of picomolar platelet-derived growth factor directly in blood serum. Anal. Chem. 2007, 79, 229–233. [DOI] [PubMed] [Google Scholar]
- Patterson A.; Caprio F.; Vallée-Bélisle A.; Moscone D.; Plaxco K. W.; Palleschi G.; Ricci F. Using triplex-forming oligonucleotide probes for the reagentless, electrochemical detection of double-stranded DNA. Anal. Chem. 2010, 82, 9109–9115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci F.; Lai R. Y.; Plaxco K. W. Linear, redox modified DNA probes as electrochemical DNA sensors. Chem. Commun. 2007, 3768–3770. [DOI] [PubMed] [Google Scholar]
- Ricci F.; Plaxco K. E-DNA sensors for convenient, label-free electrochemical detection of hybridization. Microchim. Acta 2008, 163, 149–155. [Google Scholar]
- Schoukroun-Barnes L. R.; Wagan S.; White R. J. Enhancing the Analytical Performance of Electrochemical RNA Aptamer-Based Sensors for Sensitive Detection of Aminoglycoside Antibiotics. Anal. Chem. 2014, 86, 1131–1137. [DOI] [PubMed] [Google Scholar]
- White R. J.; Phares N.; Lubin A. A.; Xiao Y.; Plaxco K. W. Optimization of electrochemical aptamer-based sensors via optimization of probe packing density and surface chemistry. Langmuir 2008, 24, 10513–10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.; Lubin A. A.; Heeger A. J.; Plaxco K. W. Label-free electronic detection of thrombin in blood serum by using an aptamer-based sensor. Angew. Chem. 2005, 117, 5592–5595. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Qu X.; Plaxco K. W.; Heeger A. J. Label-free electrochemical detection of DNA in blood serum via target-induced resolution of an electrode-bound DNA pseudoknot. J. Am. Chem. Soc. 2007, 129, 11896–11897. [DOI] [PubMed] [Google Scholar]
- Lubin A. A.; Plaxco K. W. Folding-Based Electrochemical Biosensors: The Case for Responsive Nucleic Acid Architectures. Acc. Chem. Res. 2010, 43, 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci F.; Bonham A. J.; Mason A. C.; Reich N. O.; Plaxco K. W. Reagentless, electrochemical approach for the specific detection of double- and single-stranded DNA binding proteins. Anal. Chem. 2009, 81, 1608–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Morris M. D.; Macazo F. C.; Schoukroun-Barnes L. R.; White R. J. The Current and Future Role of Aptamers in Electroanalysis. J. Electrochem. Soc. 2014, 161, H301–H313. [Google Scholar]
- Xiao Y.; Lai R. Y.; Plaxco K. W. Preparation of electrode-immobilized, redox-modified oligonucleotides for electrochemical DNA and aptamer-based sensing. Nat. Protoc. 2007, 2, 2875–2880. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Uzawa T.; White R. J.; DeMartini D.; Plaxco K. W. On the Signaling of Electrochemical Aptamer-Based Sensors: Collision- and Folding-Based Mechanisms. Electroanalysis 2009, 21, 1267–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R. J.; Plaxco K. W. Exploiting binding-induced changes in probe flexibility for the optimization of electrochemical biosensors. Anal. Chem. 2009, 82, 73–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K.-C.; White R. J. Random Walk on a Leash: A Simple Single-Molecule Diffusion Model for Surface-Tethered Redox Molecules with Flexible Linkers. J. Am. Chem. Soc. 2013, 135, 12808–12817. [DOI] [PubMed] [Google Scholar]
- Ferguson B. S.; Hoggarth D. A.; Maliniak D.; Ploense K.; White R. J.; Woodward N.; Hsieh K.; Bonham A. J.; Eisenstein M.; Kippin T. E.; Plaxco K. W.; Soh H. T. Real-Time, Aptamer-Based Tracking of Circulating Therapeutic Agents in Living Animals. Science Translat. Med. 2013, 5, 213–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon E. M.; Salas J. E.; Barton J. K. An electrical probe of protein–DNA interactions on DNA-modified surfaces. Nat. Biotechnol. 2002, 20, 282–286. [DOI] [PubMed] [Google Scholar]
- Meunier-Prest R.; Bouyon A.; Rampazzi E.; Raveau S.; Andreoletti P.; Cherkaoui-Malki M. Electrochemical probe for the monitoring of DNA–protein interactions. Biosens. Bioelectron. 2010, 25, 2598–2602. [DOI] [PubMed] [Google Scholar]
- Ban C.; Chung S.; Park D.-S.; Shim Y.-B. Detection of protein–DNA interaction with a DNA probe: distinction between single-strand and double-strand DNA–protein interaction. Nucleic Acids Res. 2004, 32, e110–e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorodetsky A. A.; Ebrahim A.; Barton J. K. Electrical detection of TATA binding protein at DNA-modified microelectrodes. J. Am. Chem. Soc. 2008, 130, 2924–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRosa M. C.; Sancar A.; Barton J. K. Electrically monitoring DNA repair by photolyase. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 10788–10792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.-Z.; Long Y.-T.; Lee J. S.; Kraatz H.-B. Protein–DNA interaction: impedance study of MutS binding to a DNA mismatch. Chem. Commun. 2004, 574–575. [DOI] [PubMed] [Google Scholar]
- Wu M.; Flynn E. K.; Karpel R. L. Details of the nucleic acid binding site of T4 gene 32 protein revealed by proteolysis and DNA Tm depression methods. J. Mol. Biol. 1999, 286, 1107–1121. [DOI] [PubMed] [Google Scholar]
- Karpel R. L.T4 bacteriophage gene 32 protein. The Biology of Non-Specific DNA–Protein Interactions; CRC Press: Boca Raton, FL, 1990; pp 103–130. [Google Scholar]
- Kowalczykowski S. C.; Lonberg N.; Newport J. W.; von Hippel P. H. Interactions of bacteriophage T4-coded gene 32 protein with nucleic acids. I. Characterization of the binding interactions. J. Mol. Biol. 1981, 145, 75–104. [DOI] [PubMed] [Google Scholar]
- Bittner M.; Burke R. L.; Alberts B. M. Purification of the T4 gene 32 protein free from detectable deoxyribonuclease activities. J. Biol. Chem. 1979, 254, 9565–9572. [PubMed] [Google Scholar]
- Shamoo Y.; Adari H.; Konigsberg W. H.; Williams K. R.; Chase J. W. Cloning of T4 gene 32 and expression of the wild-type protein under lambda promoter PL regulation in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 8844–8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamoo Y.; Friedman A. M.; Parsons M. R.; Konigsberg W. H.; Steitz T. A. Crystal structure of a replication fork single-stranded DNA binding protein (T4 gp32) complexed to DNA. Nature 1995, 376, 362–366. [DOI] [PubMed] [Google Scholar]
- Jensen D. E.; Kelly R. C.; von Hippel P. H. DNA “melting” proteins. II. Effects of bacteriophage T4 gene 32-protein binding on the conformation and stability of nucleic acid structures. J. Biol. Chem. 1976, 251, 7215–7228. [PubMed] [Google Scholar]
- Josephs E. A.; Ye T. Nanoscale Spatial Distribution of Thiolated DNA on Model Nucleic Acid Sensor Surfaces. ACS Nano 2013, 7, 3653–3660. [DOI] [PubMed] [Google Scholar]
- Steel A. B.; Herne T. M.; Tarlov M. J. Electrochemical quantitation of DNA immobilized on gold. Anal. Chem. 1998, 70, 4670–4677. [DOI] [PubMed] [Google Scholar]
- Kozinski A. W.; Felgenhauer Z. Z. Molecular recombination in T4 bacteriophage deoxyribonucleic acid. II. Single-strand breaks and exposure of uncomplemented areas as a prerequisite for recombination. J. Virol. 1967, 1, 1193–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonberg N.; Kowalczykowski S. C.; Paul L. S.; von Hippel P. H. Interactions of bacteriophage T4-coded gene 32 protein with nucleic acids. III. Binding properties of two specific proteolytic digestion products of the protein (G32P*I and G32P*III). J. Mol. Biol. 1981, 145, 123–138. [DOI] [PubMed] [Google Scholar]
- Cash K. J.; Ricci F.; Plaxco K. W. An Electrochemical Sensor for the Detection of Protein–Small Molecule Interactions Directly in Serum and Other Complex Matrices. J. Am. Chem. Soc. 2009, 131, 6955–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malovichko M. V.; Sabo T. M.; Maurer M. C. Ligand binding to anion-binding exosites regulates conformational properties of thrombin. J. Biol. Chem. 2013, 288, 8667–8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode W.; Turk D.; Karshikov A. The refined 1.9-Å X-ray crystal structure of d-Phe-Pro-Arg chloromethylketone-inhibited human α-thrombin: Structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure–function relationships. Protein Sci. 1992, 1, 426–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban Fernandez de Avila B.; Watkins H. M.; Pingarron J. M.; Plaxco K. W.; Palleschi G.; Ricci F. Determinants of the Detection Limit and Specificity of Surface-Based Biosensors. Anal. Chem. 2013, 85, 6593–6597. [DOI] [PubMed] [Google Scholar]
- Kowalczykowski S. C.; Paul L. S.; Lonberg N.; Newport J. W.; McSwiggen J. A.; Von Hippel P. H. Cooperative and noncooperative binding of protein ligands to nucleic acid lattices: experimental approaches to the determination of thermodynamic parameters. Biochemistry 1986, 25, 1226–1240. [DOI] [PubMed] [Google Scholar]
- McGhee J. D.; von Hippel P. H. Theoretical aspects of DNA–protein interactions: co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974, 86, 469–489. [DOI] [PubMed] [Google Scholar]
- Tsodikov O. V.; Holbrook J. A.; Shkel I. A.; Record M. T. Jr Analytic binding isotherms describing competitive interactions of a protein ligand with specific and nonspecific sites on the same DNA oligomer. Biophys. J. 2001, 81, 1960–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.