

Abstract
As a driver for many biological processes, phosphorylation remains an area of intense research interest. Advances in multiplexed quantitation utilizing isobaric tags (e.g., TMT and iTRAQ) have the potential to create a new paradigm in quantitative proteomics. New instrumentation and software are propelling these multiplexed workflows forward, which results in more accurate, sensitive, and reproducible quantitation across tens of thousands of phosphopeptides. This study assesses the performance of multiplexed quantitative phosphoproteomics on the Orbitrap Fusion mass spectrometer. Utilizing a two-phosphoproteome model of precursor ion interference, we assessed the accuracy of phosphopeptide quantitation across a variety of experimental approaches. These methods included the use of synchronous precursor selection (SPS) to enhance TMT reporter ion intensity and accuracy. We found that (i) ratio distortion remained a problem for phosphopeptide analysis in multiplexed quantitative workflows, (ii) ratio distortion can be overcome by the use of an SPS-MS3 scan, (iii) interfering ions generally possessed a different charge state than the target precursor, and (iv) selecting only the phosphate neutral loss peak (single notch) for the MS3 scan still provided accurate ratio measurements. Remarkably, these data suggest that the underlying cause of interference may not be due to coeluting and cofragmented peptides but instead from consistent, low level background fragmentation. Finally, as a proof-of-concept 10-plex experiment, we compared phosphopeptide levels from five murine brains to five livers. In total, the SPS-MS3 method quantified 38 247 phosphopeptides, corresponding to 11 000 phosphorylation sites. With 10 measurements recorded for each phosphopeptide, this equates to more than 628 000 binary comparisons collected in less than 48 h.
As a key mediator of cellular signaling, phosphorylation remains a principal target for biological interrogation.1 Identifying and quantifying the phosphorylation state of proteins involved in cell progression, metabolism, growth, and disease is critical for the continued elucidation of cellular function.2 Global phosphoproteome characterization is challenging due to the estimated large volume of phosphorylation sites in eukaryotic cells and the often low abundance/stoichiometry of the phosphoproteome.3,4 Continuing technological and methodological advancements have resulted in the characterization of tens of thousands of phosphorylation sites across numerous species, but it is apparent that only a fraction of all phosphorylation events have been characterized.5−11 Furthermore, phosphorylation dynamics, assessed via relative quantification, have historically been limited to binary or ternary comparisons, further limiting the breadth and depth of phosphopeptide analysis.12−17 Novel methodologies are needed in order to overcome the current shortcomings of phosphoproteome characterization.
Mass spectrometry remains an unmatched platform for comprehensive phosphoproteome analysis. Coupling deep identification with relative quantification has provided valuable biological insights that would be otherwise unobtainable by traditional biochemical techniques.18−24 Isobaric tags for relative and absolute quantitation (iTRAQ) and tandem-mass-tag (TMT) based methodologies permit the simultaneous comparison of up to 8 (iTRAQ) or 10 (TMT) samples, facilitating complex experimental designs and the inclusion of biological replicates within the same experiment.
A primary hurdle for isobaric based quantification technologies is the presence of interfering coisolated species that result in distorted reporter ion intensities. A number of publications have documented this phenomenon, and several have demonstrated approaches to alleviate the interference.25−31 One such approach was the inclusion of a quantitative MS3 spectrum.32
Recently, the sensitivity of the MS3 method was dramatically improved by isolating multiple fragment ions in the MS2 spectrum using isolation waveforms with multiple notches (e.g., synchronous precursor selection, SPS).33 The SPS-MS3 method is available on the Orbitrap Fusion, which leverages advancements in software and hardware to provide increased scan rates and improved sensitivity, resolution, and quantitative accuracy. Furthermore, a unique architecture expands the concept of a hybrid mass spectrometer by incorporating three mass analyzers (i.e., quadrupole mass filter, quadrupole ion trap, and Orbitrap) operating in a task parallelized manner.
Here, we assessed the performance of the SPS-MS3 method on two different phosphoproteome samples. We utilized a 2-phosphoproteome model of interference to characterize the quantitative accuracy of various SPS-MS3 and MS2 methods on the Orbitrap Fusion. We observed that known ratios were distorted for the MS2 method compared to the SPS-MS3 method. In a large-scale demonstration of the method, we performed a proteome-wide phosphorylation analysis in 48 h, which compared brain and liver phosphorylation level differences from five mice in a single 10-plex experiment.
Experimental Procedures
Protein Extraction and Digestion
Five murine brains and livers were harvested from CO2 asphyxiated 3-week-old male Swiss-Webster mice (Jackson Lab, Bar Harbor, ME). Saccharomyces cerevisiae cells were grown to an OD of 1.0, washed with ice cold PBS and snap frozen in liquid N2 until further use. Brain/liver tissues and yeast cells were mechanically lysed with a homogenizer in SDS lysis buffer [2.0% SDS w/v, 250 mM NaCl, PhosSTOP (Roche, Madison, WI) phosphatase inhibitors, 2 mM sodium vanadate, EDTA free protease inhibitor cocktail (Promega, Madison, WI), and 50 mM HEPES, pH 8.5]. Lysates were reduced with 5 mM DTT and cysteine residues alkylated with iodoacetamide (14 mM) in the dark (30 min). Protein was extracted by methanol–chloroform precipitation and subsequent ice cold acetone washes. Pellets were dried and resuspended in 8 M urea containing 50 mM HEPES (pH 8.5). Protein concentrations were measured by BCA assay (Thermo Scientific, Rockford, IL) prior to protease digestion. Protein lysates were diluted to 4 M urea and digested with LysC (Wako, Japan) in a 1/200 enzyme/protein ratio overnight. Protein extracts were diluted further to a 1.5 M urea concentration, and trypsin (Promega, Madison, WI) was added to a final 1/250 enzyme/protein ratio for 6 h at 37 °C. Digests were acidified with 200 μL of 20% formic acid (FA) to a pH ∼2 and subjected to C18 solid-phase extraction (SPE) (Sep-Pak, Waters, Milford, MA).
Phosphopeptide Enrichment
As the proportion of phosphopeptides identified from nonphosphopeptide enriched proteomes is routinely <5%, a sufficient amount of starting material is necessary in order to produce a phosphopeptide population amenable to LC–MS. Given the amount of starting material, it is prudent to consider the costs of TMT reagents. The methods have been optimized to incorporate TMT labeling post phosphopeptide enrichment, resulting in a significant cost reduction. For example, TMT labeling post phosphopeptide enrichment results in at least a 40-fold reduction in peptide concentration, providing a corresponding savings in TMT reagent cost.
Enrichment proceeded with some modifications to the method of Kettenbach et al.34 Tryptic peptides (∼10 mg per TMT channel) were resuspended in 1 mL of 2 M lactic acid/50% acetonitrile (ACN) and centrifuged at 15 000g for 20 min. Supernatants were removed, placed in an Eppendorf tube containing 15 mg of titanium dioxide beads (GL Sciences, Japan), and vortexed for 1 h at room temperature. Beads were washed twice with 2 M lactic acid/50% ACN and once with 0.1% TFA in 50% ACN. Phosphopeptides were eluted twice with 150 μL of 50 mM HK2PO4, pH 10, acidified with 40 μL of 20% formic acid, and subjected to C18 StageTip desalting (3M Empore, South Eagan, MN).
Tandem Mass Tagging Labeling
Isobaric labeling of the enriched phosphopeptides was performed using either the 6-plex or 10-plex tandem mass tag (TMT) reagents (Thermo Fisher Scientific, Rockford, IL). TMT reagents (0.8 mg) were dissolved in 40 μL of dry acetonitrile (ACN), and 10 μL was added to 100 μg (Micro BCA, Thermo Scientific, Rockford, IL) of phosphopeptides dissolved in 100 μL of 200 mM HEPES, pH 8.5. After 1 h (RT), the reaction was quenched by adding 8 μL of 5% hydroxylamine. Labeled peptides were combined, acidified with 20 μL of 20% FA (pH ∼2), and concentrated via C18 SPE on Sep-Pak cartridges (50 mg bed volume). All previously described sample preparation proceeded the same for both the 6-plex and the 10-plex experiments. Additional details regarding the 6-plex sample preparation is highlighted below.
Two-Proteome Interference Model
Following phosphopeptide labeling, the brain and yeast samples were combined. Brain phosphopeptides were mixed together at 10:2:1:1:2:10 (TMT channels 126–131), and yeast phosphopeptides were mixed at 10:10:10:0:0:0 (TMT channels 126–128) (Figure 1A). After combining the phosphopeptides, samples were subjected to C18 solid-phase extraction (50 mg vide supra).
Figure 1.

(A) Murine brain and yeast lysates were digested and phosphopeptides enriched by TiO2. The mouse phosphopeptides were labeled with TMT and mixed at a relative concentration of 10:2:1:1:2:10. Yeast phosphopeptides were split into three samples, labeled with three TMT reagents, and mixed 10:10:10:0:0:0. The mouse and yeast phosphopeptides were mixed (1:1, w:w). If present, interference from the yeast background would perturb mouse ratios. For all mouse phosphopeptides, the TMT channels lacking any yeast interference provided control intensities and ratios. (B) SPS-MS3 method overview. Synchronous precursor selection (SPS) enables the simultaneous isolation of multiple MS2 fragment ions increasing TMT reporter ion signal in the MS3 scan.
Basic pH Reverse-Phase HPLC (bpHrp)
TMT labeled brain and liver phosphopeptides were subjected to orthogonal basic-pH reverse phase (bpHrp) fractionation. Labeled phosphopeptides were solubilized in buffer A (5% ACN, 10 mM ammonium bicarbonate, pH 8.0) and separated on an Agilent 300 Extend C18 column (5 μm particles, 4.6 mm i.d. and 220 mm in length). Using an Agilent 1100 binary pump equipped with a degasser and a photodiode array (PDA) detector (Thermo Scientific, San Jose, CA), a 45 min linear gradient from 8% to 35% acetonitrile in 10 mM ammonium bicarbonate pH 8 (flow rate of 0.8 mL/min) separated the peptide mixtures into a total of 96 fractions. The 96 fractions were consolidated into 24 samples in a checkerboard manner, acidified with 10 μL of 20% formic acid and vacuum-dried. Each sample was redissolved in 5% formic acid, desalted via StageTip, dried via vacuum centrifugation, and reconstituted for LC–MS/MS analysis.
Orbitrap Fusion Parameters
All spectra were acquired on an Oribtrap Fusion (Thermo Fischer Scientific) coupled to an Easy-nLC 1000 (Thermo Fisher Scientific) ultrahigh pressure liquid chromatography (UHPLC) pump. Peptides were separated on an in-house packed 100 μM inner diameter column containing 0.5 cm of Magic C4 resin (5 μm, 100 Å, Michrom Bioresources), serving as a frit, followed by 25 cm of Sepax Technologies GP-C18 resin (1.8 μm, 120 Å, Newark, DE) with a gradient consisting of 3–22% (ACN, 0.125% FA) over 165 min at ∼300 nL/min.
For all experiments, the instrument was operated in the data-dependent mode. We collected FTMS1 spectra at a resolution of 120 000, with an automated gain control (AGC) target of 200 000, and a max injection time of 100 ms. The 10 most intense ions were selected for MS/MS. Precursors were filtered according to charge state (required >1 z), and monoisotopic peak assignment. Previously interrogated precursors were excluded using a dynamic window (75 s ± 10 ppm). The MS2 precursors were isolated with a quadrupole mass filter set to a width of 0.5 m/z.
During the experiment where precursors were only analyzed by FTMS2, the Orbitrap was operated at 60 000 resolution, with an AGC target of 50 000 and a max injection time of 250 ms. Precursors were fragmented by high-energy collision dissociation (HCD) at a normalized collision energy (NCE) of 37.5%.
During the method with FTMS3 analysis, ITMS2 spectra were collected at an AGC of 4 000, max injection time of 150 ms, and CID collision energy of 35%. FTMS3 spectra utilized the same Orbitrap parameters as the FTMS2 method, except HCD collision energy was increased to 55% to ensure maximal TMT reporter ion yield. Depending on the experiment, synchronous-precursor-selection (SPS) was enabled to include up to 3, 6, or 10 MS2 fragment ions in the FTMS3 scan.
Data Processing and Spectra Assignment
A compilation of in-house software was used to convert mass spectrometric data (Thermo “.raw” files) to mzXML format as well as to correct monoisotopic m/z measurements and erroneous peptide charge state assignments. Assignment of MS/MS spectra was performed using the SEQUEST algorithm.35 The 2-phosphoproteome experiment utilized a protein sequence database that was a combination of the Mouse UniProt database (downloaded 08/02/2011) and the S. cerevisiae ORF database (downloaded 02/16/2010). All other experiments utilized only the Mouse UniProt database. In each case, reversed protein sequences were appended as well as known contaminants such as human keratins. In order to prevent inaccurate interference measurements from peptides shared by yeast and mouse, the 2-phosphoproteome FASTA database was ordered such that protein sequences from yeast were listed first, thus ensuring that peptides matching both yeast and mouse proteins would be assigned to a yeast protein. SEQUEST searches were performed using a 50 ppm precursor ion tolerance, while requiring each peptide’s N/C terminus to have trypsin protease specificity and allowing up to two missed cleavages. TMT tags on peptide N termini/lysine residues (+229.162932 Da) and carbamidomethylation of cysteine residues (+57.02146 Da) were set as static modifications, while methionine oxidation (+15.99492 Da) and serine, threonine, and tyrosine phosphorylation (+79.96633 Da) were set as variable modifications. An MS2 spectra assignment false discovery rate (FDR) of less than 1% was achieved by applying the target-decoy database search strategy.36 Filtering was performed exactly as previously described.33
We used a modified version of the Ascore algorithm to quantify the confidence with which each phosphorylation site could be assigned to a particular residue. Phosphorylation sites with Ascore values >13 (P ≤ 0.05) were considered confidently localized to a particular residue.11
Determination of TMT Reporter Ion Intensities and Quantitative Data Analysis
For quantification, a 0.03 m/z (6-plex TMT) or 0.003 m/z (10-plex TMT) window centered on the theoretical m/z value of each reporter ion was queried for the nearest signal intensity. Reporter ion intensities were adjusted to correct for the isotopic impurities of the different TMT reagents (manufacturer specifications). The signal-to-noise values for all peptides were summed within each TMT channel, and each channel was scaled according to the interchannel difference of these sums to account for differences in sample handling. For each peptide, a total minimum sum signal-to-noise value of 400 and an isolation purity greater than 75% was required.33
Neutral loss fragments were identified by parsing MS2 spectra for fragments within 0.2 m/z of the expected mass (based on precursor m/z and charge state) and with an intensity greater than 10% of the base peak intensity.
t tests with Welch’s correction for unequal variances were performed for all mouse brain and liver phosphopeptide biological replicates. Multiple test correction was performed by adjusting the calculated p-values according to Benjamini–Hochberg.37 Phosphopeptides with an adjusted p-value < 0.01 were classified as brain enriched, liver enriched, or commonly expressed. Gene ontology term enrichment was performed by submitting the three classes described above to DAVID, utilizing the complete set of quantified phosphopeptides as a background.38 All data analysis was performed using R (R Core Team, Vienna, Austria, http://www.R-project.org).
Results
Constructing a 2-Phosphoproteome Model of Interference
To assess the accuracy of TMT-based quantitative phosphoproteomics on the Orbitrap Fusion, we constructed a 2-phosphoproteome sample that contains TMT channels with and without interfering phosphopeptides. Figure 1A illustrates the preparation of the 2-phosphoproteome model. Following tissue lysis, digestion, and phosphopeptide enrichment, the mouse brain phosphopeptides were combined at a concentration of 10:2:1:1:2:10. To introduce interference, yeast phosphopeptides were mixed at a concentration of 10:10:10:0:0:0 and added to the mouse phosphopeptide dilution series (1:1 w/w). The resulting sample of mouse phosphopeptides contained three channels that might display interference from coisolated yeast phosphopeptides (m/z 126, 127, and 128) and three channels that were free of any yeast interference (m/z 129, 130, and 131). If present, interference would distort the expected ratios between channels 126 and 127 (5:1) and 126 and 128 (10:1). The SPS-MS3 method implemented here incorporated a notched isolation waveform (up to 10 notches) that isolated MS2 fragment ions based upon their relative intensity (Figure 1B).
Assessing Quantitative Phosphoproteome Accuracy
We used the 2-phosphoproteome model to assess the quantitative accuracy of several MS methods for phosphorylation analysis. Figure 2 shows the normalized TMT ratios for the 5:1 comparison with and without interference (red trace 126/127, blue trace 131/130, respectively). Yeast peptides are plotted in green and were expected to have a 1:1 ratio.
Figure 2.
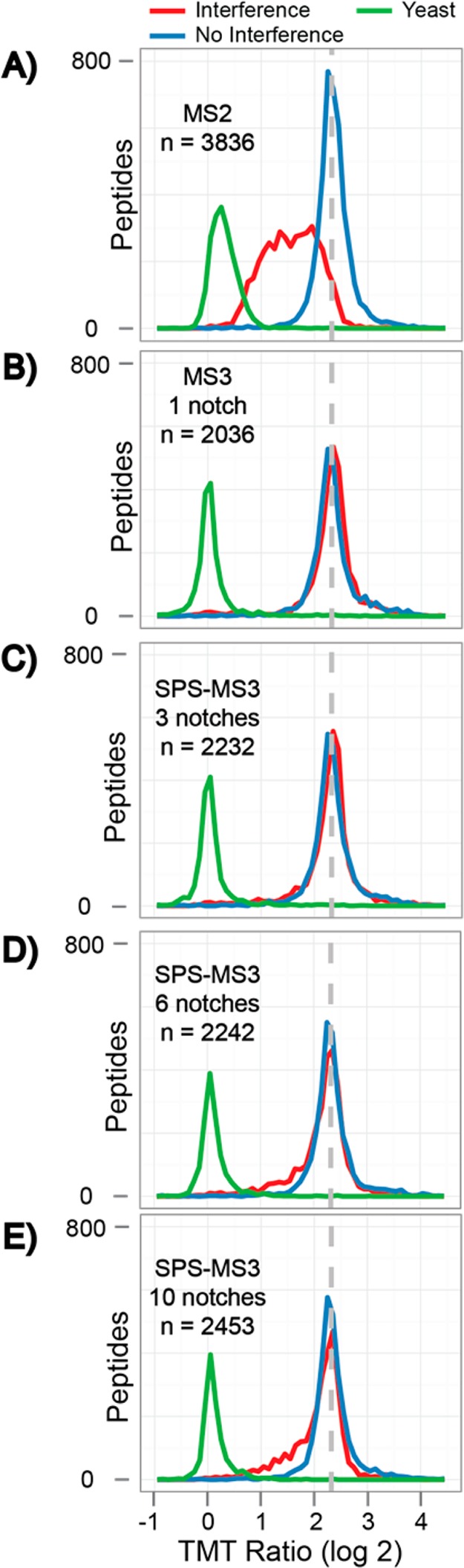
Phosphopeptide mixture was analyzed by LC–MS and quantified by MS2 and SPS-MS3 methods. SPS-MS3 performance was further investigated by varying the number of MS2 fragment ions included in the quantitative MS3 spectrum (i.e., 1, 3, 6, and 10 precursor ions). (A–E) Distributions of ratios corresponding to yeast phosphopeptides (TMT channels 126/127, green trace), mouse phosphopeptides with interference (TMT channels 126/127, red trace), and mouse phosphopeptides without interference (TMT channels 131/130, blue trace). Yeast phosphopeptides (green) are expected at a 1:1 ratio while the mouse phosphopeptides were mixed at a 5:1 ratio (red and blue). The dashed line depicts the expected ratio of 5:1. The number of quantified mouse phosphopeptides is displayed for each method. Quantification via an MS2 method (A) resulted in significant ratio distortion with a wide distribution of ratios. Utilizing a MS3 method (B–E) dramatically improves the accuracy and precision (fwhm) of phosphopeptide quantification.
As has been observed in previous studies,29−32 TMT reporter ion intensities derived from MS2 spectra were largely distorted and inaccurate (Figure 2A). We observed that a narrow quadrupole mass filter isolation of 0.5 m/z did not effectively correct interference. Furthermore, the ratio between yeast phosphopeptides, which represented the most intense and easiest measurement at 1:1, was also distorted, by the mouse phosphopeptides, to a median of 1.2:1.
For each SPS method, the distribution of ratios for the interference channel followed that of the MS3 single notch method (Figure 2B). Distributions were tight and centered about the expected ratio of 5:1. However, as more of the MS2 spectral space was included in the MS3 scan, we observed a subtle shift in the distributions of ratios (Figure 2C–E). This is apparent as the difference between the noninterference distribution (blue) and the interference distribution (red) increased as the number of notches increased. The correlation between the increase in TMT reporter ion distortion and the number of notches is likely a phenomenon specific to phosphopeptide analyses (see below).
Neutral Loss and TMT Accuracy
A common characteristic of phosphopeptide analysis is the presence of a dominant neutral loss peak following CID fragmentation. We examined the 2-phosphoproteome data set to assess the impact, if any, a dominant phosphate neutral loss fragment plays in quantitative accuracy. Identification of typical phosphate neutral loss fragments was accomplished by searching MS2 spectra for fragment ions corresponding to the expected neutral loss masses based on precursor charge state and m/z (see Experimental Procedures).
Figure 3A depicts the proportion of mouse phosphopeptides with and without a neutral loss fragment (neutral loss greater than 10% of base peak intensity). An overwhelming 82% of phosphopeptides exhibited a neutral loss fragment following CID fragmentation. Among those phosphopeptides that produced a neutral loss, 70% of the neutral loss fragments were identified as the most intense fragment in the MS2 spectrum, which translates to 57% of all phosphopeptides. Across phosphopeptide charge states 2–5, neutral loss fragments are consistently observed; although, as previously reported the intensity of the neutral loss decreases as charge state increases (Figure 3A, inset).39
Figure 3.
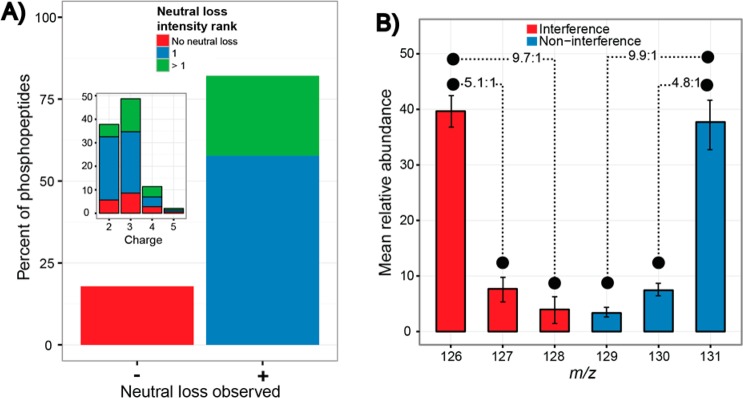
(A) Production of a neutral loss fragment from the single notch MS3 experiment was assessed and determined that 82% of mouse phosphopeptides yielded a neutral loss of phosphoric acid. In total, 50% of the neutral loss fragments were the most intense ion in the spectrum. (Inset) A neutral loss fragment was routinely present among charge states two and three. The intensity of the neutral loss fragment decreased as the charge state increased. (B) Phosphopeptides containing a rank one neutral loss, from the single notch MS3 method, were selected, and the mean relative TMT abundances were determined for each channel (error bars ± 1 SD). Channels containing interference (m/z 126/127/128) and without interference (m/z 129/130/131) are shown. Ratios between channels with and without interference were near expected values (10:1, 5:1), which indicated that the neutral loss fragment ion was a viable MS3 precursor ion that maintains the quantitative accuracy of the method.
For the SPS methods, we observed an increase in the degree of TMT reporter inaccuracy as the number of MS2 fragment ions included in the MS3 analysis was increased. The SPS-MS3 approach utilizes an intensity based rank order when selecting MS2 fragments ions. That is, for a single notch MS3 method, the most intense ion will be isolated for subsequent MS3; for a three notch SPS method, the three most intense fragment ions will be selected and so on. We believe that the increase in interference may be due to the neutral loss retaining the bulk of the precursor ion population, as observed by the rank order intensity of the neutral loss. The inclusion of additional MS2 fragment ions via the SPS-MS3 method results in an increase in the amount of MS2 m/z space included in the MS3 spectrum without a corresponding increase in the target precursor ion population. This contrasts directly with nonphosphorylated peptides, which exhibit heterogeneous fragmentation, where additional notches in the SPS-MS3 method will likely correspond to additional fragment ions from the intended precursor. For nonphosphorylated peptides this ultimately provides a proportionate increase of m/z space and target precursor ion current (Supp. Figure 1 in the Supporting Information). Therefore, a future iteration of the SPS-MS3 method could include an online, dynamic adjustment of the number of notches in order to account for the heterogeneity of the MS2 fragment ion population.
In order to isolate the effect, if any, of the neutral loss fragment on quantitative accuracy, we only looked at the quantitative accuracy of the population of phosphopeptides where the most intense fragment ion corresponded to the neutral loss of the phosphate group. Figure 3B displays a summary of TMT reporter ion intensities for this select group of phosphopeptides from the single notch MS3 experiment. The noninterference channels (blue bars) produced ratios of 9.9:1 (10:1 expected) and 4.8:1 (5:1 expected). The interference channels (red bars) produced remarkably similar ratios of 9.7:1 and 5.1:1 (10:1 and 5:1 expected, respectively). This suggests that the inclusion of a neutral loss fragment within the MS3 method does not significantly affect the accuracy of the TMT quantitation. Furthermore, this supports the notion that reporter ion interference may be caused by species that are not the same charge state and mass as the target precursor.30,31
Large-Scale Comparison of Mouse Brain and Liver Phosphorylation Levels
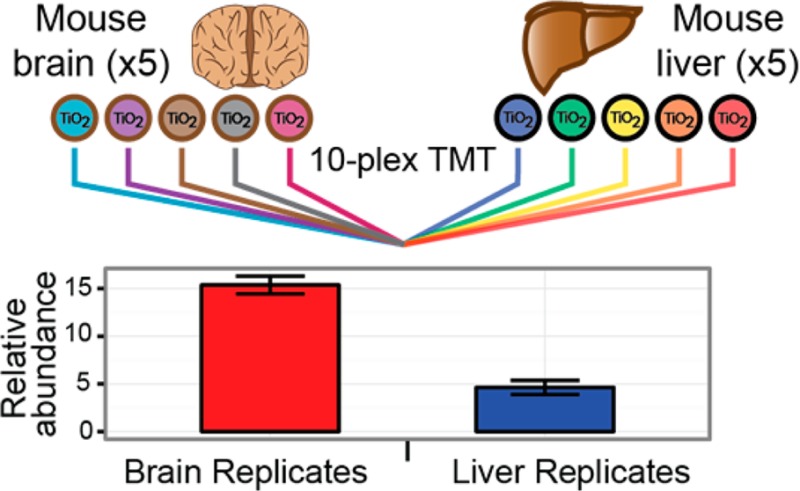
In order to more fully assess the performance of the Orbitrap Fusion, when applied to a TMT-based quantitative phosphoproteomics experiment, we constructed a 10-plex sample comprised of the brains and livers of five mice (Figure 4A). In addition to assessing the depth of the phosphoproteome characterization, the replicate tissue measurements also provided a means of assessing the overall utility of the entire quantitative phosphoproteomics workflow.
Figure 4.

(A) 10-plex TMT phosphopeptide preparation. Following proteolytic digestion, phosphopeptides were enriched by TiO2 and labeled with the 10-plex TMT reagents. Subsequent offline, basic pH reversed-phase fractionation was employed. (B) The instrument interrogated each sample using a data-dependent, SPS-MS3 method. Following ITMS2 analysis of the precursor ions, up to 10 MS2 fragments (light gray bars) were isolated and further fragmented to provide the quantitative MS3 spectrum. Reporter ion intensities corresponding to the 10 TMT channels were normalized, scaled, and summarized for the five brain and liver replicates.
Figure 4B depicts the scan sequence by which each phosphopeptide precursor is interrogated. First the precursor ion is interrogated using an ion trap MS2 scan (ITMS2). The precursor ions are isolated with the quadrupole mass filter and then fragmented by CID. Online, up to 10 fragment ions (prioritized by intensity) are noted in the MS2 spectrum for interrogation by SPS-MS3 in the subsequent scan. Following a reinjection, isolation, and fragmentation of the MS1 precursor ion, the previously determined MS2 ions are isolated via a notched isolation waveform. This collection of fragment ions is then further fragmented via HCD and passed to the Orbitrap (SPS-MS3 or FTMS3). Quantitation occurs by measuring the signal-to-noise (as a proxy for the number of ions) for each of the TMT reporters.40
In total, we quantified more than 38 000 phosphopeptides in this analysis. Because of filtering for minimum TMT signal (see Experimental Procedures), the identification rate was higher such that ∼80% of all the identified unique phosphopeptides passed all thresholds. This resulted in 11 015 phosphorylation sites and 2 958 composite phosphorylation sites quantified, providing 13 973 total quantified phosphorylation forms (Supplementary Table 1 in the Supporting Information).
In addition to assessing the depth of the phosphoproteome coverage, we assessed the level of quantitative reproducibility among biological replicates. An example of the intratissue reproducibility is highlighted in Figure 5A. For each tissue, the ratio of one biological replicate to all other biological replicates was plotted, yielding a narrow distribution of ratios with an apex centered at a ratio of one. To further highlight global reproducibility, median correlation coefficients (Pearson) for both the brain and liver replicates were determined to exceed 0.85. Finally, replicate biological comparisons displayed remarkable consistency with strong linear relationships (brain mean r2 = 0.92, liver mean r2 = 0.88) (Supplementary Figure 2 in the Supporting Information).
Figure 5.
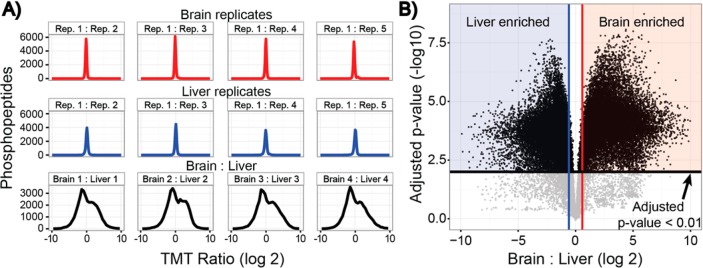
(A) Biological replicates were assessed for reproducibility. One biological replicate from brain (red) and liver (blue) was compared to all other biological replicates. The tight distributions of ratios centered about 0 (log 2) indicated good reproducibility. In contrast, ratios of brain to liver (black) display a wide distribution, highlighting the phosphoproteome diversity between the tissues. (B) Reproducible and accurate measurements across all 10 samples permitted the stringent filtering of tissue-enriched phosphopeptides. Liver (blue) and brain (red) enriched phosphopeptides were identified through a Welch’s corrected t test. Vertical lines (blue and red) represent a 1.5-fold change in expression. In all, 83% of phosphopeptides were significantly enriched in the liver or brain (adjusted p-value < 0.01).
Obtaining accurate measurements across all 10 tissues provided the opportunity to stringently assess the phosphoproteome differences between the brain and liver. Following a Welch’s corrected t test for unequal variances and Benjamini–Hochberg p-value correction for multiple testing, we observed that 83% of phosphopeptides were significantly enriched in either the brain or liver (adjusted p-value < 0.01, Figure 5B). The median fold change for brain-enriched peptides was 8-fold, while liver-enriched peptides was 5-fold.
Scaled abundances for all quantified phosphopeptides were compared among brain and liver replicates and plotted in Figure 6A. Nearly half (45%) of phosphopeptides exhibited significant expression in the brain, 38% exhibited significant expression in the liver, and 17% had consistent expression across both tissues. Gene ontology (GO) term enrichment was employed to highlight the fidelity of the measurements, and significantly enriched terms are highlighted in Figure 6B. Brain enriched terms included Synapse (cellular component) and ion channel activity (molecular function), while the liver specific phosphopeptides matched to GO terms: Transition metal ion binding (molecular function) and carboxylic acid catabolic process (biological process). GO terms corresponding to the subset of commonly expressed phosphopeptides included actin (cellular component) and DNA binding (molecular function).
Figure 6.

(A) Relative abundances of 38 247 phosphopeptides from quintuplicate biological replicates of mouse livers and brains were plotted. Statistical comparison (t test, adjusted p-values < 0.01) provided three clusters representing phosphopeptides enriched in the brain, the liver, or those that were commonly expressed in both tissues. (B) GO term enrichment was performed on each cluster resulting in descriptive terms consistent with the originating tissue. The dashed line represents the term frequency across all phosphopeptides. (C,D) Tissue specific pathways highlight the phosphopeptide coverage and quantitative reproducibility of the sample preparation and method.
With this level of phosphoproteome coverage, it was possible to highlight pathways corresponding to phosphopeptides enriched in either the brain or liver (Figure 6C,D). For each phosphopeptide, the relative abundance in the brain or liver replicates is plotted. Figure 6C illustrates a calcium signaling pathway that was observed to be highly brain specific. As displayed, Grm5, a G-protein coupled glutamate receptor initiates a signaling cascade through Gnaq, a signaling transducer, and production of inositol 1,4,5-trisphosphate (IP3) via Plcb1 and Plcd1. Stimulation of Itpr1 results in calcium release from the endoplasmic reticulum and activation of Calm1, ultimately resulting in the control of a number of biological processes including long-term depression/potentiation, MAPK signaling, and apoptosis. A liver specific pathway, PPARα signaling, is displayed in Figure 6D. Long chain fatty acids are transported via Slc27a2 and Fabp1. The RXR/PPARα heterodimer is activated by 9-cis retinoic acid, and the presence of fatty acids results in transcriptional activation of a number of downstream targets, including those involved in fatty acid transport (ACSL1 and Dbi) and cholesterol metabolism (Cyp8b1 and Nr1h3). For all peptides, the site of phosphorylation was localized via the Ascore algorithm.
Discussion
Phosphorylation analysis differs from proteome analysis in at least two ways. First, phosphopeptide enrichment creates a sample of dramatically reduced complexity (and potential interference) since most peptides do not contain a phosphate molecule. Nevertheless, we detected significant distortion using the 2-proteome model where both proteomes were made up of phosphopeptides, demonstrating that interference remains a problem for phosphorylation analysis. The starting amounts for phosphorylation analysis are up to 100 times greater than for proteome analysis. Thus, the phospho-enriched mixture from 10 mg of starting material may actually be of similar complexity to the 100 μg of proteome material.
Second, MS/MS spectra derived from phosphopeptides are commonly dominated by an intense fragment ion corresponding to the neutral loss (NL) of phosphoric acid. Prior to this work, there was some concern that the NL peak might correspond with a significant population of interfering ions. However, excluding these peaks was not an option because it would limit the resulting TMT-MS3 reporter ion population too much. We found that ratio accuracy was only minimally affected by including the NL peak. Indeed, using a single MS3 notch, which selected the NL peak as the only source for MS3 ions, did not result in significant ratio distortion. These results strongly suggest that ratio distortion is caused by species that are coisolated at the MS1 level and that differ in charge state from the target ion. This finding is in agreement with the previous work by Coon and colleagues that demonstrated that an alternative TMT purification technique based on a proton transfer reaction (PTR) also improves quantitative accuracy.30,31 On the basis of these results, we no longer believe that ratio distortion is caused by coeluting peptides, which are coisolated and cofragmented. Rather, we suspect that the majority of interference is likely caused by sustained, low levels of singly charged fragment ions created through the electrospray process (e.g., in source dissociation), which are in-turn coisolated and cofragmented with the target precursor peptide.
Multiplexing experiments with isobaric tags have the advantage that within one experiment there are no missing values. This is in contrast to binary comparisons where multiple experiments are required to compare all samples. For example, we quantified more than 38 000 phosphopeptides. Each phosphopeptide quantified produced 10 measurements from the 10 reporter ions, greatly facilitating the statistical analyses. Phosphorylation differences between brain and liver tissue were very reproducible across biological replicates. Given that the phosphopeptide enrichment step was performed prior to TMT labeling and mixing, the enrichment step was also reproducible. This is important because the alternative would be to label 1 mg or more of peptides with each reagent and then to combine them prior to enrichment, which would have dramatically increased the cost of the experiment.
We found that most phosphopeptides were present at differing levels depending on their tissue source. Only 17% of phosphorylation sites were not assigned as significantly enriched in either the brain or the liver. Sites assigned to the liver or brain were significantly enriched for gene ontology categories representative of the tissue of origin. For example, categories such as “synapse” and “ion channel activity” were frequently identified in brain and they are representative of the underlying difference in signaling.
Conclusion
Quantitative multiplexed phosphoproteome characterization, utilizing TMT reagents, presents an opportunity for unbiased biological discovery. The ability to multiplex up to 10 replicates or conditions in a single sample signifies a landmark shift in throughput, reproducibility, and robustness of these workflows. While MS2-based quantification of phosphopeptides via isobaric tagging was found to have distorted accuracy, utilizing an SPS-MS3 scan dramatically improved phosphopeptide quantitative accuracy. We demonstrated the technique via the quantification of tens of thousands of phosphopeptides from five mouse livers and brains.
Acknowledgments
We thank all members of the Gygi lab for support and discussion. This work was supported in part by the NIH (Grant GM67945). G.C.M. was supported in part by an industry-sponsored research project from ThermoFisher Scientific. M.P.J. was supported in part by the NIH (Grants GM103785 and HD073104).
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Notes
Data Availability: All raw files will be provided upon request.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Graves J. D.; Krebs E. G. Pharmacol. Ther. 1999, 82, 111–121. [DOI] [PubMed] [Google Scholar]
- Grimsrud P. A.; Swaney D. L.; Wenger C. D.; Beauchene N. A.; Coon J. J. ACS Chem. Biol. 2010, 5, 105–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemeer S.; Heck A. J. Curr. Opin. Chem. Biol. 2009, 13, 414–420. [DOI] [PubMed] [Google Scholar]
- Ubersax J. A.; Ferrell J. E. Jr Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [DOI] [PubMed] [Google Scholar]
- Beausoleil S. A.; Villén J.; Gerber S. A.; Rush J.; Gygi S. P. Nat. Biotechnol. 2006, 24, 1285–1292. [DOI] [PubMed] [Google Scholar]
- Villen J.; Beausoleil S. A.; Gerber S. A.; Gygi S. P. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 1488–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina H.; Horn D. M.; Tang N.; Mathivanan S.; Pandey A. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 2199–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai B.; Villén J.; Beausoleil S. A.; Mintseris J.; Gygi S. P. J. Proteome Res. 2008, 7, 1675–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villén J.; Gygi S. P. Nat. Protoc. 2008, 3, 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt L. J.; Tuch B. B.; Villén J.; Johnson A. D.; Gygi S. P.; Morgan D. O. Science 2009, 325, 1682–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin E. L.; Jedrychowski M. P.; Elias J. E.; Goswami T.; Rad R.; Beausoleil S. A.; Villén J.; Haas W.; Sowa M. E.; Gygi S. P. Cell 2010, 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruhler A.; Olsen J. V.; Mohammed S.; Mortensen P.; Færgeman N. J.; Mann M.; Jensen O. N. Mol. Cell. Proteomics 2005, 4, 310–327. [DOI] [PubMed] [Google Scholar]
- Olsen J. V.; Blagoev B.; Gnad F.; Macek B.; Kumar C.; Mortensen P.; Mann M. Cell 2006, 127, 635–648. [DOI] [PubMed] [Google Scholar]
- Dephoure N.; Zhou C.; Villen J.; Beausoleil S. A.; Bakalarski C. E.; Elledge S. J.; Gygi S. P. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 10762–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub H.; Olsen J. V.; Bairlein M.; Gnad F.; Oppermann F. S.; Körner R.; Greff Z.; Kéri G.; Stemmann O.; Mann M. Mol. Cell 2008, 31, 438–448. [DOI] [PubMed] [Google Scholar]
- Olsen J. V.; Vermeulen M.; Santamaria A.; Kumar C.; Miller M. L.; Jensen L. J.; Gnad F.; Cox J.; Jensen T. S.; Nigg E. A.; Brunak S.; Mann M. Sci. Signal. 2010, 3, ra3. [DOI] [PubMed] [Google Scholar]
- Lundby A.; Secher A.; Lage K.; Nordsborg N. B.; Dmytriyev A.; Lundby C.; Olsen J. V. Nat. Commun. 2012, 3, 876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P. P.; Kang S. A.; Rameseder J.; Zhang Y.; Ottina K. A.; Lim D.; Peterson T. R.; Choi Y.; Gray N. S.; Yaffe M. B.; Marto J. A.; Sabatini D. M. Science 2011, 332, 1317–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Yoon S.-O.; Poulogiannis G.; Yang Q.; Ma X. M.; Villén J.; Kubica N.; Hoffman G. R.; Cantley L. C.; Gygi S. P.; Blenis J. Science 2011, 332, 1322–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R.; Haas W.; Dephoure N.; Huttlin E. L.; Zhai B.; Sowa M. E.; Gygi S. P. Nat. Methods 2011, 8, 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille A. M.; Christen S.; Shimobayashi M.; Cornu M.; Fava L. L.; Moes S.; Prescianotto-Baschong C.; Sauer U.; Jenoe P.; Hall M. N. Science 2013, 339, 1320–1323. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Zhang C.; Croucher D. R.; Soliman M. A.; St-Denis N.; Pasculescu A.; Taylor L.; Tate S. A.; Hardy W. R.; Colwill K.; Dai A. Y.; Bagshaw R.; Dennis J. W.; Gingras A.-C.; Daly R. J.; Pawson T. Nature 2013, 499, 166–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courcelles M.; Frémin C.; Voisin L.; Lemieux S.; Meloche S.; Thibault P. Mol. Syst. Biol. 2013, 9, n/a–n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-Y.; Welsh E. A.; Oguz U.; Fang B.; Bai Y.; Kinose F.; Bronk C.; Remsing Rix L. L.; Beg A. A.; Rix U.; Eschrich S. A.; Koomen J. M.; Haura E. B. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 12414–12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M.; Boesche M.; Eberhard D.; Matthieson T.; Sweetman G.; Kuster B. Mol. Cell. Proteomics 2008, 7, 1702–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ow S. Y.; Salim M.; Noirel J.; Evans C.; Rehman I.; Wright P. C. J. Proteome Res. 2009, 8, 5347–5355. [DOI] [PubMed] [Google Scholar]
- Lu R.; Markowetz F.; Unwin R. D.; Leek J. T.; Airoldi E. M.; MacArthur B. D.; Lachmann A.; Rozov R.; Ma’ayan A.; Boyer L. A.; Troyanskaya O. G.; Whetton A. D.; Lemischka I. R. Nature 2009, 462, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirran S. L.; Botting C. H. J. Proteomics 2010, 73, 1391–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp N. A.; Huber W.; Sadowski P. G.; Charles P. D.; Hester S. V.; Lilley K. S. Mol. Cell. Proteomics 2010, 9, 1885–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger C. D.; Lee M. V.; Hebert A. S.; McAlister G. C.; Phanstiel D. H.; Westphall M. S.; Coon J. J. Nat. Methods 2011, 8, 933–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent C. E.; Rensvold J. W.; Westphall M. S.; Pagliarini D. J.; Coon J. J. Anal. Chem. 2013, 85, 2079–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L.; Rad R.; Gygi S. P.; Haas W. Nat. Methods 2011, 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister G. C.; Nusinow D. P.; Jedrychowski M. P.; Wühr M.; Huttlin E. L.; Erickson B. K.; Rad R.; Haas W.; Gygi S. P. Anal. Chem. 2014, 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenbach A. N.; Gerber S. A. Anal. Chem. 2011, 83, 7635–7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng J. K.; McCormack A. L.; Yates J. R. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Elias J. E.; Gygi S. P. Nat. Methods 2007, 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Benjamini Y.; Hochberg Y. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar]
- Huang D. W.; Sherman B. T.; Lempicki R. A. Nat. Protoc. 2008, 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Martin D. B.; Eng J. K.; Nesvizhskii A. I.; Gemmill A.; Aebersold R. Anal. Chem. 2005, 77, 4870–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov A.; Denisov E. J. Am. Soc. Mass Spectrom. 2009, 20, 1486–1495. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.