

Abstract
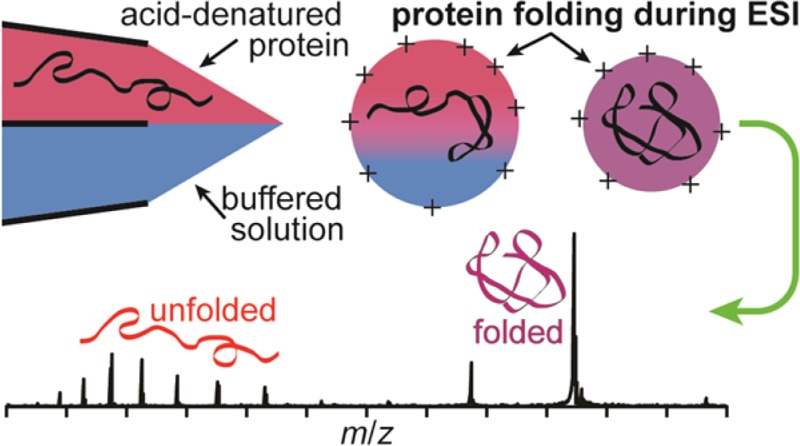
Theta-glass emitters are used to rapidly mix two solutions to induce either protein folding or unfolding during nanoelectrospray (nanoESI). Mixing acid-denatured myoglobin with an aqueous ammonium acetate solution to increase solution pH results in protein folding during nanoESI. A reaction time and upper limit to the droplet lifetime of 9 ± 2 μs is obtained from the relative abundance of the folded conformer in these rapid mixing experiments compared to that obtained from solutions at equilibrium and a folding time constant of 7 μs. Heme reincorporation does not occur, consistent with the short droplet lifetime and the much longer time constant for this process. Similar mixing experiments with acid-denatured cytochrome c and the resulting folding during nanoESI indicate a reaction time of between 7 and 25 μs depending on the solution composition. The extent of unfolding of holo-myoglobin upon rapid mixing with theta-glass emitters is less than that reported previously (Fisher et al. Anal. Chem. 2014, 86, 4581−4588), a result that is attributed to the much smaller, ∼1.5 μm, average o.d. tips used here. These results indicate that the time frame during which protein folding or unfolding can occur during nanoESI depends both on the initial droplet size, which can be varied by changing the emitter tip diameter, and on the solution composition. This study demonstrates that protein folding or unfolding processes that occur on the ∼10 μs time scale can be readily investigated using rapid mixing with theta-glass emitters combined with mass spectrometry.
Electrospray ionization (ESI) mass spectrometry (MS) is widely used to obtain information about protein structure, including protein identification and analysis of posttranslational modifications.1−3 Information about protein conformation can also be obtained from charge state distributions in ESI mass spectra.4−8 Gaseous ions formed from solutions in which a protein has a native or globular conformation are less highly charged than the corresponding ions formed from solutions in which the protein is unfolded. The relative abundances of different conformations of a protein in solution have been obtained by modeling the resulting charge-state distributions observed in ESI mass spectra.6 Charge-state distributions have been used to monitor protein unfolding both as a function of temperature by heating the sample solution during ESI4,7,8 and as a function of solution pH by inducing a pH change in the sample solution using the electrolytic oxidation of water that occurs at the metal–liquid interface in an ESI emitter.5
Protein conformational changes can also take place during the ESI process. McLuckey and co-workers demonstrated that exposing aqueous ESI droplets containing folded proteins to either gaseous acids9 or bases10 in an ESI interface can result in a bimodal charge-state distribution of ions, indicating that a fraction of the protein population unfolds in the ESI droplet. ESI droplets generated from “native” protein solutions have also been exposed to acids and supercharging reagents in solution by flowing a continuous stream of solution from a hypodermic needle into the ESI plume.11 The stream–droplet interactions in these experiments result in bimodal charge-state distributions of the resulting protein ions, indicative of protein unfolding. Protein unfolding in ESI droplets can also be induced thermally, which is the likely mechanism of electrothermal supercharging,12,13 wherein high charge states are obtained from buffered aqueous protein solutions by raising the electrospray potential to collisionally activate and heat the ESI droplets.
In contrast to the many studies of protein unfolding during ESI, there are fewer reports of protein folding during ESI. Acid-denatured proteins in ESI droplets have been exposed to gaseous bases in an ESI interface, resulting in a bimodal distribution of charge states for some proteins.14 Although gas-phase proton transfer reactions may contribute to lower charging in these experiments, the bimodal charge-state distributions indicate that a fraction of the protein population folds as a result of an increase in the droplet pH. However, comparisons of the results obtained from these experiments to those from bulk-solution experiments are complicated because of the dynamic nature of ESI. A low charge-state distribution of myoglobin that was thermally denatured was observed in nanoESI, which suggests that protein folding occurred in the droplets upon rapid evaporative cooling.8
Quantitative information about protein folding kinetics can be obtained in mixing experiments, and ESI MS has been combined with a variety of rapid mixers, including continuous,15,16 stopped,17 and laminar flow18,19 mixers, to measure the kinetics of protein folding17−19 and unfolding16 and the reincorporation rates of noncovalent cofactors into the protein structure.15 In conventional mixers, faster kinetic processes can be measured by increasing the flow rates of the solutions. A dead time of 200 μs has been reported for a laminar flow mixer coupled with MS operating at a flow rate of 10 μL/s.19 This setup was used to study the subms folding steps that occur after the initial collapse, or “burst-phase,” of apo-myoglobin that takes place on an even shorter time scale (∼7 μs time constant).20 Rapid mixing of two solutions during ESI has also been used to unfold proteins during ESI. Microfabricated dual sprayers have been used to mix solutions containing peptides or proteins with solutions containing supercharging reagents,21 resulting in increased charging indicative of protein unfolding.
Double-barrel wire-in-a-capillary emitters made from theta glass (theta-glass emitters) have also been used to mix solutions during ESI. Rapid mixing from theta-glass emitters was first used to form a noncovalent complex between a short peptide (KAA) and a glycopeptide (vancomycin) and to exchange hydrogen and deuterium with undeuterated and partially deuterated vancomycin during ESI.22 Substantial mixing was reported to occur in the Taylor cone prior to droplet formation.22 Solutions containing folded proteins were mixed with acidic solutions using theta-glass emitters, which produced bimodal charge-state distributions and loss of the heme group for myoglobin.23 These results indicate that protein unfolding occurred during ESI.23 Supercharging reagents were mixed into solutions containing folded proteins during ESI using theta-glass emitters which resulted in higher charging and loss of heme for myoglobin, consistent with unfolding of the proteins in the ESI droplets induced by the supercharging reagents.23 A fast complexation reaction was used to show that complete mixing can occur between two different solutions in the theta-glass emitters during nanoESI.24 An estimate of the droplet lifetime of <27 μs was determined24 from the reaction kinetics between l-ascorbic acid and 2,6-dichloroindophenol using theta-glass emitters with ∼1.5 μm o.d. tips and from the faster reaction rates observed in droplets over those in bulk solution.25,26 The very short time scale for mixing and droplet lifetimes indicates that fast protein folding could be observed by rapidly mixing solutions in theta-glass emitters coupled with MS.24 Investigation of protein folding by mixing solutions using theta-glass emitters has the advantage of very low flow rates, e.g., as low as 1.4 nL/s, resulting in low sample consumption.24
Here, theta-glass emitters are used to investigate protein folding and unfolding during nanoESI by inducing a pH change in the electrospray solution by rapidly mixing two solutions at the emitter tip. The relative flow rates of these solutions are measured using peptides as standards. From the measured extents of myoglobin and cytochrome c folding and known protein-folding time constants, a time of between 7 and 25 μs, depending on solution composition, is obtained as an upper limit for the lifetime of nanoESI droplets. Significantly fewer sodium ions are adducted to the ions generated from the native-like conformer when native myoglobin is mixed with acid using the theta-glass emitters than to these ions generated from a native solution. This is likely the result of a higher concentration of protons and the short time available for protein unfolding to occur. These results suggest that rapid mixing from theta-glass emitters may be useful in reducing sodium adduction in native MS.
Experimental Section
Experiments were performed using a 9.4 T Fourier-transform ion cyclotron resonance mass spectrometer that is described elsewhere.27 Theta glass capillaries (Warner Instruments, LLC, Hamden, CT) were pulled using a model p-87 Flaming/Brown micropipette puller (Sutter Instruments Co., Novato, CA) into tips with an o.d. of 1.36 ± 0.02 μm parallel to and 1.71 ± 0.04 μm perpendicular to the central divider.24 A grounded platinum wire is brought into contact with the solution that is loaded into each barrel, and a backing pressure of ∼10 psi is applied to the back end of the capillary. Ion formation is initiated by applying a potential of ∼−700 V to the heated capillary of the mass spectrometer interface. All data were acquired using a Predator data station (National High Magnetic Field Laboratory, Tallahassee, FL), and mass spectra were background subtracted. Average charge states are computed as the abundance weighted sum of individual charge states in a distribution. All reported uncertainties are standard deviations determined from three replicate measurements.
Leu-enkephalin acetate salt hydrate, Met-enkephalin acetate salt hydrate, ammonium acetate, formic acid, equine apo- and holo-myoglobin, equine cytochrome c, and polypropylenimine hexadecaamine dendrimer, generation 3.0, were obtained from Sigma-Aldrich (St. Louis, MO). Glacial acetic acid was obtained from Fisher Scientific (Fair Lawn, NJ). Solutions were prepared with an analyte concentration of 10 μM in 18.2 MΩ water from a Milli-Q water purification system (Millipore, Billerica, MA).
The initial pH of droplets formed when two solutions are mixed using the theta-glass emitters is determined from the pKb value of ammonia (4.8), the pKa values of acetic and formic acid (4.8 and 3.8, respectively; all values at 25 °C),28 and the initial concentrations of these species in the droplets. Initial concentrations are determined from the initial concentrations of the analytes in the solutions in both barrels of the theta-glass emitters and from the relative flow rates of these solutions during nanoESI. Relative flow rates are measured using Leu-enkephalin and Met-enkephalin as internal standards (1.0 μM) as described elsewhere.24
Results and Discussion
Unfolding Holo-Myoglobin During NanoESI
In low-ionic-strength aqueous solutions (salt concentrations of less than ∼0.02 M), holo-myoglobin (holo-Mb) has a native conformation between pH = 5 and 7,29 a less-compact globular structure around pH = 3,29 and an unfolded structure with no heme group attached at lower pH.30 A representative nanoESI mass spectrum of holo-Mb in a 1.0 mM aqueous ammonium acetate solution (pH = 6.4) is shown in Figure 1a. The charge states (7–9+) are indicative of forming these ions from an aqueous solution in which the protein has a native structure.6 A nanoESI mass spectrum obtained by mixing this solution with a 1.0 M aqueous acetic acid solution at a 1:1 ratio (pH = 2.6) is shown in Figure 1b. The 10–26+ charge states of apo-myoglobin (apo-Mb) are formed and no holo-Mb is observed, indicating that extensive unfolding of the protein and concomitant loss of the heme group has occurred in solution.
Figure 1.
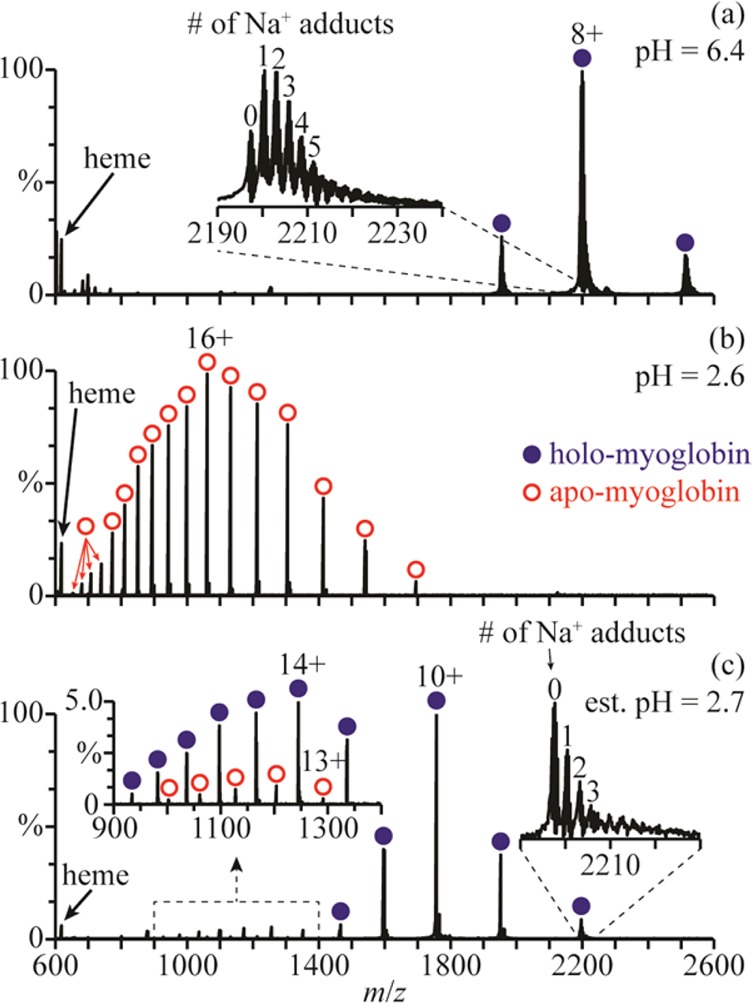
NanoESI mass spectra of (a) holo-Mb in a 1.0 mM aqueous ammonium acetate solution (pH = 6.4), (b) this ammonium acetate solution mixed with a 1.0 M aqueous acetic acid solution at a 1:1 ratio (pH = 2.6), and (c) this ammonium acetate solution mixed with the acetic acid solution using a theta-glass emitter (estimated pH = 2.7). Insets in (a) and (c) show the extent of Na+ adduction to the 8+ charge states and, in (c), the distributions of apo- and holo-Mb between m/z 900 and 1400.
A nanoESI mass spectrum resulting from mixing these two solutions using the theta-glass emitters is shown in Figure 1c. The charge-state distribution of holo-Mb is bimodal, with a high abundance distribution between 8+ and 12+ (comprising 86 ± 3% of myoglobin) and a low abundance distribution between 13+ and 19+ (comprising 11 ± 1% of myoglobin). The distribution between 13+ and 19+ corresponds to more open structures resulting from protein unfolding during nanoESI. The 13–17+ charge states of apo-Mb are also observed (comprising 3 ± 2% of myoglobin), indicating that some heme loss accompanies unfolding of the protein in the nanoESI droplet.
The average charge state of the distribution between 8+ and 12+ in the rapid mixing experiments (9.8 ± 0.2) is higher than the average charge state of holo-Mb in the mass spectra obtained from the pH = 6.4 solution (8.0 ± 0.1). The higher charging obtained for the folded form of the protein in the rapid mixing experiments may be a result of a small change in the conformation, or it could be due to effects of solution composition on charging. To determine the role of solution composition on charging in these experiments, nanoESI mass spectra of polypropylenimine hexadecaamine dendrimer, generation 3.0 (DAB-16), were obtained under similar conditions. NanoESI of DAB-16 in a 1.0 mM aqueous ammonium acetate solution adjusted to pH = 6.4 with acetic acid results in an average charge state of 3.6 ± 0.1 (Figure 2a). This solution was mixed with a 1.0 M aqueous acetic acid solution at a 1:1 ratio (pH = 2.6), and in the nanoESI mass spectra of the resulting solution, the average charge state is 4.0 ± 0.1 (Figure 2b). Results from small-angle neutron scattering, viscosimetry, and molecular dynamics studies indicate that the conformation of DAB-16 does not strongly depend on solution composition.31 Thus, the slightly higher charging obtained from this solution is likely a result of the different solution composition, although it may also reflect small changes to the shape of DAB-16. These results suggest that the shift in charging of the folded form of myoglobin in the rapid mixing experiments may be due, at least in part, to the different solution composition as well as any potential change to the native protein structure.
Figure 2.

NanoESI mass spectra of (a) DAB-16 in a 1.0 mM aqueous ammonium acetate solution adjusted to pH = 6.4 with acetic acid and (b) this solution adjusted to pH = 2.6 by mixing with a 1.0 M aqueous acetic acid solution at a 1:1 ratio. qave denotes the average charge state.
Fisher et al.23 also reported that myoglobin unfolds when aqueous holo-Mb is mixed with an aqueous solution of 1% acetic acid (∼174 mM) using theta-glass emitters, resulting in the formation of 7% apo-Mb. In our experiment, a 5-fold higher concentration of acetic acid is used, yet only 3% apo-Mb is produced (Figure 1c). The different extents of unfolding and heme loss in these two experiments are likely a result of different reaction times. The reaction time is limited by the droplet lifetime, which depends on the initial droplet diameter32 and, thus, on the diameter of the electrospray capillary.33 Fisher et al. used theta-glass emitters with ∼10 μm o.d. tips, whereas ∼1.5 μm o.d. tips were used here, which results in smaller initial droplets and less time for protein unfolding to occur.
It is also interesting that there is less sodium ion adduction on the folded holo-Mb ions formed after mixing the two solutions using the theta-glass emitters (Figure 1c) than on those formed directly from the ammonium acetate solution (Figure 1a). For example, the average number of sodium ions adducted to the 8+ charge state is 3.1 ± 0.8 in the nanoESI mass spectra of the ammonium acetate solution and only 0.7 ± 0.2 in those resulting from the solutions mixed using the theta-glass emitters. The lower average number of sodium adducts upon mixing likely results from the >1000-fold increase in the initial concentration of protons in the droplet, which can displace sodium ions near the surface of the protein during droplet evaporation.
Folding Apo-Myoglobin During NanoESI
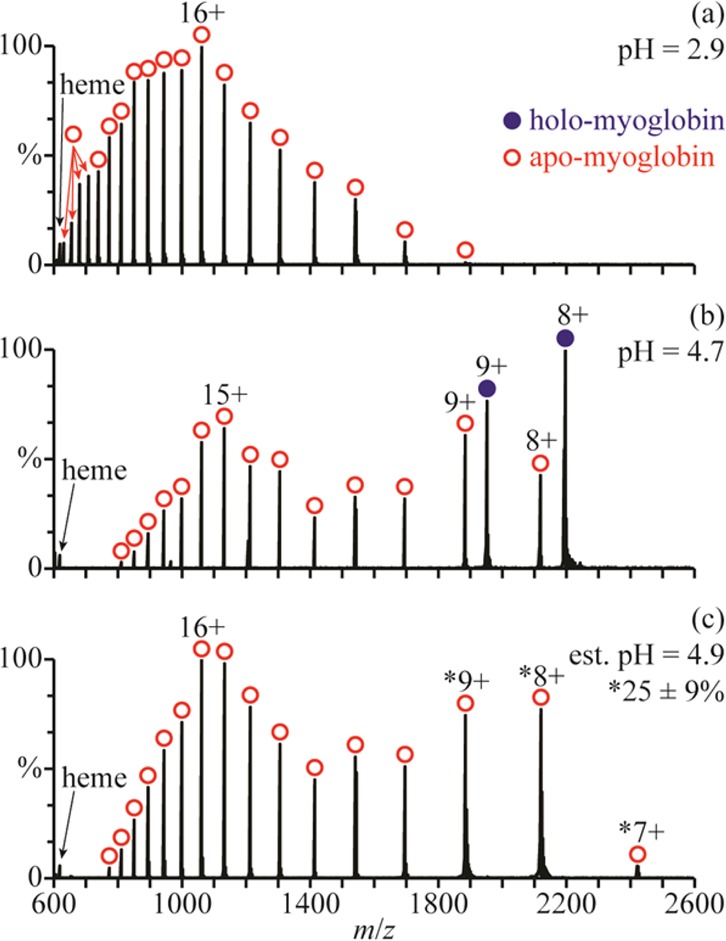
There are at least three conformers of apo-myoglobin (apo-Mb) that exist in aqueous solution. Between pH = 5 and 7, apo-Mb adopts a conformation similar to that of native holo-Mb, at pH = 4, it has a less compact globular structure, and below pH = 3, it is unfolded.34 NanoESI of acid-denatured apo-Mb (pH = 2.9; Figure 3a) results in a charge-state distribution between 11+ and 28+. This high charging is consistent with an unfolded form of apo-Mb in solution. Results from raising the pH of this solution using a 100 mM aqueous ammonium acetate solution are shown in Figure 3b–d. The addition of ammonium acetate results in the elimination of the highest charge states, and a second charge-state distribution between 7+ and 9+ becomes more abundant with increasing pH. This charge-state distribution is consistent with a globular conformer in solution and comprises 13 ± 4%, 25 ± 5%, and 35 ± 7% of myoglobin at pH = 3.9, 4.4, and 4.7, respectively (Figure 3b–d). The 11+ charge state is slightly more abundant than the 10+ and 12+ charge states, suggesting that a partially folded intermediate structure may also be present.
Figure 3.

NanoESI mass spectra of (a) acid-denatured apo-Mb (pH = 2.9), acid-denatured apo-Mb mixed with a 100 mM aqueous ammonium acetate solution at ratios of (b) 10:1 (pH = 3.9), (c) 2:1 (pH = 4.4), and (d) 1:1 (pH = 4.7), and (e) acid-denatured apo-Mb mixed with this ammonium acetate solution using a theta-glass emitter (estimated pH = 4.7). Percentages are the relative abundances of the folded fractions (7–9+ charge states) of apo-Mb.
Mixing the initial acidified solution (Figure 3a) with the 100 mM aqueous ammonium acetate solution using the theta-glass emitters produces droplets with an initial pH of ∼4.7. In the nanoESI mass spectra resulting from this experiment (Figure 3e), there are at least two charge-state distributions for apo-Mb: one between 10+ and 21+ corresponding to an unfolded conformer and another between 7+ and 9+ corresponding to the fraction of apo-Mb that is folded into a globular conformer during nanoESI (25 ± 2% of apo-Mb). The 11+ charge state is again more abundant than the 10+ and 12+, possibly suggesting the presence of a partially folded intermediate structure. At pH = 4.7, the equilibrium distribution of folded protein (7−9+) constitutes 35 ± 7% of apo-Mb (Figure 3d). However, the abundance of this distribution in the rapid mixing experiments (estimated initial droplet pH = 4.7) more closely resembles that in the nanoESI mass spectra of a pH = 4.4 solution at equilibrium, wherein it compromises 25 ± 5% of apo-Mb (Figure 3c). This result indicates that the droplet lifetime is sufficiently short so that equilibrium is not reached during nanoESI and that protein folding is incomplete.
The initial collapse of apo-Mb from an unfolded to a globular structure occurs with a time constant of ∼7 μs,20 and the subsequent formation of a structure similar to that of native holo-Mb takes more than a ms to occur.35 In previous mixing experiments using these same theta-glass emitters, a <27 μs droplet lifetime was deduced from the extent of product formation of a fast reaction with a known rate constant in bulk solution.24 The short droplet lifetime established in those experiments indicates that only the initial collapse of apo-Mb is likely to occur to a significant extent in the droplets.
An estimate of the time scale for protein folding in this study is obtained by modeling the initial collapse of apo-Mb as an independent, two-state folding reaction.36,37 The integrated rate law for a two-state folding reaction is given in eq 1:
| 1 |
where t is the reaction time, τ is the protein folding time constant, Ae is the abundance of the folded protein conformer at equilibrium, and Ao and At are the abundances of the folded protein conformer at times 0 and t, respectively. From the relative abundances of the globular apo-Mb conformer in the unmixed, equilibrium, and rapid mixing experiments (Figure 3a,d,e, respectively) and the 7 μs time constant of the initial collapse of apo-Mb,20 a reaction time of 9 ± 2 μs is obtained. This reaction time is consistent with the <27 μs droplet lifetime reported previously that was deduced on the basis of the extent of product formation for a bimolecular reaction and a known solution reaction rate constant.24 Because the concentrations of the reagents increase to an unknown extent as droplets evaporate and bimolecular reaction rates depend on concentration, there is a significant uncertainty in the true droplet lifetime obtained from previous measurements. In contrast, folding of apo-Mb is a unimolecular process that does not depend on protein concentration at the concentrations used. Surface effects as a result of the high surface-to-volume ratios of small droplets compared to that of bulk solution may influence protein folding rates in droplets.26 However, similarity between the droplet lifetimes obtained by both methods indicate that surface effects may be small in these protein folding experiments.
Folding and Reincorporation of the Heme
Although folding of apo-Mb occurs quickly,20,35 reincorporation of the heme group into the protein structure requires considerably more time (hundreds of milliseconds to seconds).15 In order to determine if heme reincorporation can occur during nanoESI, solutions that result in heme reincorporation at equilibrium when mixed at a 1:1 ratio were mixed using the theta-glass emitters. A mass spectrum resulting from mixing a solution of acid-denatured holo-Mb (pH = 2.9; Figure 4a) and a solution of 100 mM aqueous ammonium acetate at a 1:1 ratio is shown in Figure 4b. There is a bimodal charge-state distribution of apo-Mb centered at 15+ and at 9+ as well as a distribution of holo-Mb in the 9+ and 8+ charge states. The relative abundances of unfolded and folded apo-Mb and folded holo-Mb are 50 ± 3%, 13 ± 3%, and 37 ± 4%, respectively.
Figure 4.
Mass spectra of (a) acid-denatured holo-Mb (pH = 2.9), (b) acid-denatured holo-Mb mixed with a 100 mM aqueous ammonium acetate solution at a 1:1 ratio (pH = 4.7), and (c) acid-denatured holo-Mb mixed with the ammonium acetate solution using a theta-glass emitter (estimated pH = 4.9). The percentage in (c) is the relative abundance of the folded fraction (7–9+ charge states) of apo-Mb.
A nanoESI mass spectrum resulting from mixing the acidified solution (Figure 4a) with the 100 mM aqueous ammonium acetate solution using a theta-glass emitter is shown in Figure 4c. There is a bimodal charge-state distribution of apo-Mb centered at 16+ and ∼8+, corresponding to unfolded and folded forms of the protein, respectively. The initial pH of the mixed droplets is ∼4.9, and the folded form of the protein comprises 25 ± 9% of myoglobin, consistent with the results from solutions without heme present (25 ± 2%, Figure 3e). No holo-Mb is observed despite the presence of both a globular apo-Mb conformer and the heme group in solution. This result indicates that heme reincorporation does not occur to an appreciable extent within the nanoESI droplet, which is consistent with the droplet lifetime of ∼9 μs measured using the refolding of apo-Mb and the heme reincorporation time constant on the order of hundreds of milliseconds to seconds.15
Temperature of NanoESI Droplets
Fragments of ions formed by ESI can be observed in some ESI mass spectra, and this has led some to conclude that the ESI process itself activates ions.38−42 For example, Vékey and co-workers concluded from the extent of fragmentation of benzylpyridinium salts during ESI compared to that predicted by RRKM calculations at various temperatures that electrospray ionization produces ions with internal temperatures greater than 200 °C.38,39 The potential used to produce ions in ESI can result in droplet heating, such as in electrothermal supercharging, wherein high charge states of protein ions are formed from buffered aqueous solutions.12,13 However, extensively hydrated ions can also be directly produced using ESI, and evaporative cooling significantly reduces the temperatures of these clusters.43−46 Results for trivalent atomic ions show that these ions require approximately 18 water molecules to be stable, indicating that these clusters are formed by evaporative cooling of even larger clusters and not by condensation of water onto minimally solvated or bare ions.46 Because preservation of weakly bound water molecules can only occur in ESI droplets that are not significantly heated, these results show that the ESI process itself is not activating, although activation can occur either in the ESI droplet or after ion formation as a result of instrumental conditions.
Apo-Mb thermally denatures between about 60 and 70 °C in unbuffered aqueous solutions.20 The folding of this protein during nanoESI shown here (Figures 2e and 3c) indicates that the droplet temperature in these experiments must be less than 70 °C. This result shows that the droplet temperature in ESI can be lower than the melting point of some proteins. In sum, these results indicate that ESI droplets can have low temperatures and that ion formation by ESI is intrinsically soft under appropriate experimental conditions.
Folding Cytochrome c During NanoESI
In low ionic strength aqueous solutions, cytochrome c (cyt c) is unfolded with little secondary structure at pH = 2 but has a native folded structure between pH = 3 and 7.47,48 A globular form, known as the A state, can form at high salt concentrations (0.2 M),49,50 and a partially folded intermediate structure may also form during folding in salt-free solutions.51,52 A mass spectrum of cyt c in an aqueous solution with 2.5% formic acid (pH = 2.0) is shown in Figure 5a. The charge-state distribution is bimodal, with a low-abundance distribution between 7+ and 9+ corresponding to a folded structure (11 ± 4% of cyt c) and a high-abundance distribution between 10+ and 18+ corresponding to unfolded structures in solution. This solution was mixed with a solution of 500 mM aqueous ammonium acetate at a 1:1 ratio (pH = 4.2), and a mass spectrum of the resulting solution is shown in Figure 5b. The 6–8+ charge states are formed and comprise 98 ± 0% of cyt c in the mass spectra. The 14+ charge state is also present at low abundance. These results indicate that cyt c has predominantly adopted a folded structure in this solution at equilibrium.
Figure 5.

Mass spectra of (a) cyt c in an aqueous solution of 2.5% formic acid (pH = 2.0), (b) this formic acid solution mixed with a 500 mM aqueous ammonium acetate solution at a 1:1 ratio (pH = 4.2), and (c) this formic acid solution mixed with the ammonium acetate solution using a theta-glass emitter (estimated pH = 4.2). Mass spectra of (d) cyt c in an aqueous solution of 1.0% acetic acid (pH = 2.8), (e) this acetic acid solution mixed with water at a 1:1 ratio (pH = 2.9), and (f) this acetic acid solution mixed with water using a theta-glass emitter (estimated pH = 2.9). Percentages are the relative abundances of the fraction of cyt c in charge states corresponding to folded conformers (denoted with ∗).
A mass spectrum resulting from mixing these two solutions using a theta-glass emitter is shown in Figure 5c. The charge state distribution is bimodal, with one distribution between 6+ and 8+ corresponding to a folded structure (21 ± 1% of cyt c) and another between 9+ and 17+ corresponding to an unfolded structure. The initial folding step of cyt c occurs with a time constant of ∼57 μs,47 and other steps may also occur with time constants at or above 600 μs.52−54 The droplet lifetime of <27 μs reported previously24 and the 9 μs lifetime obtained here for the folding of apo-Mb indicate that only the 57 μs folding step of cyt c is likely to occur to a significant extent in these experiments. The abundances of the folded cyt c conformer in the unmixed, equilibrium, and rapid mixing experiments (Figure 5a–c, respectively) and the 57 μs time constant are used to obtain a reaction time of 7 ± 3 μs from eq 1. This result is remarkably similar to the reaction time of 9 ± 2 μs obtained for the folding of apo-Mb during the nanoESI process.
Slightly different solution conditions were used in these experiments than those in which the protein folding time constants were obtained, which may affect the droplet lifetimes reported here. The time constant for the initial collapse of apo-Mb was measured in a pH = 5.9 solution containing 10 mM sodium acetate20 and the folding time constant of cyt c in a pH = 4.5 solution containing 25 mM sodium phosphate and 25 mM sodium acetate.47 In this study, apo-Mb and cyt c were folded in droplets containing higher salt concentrations (∼50 and ∼250 mM ammonium acetate, respectively) and slightly lower initial pHs (pH = 4.7 and 4.2, respectively). The different solution compositions likely result in slightly different protein-folding time constants in the nanoESI droplets than were measured in those bulk solution experiments. However, the remarkable agreement in reaction times obtained for apo-Mb and cyt c in the rapid mixing experiments indicates a relatively small uncertainty in the measured reaction times despite the different solution conditions used to obtain the folding time constants.
Between pH = 2 and 3, the unfolded and globular forms of cyt c coexist in equilibrium.48 The equilibrium distribution of partially acid-denatured cyt c can be shifted toward the folded conformer by diluting the solution with water. In the nanoESI mass spectrum of cyt c at pH = 2.8 (1.0% acetic acid in water, Figure 5d), the charge-state distribution is bimodal, with one distribution centered at 8+ corresponding to a folded structure (34 ± 7% of cyt c) and another distribution centered at 16+ corresponding to an unfolded structure. Results from mixing this solution with water at a 1:1 ratio (pH = 2.9) are shown in Figure 5e. The same charge-state distributions are observed, but the relative abundances of these distributions indicate that 76 ± 5% of cyt c is in the folded form. This result shows that the equilibrium distribution between the two forms of cyt c shifts toward the globular structure in this solution.
A nanoESI mass spectrum resulting from mixing these two solutions using a theta-glass emitter is shown in Figure 5f. The same charge-state distributions are observed, and the relative abundances of these distributions indicate that the folded form comprises 49 ± 7% of cyt c. From the abundances of the globular cyt c conformer in the unmixed, equilibrium, and rapid mixing experiments (Figure 5d–f, respectively) and a folding time constant of 57 μs for cyt c,47 a reaction time of 25 ± 7 μs is obtained from eq 1. This reaction time is about 3-fold higher than the reaction times for the folding of myoglobin (9 ± 2 μs) and of cyt c from the acidified solution mixed with 500 mM aqueous ammonium acetate (7 ± 3 μs).
The shorter droplet lifetime deduced from the experiments in which ammonium acetate is used to increase the initial pH of the droplets may be due to uncertainties associated with the folding time constants measured under slightly different conditions. It may also be a result of faster water evaporation or of differences in the temperatures of the nanodrops. Emission of water-solvated ammonium ions from the droplet55,56 could result in an increased rate of droplet evaporation and, hence, a shorter droplet lifetime. The presence of ammonium acetate may also affect the droplet temperature,57 which would affect the protein-folding time constants.37,58
Conclusions
Theta-glass emitters were used to rapidly mix two solutions in order to induce either protein folding or unfolding during nanoESI. Acid-denatured myoglobin was mixed with a solution of ammonium acetate to increase pH and induce folding both with and without heme present in solution. The extents of folding in these experiments compared to those obtained at equilibrium indicates a reaction time of 9 ± 2 μs, which is an upper limit to the droplet lifetime because some folding will occur in the Taylor cone prior to droplet formation. Reincorporation of the heme into the folded protein structure does not occur, consistent with the much longer time constant of hundreds of milliseconds to seconds for this process.15 Similarly, droplet lifetimes of 7 ± 3 and 25 ± 7 μs, depending on solution compositions, were obtained from folding experiments with cyt c. These results indicate that the nanoESI droplet lifetime can be very short and that the droplets are not heated past the melting points of many proteins7,20 under appropriate experimental conditions.
The extent of unfolding of holo-Mb obtained in the rapid mixing experiments in this study is less than that reported earlier,23 a result that is consistent with the much smaller tips used in the current experiments. These results indicate that the time frame for reactions to occur during the nanoESI process can be readily controlled by changing the emitter tip diameter or other properties that affect the initial size of the nanoESI droplets. Thus, it should be possible to acquire “snapshots” of protein folding or unfolding at various time points by varying the diameter of the tips of the theta-glass emitters.
The extent of sodium ion adduction to the folded forms of protein ions resulting from rapidly mixing proteins in buffered aqueous solutions with acidified solutions using the theta-glass emitters is less than in protein ions generated from native solutions. This is likely the result of the much higher concentration of protons that can displace sodium ions at the protein surface during nanoESI. This may be an effective way to reduce sodium adduction in native MS without significantly affecting the resulting protein ion structure if the unfolding time of the protein is significantly greater than the time frame for ion formation in nanoESI.
Acknowledgments
The authors thank Catherine Cassou for helpful discussions and the National Institutes of Health for funding (R01GM097357).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Aebersold R.; Mann M. Nature 2003, 422, 198–207. [DOI] [PubMed] [Google Scholar]
- Siuti N.; Kelleher N. L. Nat. Methods 2007, 4, 817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.; Borchers C. H. Proteomics 2013, 13, 974–981. [DOI] [PubMed] [Google Scholar]
- Mirza U.; Cohen S.; Chait B. Anal. Chem. 1993, 65, 1–6. [DOI] [PubMed] [Google Scholar]
- Konermann L.; Silva E.; Sogbein O. Anal. Chem. 2001, 73, 4836–4844. [DOI] [PubMed] [Google Scholar]
- Kaltashov I.; Eyles S. Mass Spectrom. Rev. 2002, 21, 37–71. [DOI] [PubMed] [Google Scholar]
- Liu J.; Konermann L. J. Am. Soc. Mass Spectrom. 2009, 20, 819–828. [DOI] [PubMed] [Google Scholar]
- Sterling H. J.; Williams E. R. J. Am. Soc. Mass Spectrom. 2009, 20, 1933–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharlamova A.; Prentice B. M.; Huang T.; McLuckey S. A. Anal. Chem. 2010, 82, 7422–7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharlamova A.; McLuckey S. A. Anal. Chem. 2011, 83, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. H.; Wijeratne A. B.; Li L.; Edwards B. L.; Schug K. A. Anal. Chem. 2011, 83, 643–647. [DOI] [PubMed] [Google Scholar]
- Sterling H. J.; Cassou C. A.; Susa A. C.; Williams E. R. Anal. Chem. 2012, 84, 3795–3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassou C. A.; Sterling H. J.; Susa A. C.; Williams E. R. Anal. Chem. 2013, 85, 138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharlamova A.; DeMuth J. C.; McLuckey S. A. J. Am. Soc. Mass Spectrom. 2012, 23, 88–101. [DOI] [PubMed] [Google Scholar]
- Simmons D. A.; Konermann L. Biochemistry 2002, 41, 1906–1914. [DOI] [PubMed] [Google Scholar]
- Zinck N.; Stark A.; Wilson D. J.; Sharon M. ChemistryOpen 2014, 3, 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakowski B.; Konermann L. Anal. Biochem. 2001, 292, 107–114. [DOI] [PubMed] [Google Scholar]
- Wu L.; Lapidus L. J. Anal. Chem. 2013, 85, 4920–4924. [DOI] [PubMed] [Google Scholar]
- Vahidi S.; Stocks B. B.; Liaghati-Mobarhan Y.; Konermann L. Anal. Chem. 2013, 85, 8618–8625. [DOI] [PubMed] [Google Scholar]
- Ballew R.; Sabelko J.; Gruebele M. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 5759–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miladinovic S. M.; Fornelli L.; Lu Y.; Piech K. M.; Girault H. H.; Tsybin Y. O. Anal. Chem. 2012, 84, 4647–4651. [DOI] [PubMed] [Google Scholar]
- Mark L. P.; Gill M. C.; Mahutc M.; Derrick P. J. Eur. J. Mass Spectrom. 2012, 18, 439–446. [DOI] [PubMed] [Google Scholar]
- Fisher C. M.; Kharlamova A.; McLuckey S. A. Anal. Chem. 2014, 86, 4581–4588. [DOI] [PubMed] [Google Scholar]
- Mortensen D. N.; Williams E. R. Anal. Chem. 2014, 86, 9315–9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girod M.; Moyano E.; Campbell D. I.; Cooks R. G. Chem. Sci. 2011, 2, 501–510. [Google Scholar]
- Badu-Tawiah A. K.; Campbell D. I.; Cooks R. G. J. Am. Soc. Mass Spectrom. 2012, 23, 1461–1468. [DOI] [PubMed] [Google Scholar]
- Jurchen J. C.; Williams E. R. J. Am. Chem. Soc. 2002, 125, 2817–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRC Handbook of Chemistry and Physics, 55th ed.; Weast R. C., Ed.; CRC Press: Boca Raton, FL, 1974. [Google Scholar]
- Sage J. T.; Morikis D.; Champion P. M. Biochemistry 1991, 30, 1227–1237. [DOI] [PubMed] [Google Scholar]
- Griko Y. V.; Privalov P. L.; Venyaminov S. Y.; Kutyshenko V. P. J. Mol. Biol. 1988, 202, 127–138. [DOI] [PubMed] [Google Scholar]
- Scherrenberg R.; Coussens B.; van Vliet P.; Edouard G.; Brackman J.; de Brabander E.; Mortensen K. Macromolecules 1998, 31, 456–461. [Google Scholar]
- Grimm R. L.; Beauchamp J. L. Anal. Chem. 2002, 74, 6291–6297. [DOI] [PubMed] [Google Scholar]
- Schmidt A.; Karas M.; Dülcks T. J. Am. Soc. Mass Spectrom. 2003, 14, 492–500. [DOI] [PubMed] [Google Scholar]
- Goto Y.; Fink A. L. J. Mol. Biol. 1990, 214, 803–805. [DOI] [PubMed] [Google Scholar]
- Callender R.; Dyer R.; Gilmanshin R.; Woodruff W. Annu. Rev. Phys. Chem. 1998, 49, 173–202. [DOI] [PubMed] [Google Scholar]
- Gilmanshin R.; Williams S.; Callender R.; Woodruff W.; Dyer R. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 3709–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmanshin R.; Callender R.; Dyer R. Nat. Struct. Biol. 1998, 5, 363–365. [DOI] [PubMed] [Google Scholar]
- Drahos L.; Heeren R. M. A.; Collette C.; De Pauw E.; Vékey K. J. Mass Spectrom. 1999, 34, 1373–1379. [DOI] [PubMed] [Google Scholar]
- Takáts Z.; Drahos L.; Schlosser G.; Vékey K. Anal. Chem. 2002, 74, 6427–6429. [DOI] [PubMed] [Google Scholar]
- Naban-Maillet J.; Lesage D.; Bossee A.; Gimbert Y.; Sztaray J.; Vékey K.; Tabet J. J. Mass Spectrom. 2005, 40, 1–8. [DOI] [PubMed] [Google Scholar]
- Gabelica V.; De Pauw E. Mass Spectrom. Rev. 2005, 24, 566–587. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Yoon S. H.; Heron S. R.; Masselon C. D.; Edgar J. S.; Turecek F.; Goodlett D. R. J. Am. Soc. Mass Spectrom. 2012, 23, 1062–1070. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Cruz S. E.; Klassen J. S.; Williams E. R. J. Am. Soc. Mass Spectrom. 1999, 10, 958–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prell J. S.; O’Brien J. T.; Holm A. I. S.; Leib R. D.; Donald W. A.; Williams E. R. J. Am. Chem. Soc. 2008, 130, 12680–12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Freivogel P.; Schindler T.; Beauchamp J. L. J. Am. Chem. Soc. 1998, 120, 11758–11765. [Google Scholar]
- Bush M. F.; Saykally R. J.; Williams E. R. J. Am. Chem. Soc. 2008, 130, 9122–9128. [DOI] [PubMed] [Google Scholar]
- Shastry R. M. C.; Luck S. D.; Roder H. Biophys. J. 1998, 74, 2714–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno T. Protein Sci. 1998, 7, 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen E.; Babul J. Biochemistry 1975, 14, 5135–5140. [DOI] [PubMed] [Google Scholar]
- Pletneva E.; Gray H.; Winkler J. J. Am. Chem. Soc. 2005, 127, 15370–15371. [DOI] [PubMed] [Google Scholar]
- Krantz B.; Mayne L.; Rumbley J.; Englander S.; Sosnick T. J. Mol. Biol. 2002, 324, 359–371. [DOI] [PubMed] [Google Scholar]
- Kathuria S. V.; Chan A.; Graceffa R.; Nobrega R. P.; Matthews C. R.; Irving T. C.; Perot B.; Bilsel O. Biopolymers 2013, 99, 888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S.; Yeh S.; Das T.; Chan C.; Gottfried D.; Rousseau D. Nat. Struct. Biol. 1997, 4, 44–50. [DOI] [PubMed] [Google Scholar]
- Chan C.; Hu Y.; Takahashi S.; Rousseau D.; Eaton W.; Hofrichter J. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labowsky M. Rapid Commun. Mass Spectrom. 2010, 24, 3079–3091. [DOI] [PubMed] [Google Scholar]
- Ahadi E.; Konermann L. J. Am. Chem. Soc. 2011, 133, 9354–9363. [DOI] [PubMed] [Google Scholar]
- Mirza U.; Chait B. Int. J. Mass Spectrom. Ion Process. 1997, 162, 173–181. [Google Scholar]
- Pascher T. Biochemistry 2001, 40, 5812–5820. [DOI] [PubMed] [Google Scholar]