

Abstract
Integral membrane proteins comprise ∼25% of the human proteome. Yet, our understanding of their molecular physiology is still in its infancy. This can be attributed to two factors: the experimental challenges that arise from the difficult chemical nature of membrane proteins, and the unclear relationship between their activity and their native environment. New approaches are therefore required to address these challenges. Recent developments in mass spectrometry have shown that it is possible to study membrane proteins in a solvent-free environment and provide detailed insights into complex interactions, ligand binding and folding processes. Interestingly, not only detergent micelles but also lipid bilayer nanodiscs or bicelles can serve as a means for the gentle desolvation of membrane proteins in the gas phase. In this manner, as well as by direct addition of lipids, it is possible to study the effects of different membrane components on the structure and function of the protein components allowing us to add functional data to the least accessible part of the proteome.
Introduction
Cells, as the basic functional units of every higher organism, are surrounded by membranes, which in turn provide hydrophobic barriers for maintaining the out-of equilibrium states essential to life. Interactions on the outside and inside of the membrane control cellular localization and integrity. This barricade comes at a cost, however, as nutrients, signals and products have to be able to traverse the membrane in a specific fashion, often against gradients.
To be able to meet all of these requirements simultaneously, the membrane functions as a highly dynamic partition rather than just a static barrier. The foundations of this view were laid by Singer and Nicolson, who described the membrane as a fluid mosaic where proteins and lipids can diffuse laterally to facilitate occasional interactions (Singer & Nicolson, 1972). However, it was subsequently shown that lipids and proteins are not always distributed uniformly (Simons & van Meer, 1988). As a consequence, the fluid mosaic model was refined to include membrane domains rather than individual proteins as the principal components. These domains, termed lipid rafts, represent local concentrations of specific membrane constituents formed by lipid and protein interactions (Simons & Ikonen, 1997; Anderson & Jacobson, 2002).
The lipid raft model implies that a significant portion of membrane-associated proteins are subject to lateral sorting. As a result, these proteins reside in microenvironments with dynamic yet defined protein–protein and protein–lipid contacts. However, investigations of the underlying molecular interactions remain challenging. In this article, we describe how mass spectrometry (MS)-based approaches can provide unique insights into the molecular functions of membrane proteins. Furthermore, we place the most recent findings into a physiological perspective.
Membrane proteins in human physiology
Genome-wide computational screens suggest that at least ∼25% of the human proteome is composed of proteins that are inserted into the lipid bilayer (Fagerberg et al. 2010); similar values have been predicted for other genomes (von Heijne, 2011). Integral membrane proteins are of special interest for the pharmacological treatment of disease and make up an estimated 50% of current drug targets (Hopkins & Groom, 2002; Overington et al. 2006). The biomedical interest in the membrane proteome is reflected in the large, coordinated efforts that are being invested in its structural and functional elucidation (Pieper et al. 2013). However, the combination of hydrophobic transmembrane regions and hydrophilic intra- and extracellular domains makes integral membrane proteins highly amphipathic and thus challenging for existing structural biology tools such as X-ray crystallography and NMR. Currently, less than 500 are annotated as unique structures in the Structural Biology Knowledgebase (http://blanco.biomol.uci.edu/mpstruc/). Moreover these structures are often only static snapshots of dynamic conformations, creating a gap between molecular structure and physiological function. Therefore, complementary approaches for the study of membrane proteins are in high demand. Perhaps surprisingly, MS has recently emerged as a tool capable of studying multiple aspects of the dynamics and interactions that underlie membrane protein function (Barrera & Robinson, 2011; Whitelegge, 2013).
Linking protein structure and function by means of mass spectrometry
Generally, mass spectrometry denotes the analysis of desolvated, ionized molecules and is used predominantly to determine molecular masses of ions with high accuracy. Whilst MS originated in physical chemistry, the advent of techniques that allowed the analysis of biomolecules paved the way for its application in medical sciences. Following early attempts with 252-californium desorption (Sundqvist et al. 1984), the development of matrix-assisted laser desorption ionization (MALDI) and electrospray ionization (ESI) allowed the direct analysis of liquid samples, and most importantly proteins (Karas & Hillenkamp, 1988; Fenn et al. 1989). Since the prerequisite for MS analysis is the loss of solution phase interactions during transfer into the gas phase, it was originally considered suitable primarily for the study of denatured proteins. However, Chait and co-workers reported the observation of intact complexes between myoglobin and its haem group following ESI, and thus provided evidence that aspects of the native structure could be retained in the gas phase (Katta & Chait, 1991). Subsequent studies have shown that ESI reduces the impact of desolvation on the protein fold and thus facilitates the preservation of intra- and intermolecular interactions for MS analysis (Breuker & McLafferty, 2008; Hall & Robinson, 2012).
The possibility of studying folded proteins with so-called ‘native’ MS, in which proteins are introduced from their native state in solution, has spawned a range of analytical approaches designed to investigate their structural and functional links (Fig.1) (Sharon & Robinson, 2007). By measuring the masses of protein complexes and comparing the energy required for their dissociation, binding constants and ligand preferences have been extracted (Loo, 1997). A glimpse of possible tertiary and quaternary structures may be offered by ion mobility (IM)-MS, which measures the transit time of an ionized protein molecule or complex through a helium-filled drift cell. The transit time correlates with the collisional cross-section, i.e. the rotational space that the protein occupies, and in this manner provides information about its overall fold (Lanucara et al. 2014).
Figure 1.
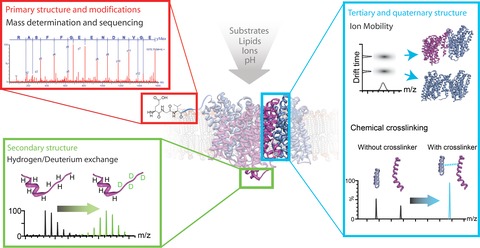
Mass spectrometry provides insights from the primary to the quaternary structure of membrane proteins
Top left: mass determination and MS-based sequencing shows the primary protein structure and attached components. Bottom left: hydrogen/deuterium exchange occurs predominantly in protein regions that lack a defined secondary structure. MS can be used to localize the incorporated deuterium ions and thus informs about the presence of secondary structure elements. Right: ion mobility and chemical crosslinking can be combined with MS to study tertiary and quaternary structures of proteins and their complexes. IM-MS measures the collisional cross-sections of desolvated proteins, while the identification of chemical crosslinks with MS reveals intra- and intermolecular distances. In the same manner, the effects of ligands and lipids or environmental changes such as altered pH or salt concentrations can be detected at all structural levels.
In addition to the study of proteins in the gas phase, MS can be integrated with chemical labelling, such as hydrogen/deuterium exchange, hydroxyl radical foot printing and chemical cross-linking, to obtain information about the solution structure. In these cases, mass determination of fragments and denatured proteins informs about the location of the labelled segments (Landreh et al. 2011). ‘Native’ and denaturing MS are often complementary, with ‘native’ MS providing information about protein complexes, e.g. architecture and stability, and denaturing MS focusing on structural details, e.g. the locations of secondary structure elements and interaction sites. Hybrid approaches, combining both forms with computational modelling, yield detailed information about the structure and dynamics of protein interactions (Politis et al. 2014).
Do observations from ‘native’ MS reflect physiological structures?
Many initial reports of folded proteins and their interactions in the gas phase were met with some skepticism. Removing water molecules from the surface of a protein represents a significant departure from any physiological environment and does certainly impact the protein structure. Structural perturbations probably occur at the atomic level, moments after desolvation, but stable secondary and tertiary structures can remain intact during the timeframe of the MS experiment (Breuker & McLafferty, 2008). Supporting evidence comes from comparisons of gas phase-derived data with high-resolution structures. For instance, the preferred sites for protein backbone cleavage in the gas phase can be correlated with the location of flexible sites in crystal structures (Zhang et al. 2013), and collisional cross-sections of protein complexes in general agree well with crystallographic data (Hall & Robinson, 2012). Such correlations are sensitive to the contribution from electrostatic interactions. Salt bridges protecting structures from rapid unfolding (Schennach & Breuker, 2014), charged side-chains form new intramolecular contacts that tether the structure (Warnke et al. 2013), and charges that are attached to the protein during the electrospray process can affect the compactness of the structure (reviewed in Hall & Robinson, 2012).
The stability of a protein fold in the gas phase will depend on the relative contributions from these factors to the overall structure. As a result folded proteins and their complexes are generally preserved well enough for structural investigations, while there are cases, particularly with unstructured and disordered domains, where collapse is observed as solvent is depleted (Pagel et al. 2013). As a consequence ‘native’ MS may not provide a full description of the protein structure in the gas phase or indeed in its physiological setting, but can reliably inform about structural features as illustrated below.
MS of intact membrane proteins
Given the versatility of MS, it comes as no surprise that it can be applied to study the vast yet elusive molecular physiology of membrane proteins. Early reports have demonstrated how MS can circumvent the detection problems posed by hydrophobic protein segments (Eichacker et al. 2004) and consequently facilitate mapping of the membrane proteome using protein fragmentation and MS analysis (Savas et al. 2011). Parallel to the broad view of physiological processes offered by membrane proteomics, the usefulness of the same techniques to investigate the architecture and function of individual membrane proteins was also recognized. In an early example, substrate binding to a membrane transporter, a protein that selectively transports molecules across the membrane barrier, was monitored by chemical modification of carboxyl groups in the cargo-binding region and identification of the modified fragments by MS (Weinglass et al. 2003). Using a less invasive approach, it was shown that the complex between a transporter and its cargo could be ionized directly from a detergent solution. Complex dissociation then released the components for MS analysis and thus allowed indirect monitoring of ligand binding (Ilag et al. 2004).
These studies hinted at the possibility of observing directly folded membrane proteins and their intact complexes. The interior of the lipid bilayer and the vacuum conditions inside the mass spectrometer both represent a low dielectric environment (Jarrold, 2007), implying that the three-dimensional structures of membrane protein could be largely preserved. In addition, electrostatic interactions contribute only modestly to the stability of membrane proteins (Joh et al. 2008), and hence, their relative strengthening in the gas phase (Daniel et al. 2002) is unlikely to skew the balance between hydrophobic and hydrophilic interactions. Yet, despite these promising circumstances, the observation of intact membrane complexes by MS proved elusive. For folded membrane proteins to be able to withstand the conditions inside the mass spectrometer, labile interactions with detergent molecules must first be lost during the desolvation process, while interactions with lipids, ligands and complex subunits must survive the flight through the mass spectrometer.
The answer to this challenge proved to be the use of detergent micelles as membrane protein shuttles during desolvation (Barrera et al. 2008). Here, the protein is electrosprayed from a solution containing detergent above the critical micelle concentration. The micelles can be maintained during ionization (Sharon et al. 2007) and transport an embedded membrane protein into the collision cell of the mass spectrometer. Subsequent activation through collisions with argon gas removes detergent molecules to release the intact complex while providing protective effects similar to those afforded by other hydrophobic electrospray additives (Borysik et al. 2013; Landreh et al. 2014a).
In an initial study, this method was shown to preserve the heterotetrameric structure and co-operative ATP binding ability of the BtuC2D2 (B12 uptake complex composed of two C and two D subunits) ATP-binding cassette transporter in the gas phase (Barrera et al. 2008). In addition, its collisional cross-section was found to correspond closely to that calculated from the crystallographic data, indicating that not only the subunit interactions, but also aspects of the overall fold remained intact (Wang et al. 2010).
Following these studies, the detergent micelle-based approach was successfully applied to a number of membrane protein complexes (reviewed in Marcoux & Robinson, 2013). However, detergents are rarely found in the natural environment of membrane proteins and can impact their stability (Sonoda et al. 2011). These problems can be alleviated by ionizing membrane proteins directly from lipid bilayer nanodiscs in place of detergent micelles (Hopper et al. 2013). As a result, proteins can be desolvated from a more native-like environment to accommodate interactions with a range of membrane components.
MS reveals specific interactions between membrane proteins and lipids
Already in early MS studies of native membrane proteins, desolvated transporter complexes were found to contain non-covalently bound lipids (Barrera et al. 2009; Lin et al. 2009; Velamakanni et al. 2009). The fact that lipid binding occurred at fixed stoichiometry and persisted even in detergent-purified complexes indicated the specificity of these interactions, and further examples have subsequently been reported (Barrera et al. 2013).
A particularly striking example of specific lipid binding was observed in the transmembrane ring of the V-type ATPase (Zhou et al. 2011). An MS analysis of the intact ATPase from Thermus thermophilus as well as the dissociated rotor domain, not only clarified its twelve-piece stoichiometry, but also revealed the presence of six phosphatidylethanolamine (PE) molecules. These lipids formed a hydrophobic lining for interactions with the stalk that translates the rotational movement to the ring. Interestingly, this lipid lining appears to be a general feature: the ten-cardiolipin central lipid plug observed in the rotor ring of Enterococcus hirae correlates with the ten subunits in the ring and with the stalk unit diameter when docked into the inside of the available high-resolution structure (Murata et al. 2005), and a similar correspondence between lipids, rotor subunits and stalk size was found in the chloroplast ATPase (Zhou et al. 2011; Schmidt et al. 2013). Together these results suggest that this lipid bushing is not only a common feature of ATPases, but can also be adapted to the individual architecture of each complex.
Lipid interactions as regulators of protein function
In recent years, MS studies have extended the range of known protein–lipid interactions significantly (Contreras et al. 2011; Barrera et al. 2013) and lent new support to the notion that such interactions do not merely stem from the convenient availability of lipids as building blocks, but can actually be of functional importance (Contreras et al. 2012). This raises the question as to whether MS can inform about the modulation of protein function by lipid interactions. In this context, three unique abilities of MS have proven useful: (1) the ability to monitor synchronized binding events and conformational changes, (2) the capacity to observe directly even small sub-populations, and (3) the potential to study multiple environmental parameters such as pH and lipid composition simultaneously.
Taking advantage of these features, we probed the effects of substrate binding to the ATP-dependent multidrug efflux pump P-glycoprotein (P-gp), a membrane transporter with multiple binding sites for nucleotides and drugs (Aller et al. 2009). The monomeric P-gp transporter recognizes and exports small hydrophobic molecules, lipids, and even peptides, and represents a major challenge in cancer therapy due to its ability to remove cytotoxic compounds from the cell (Martinez et al. 2014). However, its considerable substrate heterogeneity and the fact that its ATPase activity can be modulated by lipids and detergents have thwarted detailed investigations of its molecular action (Clay & Sharom, 2013). In ligand binding experiments using ‘native’ MS, we observed well-resolved signals for P-gp complexes with diacylglycerides, cardiolipins, ATP and the peptide ligand cyclosporin A (Marcoux et al. 2013). By exposing the protein to different combinations of these ligands, and subjecting the complexes to IM-MS, it was possible to monitor synergistic effects of concomitant binding events on the P-gp structure. We found that the presence of cyclosporin A enhances subsequent binding of cardiolipins, while simultaneous interactions with ATP induce the formation of a more compact conformer. These results illustrate how MS can be used to monitor ligand- and lipid-induced structural changes in membrane transporters (Marcoux et al. 2013).
Besides conformational changes, a much more fundamental role for protein–lipid interactions was revealed recently. In a study comparing three multimeric membrane channels, MscL, AqpZ and AmtB, different lipid-bound states could be observed for all three protein complexes (Laganowsky et al. 2014). Interestingly, these lipid-bound sub-populations proved to be significantly more resistant to gas phase unfolding while exhibiting differential binding preferences. MscL was most efficiently stabilized by phosphatidylinositol phosphate, a known regulator of its activity (Zhong & Blount, 2013). Similarly, AqpZ was found to be stabilized by cardiolipin, which can also be coupled to its function (Romantsov et al. 2010; Laganowsky et al. 2014). However, both of these proteins otherwise showed rather promiscuous binding preferences and could be stabilized to a lesser extent by non-specific lipid interactions. AmtB, on the other hand, showed a clear preference for phosphatidylglycerol, which was then revealed by X-ray analysis to engage specific lipid binding sites to induce conformational changes in the protein (Laganowsky et al. 2014). In summary, these findings demonstrate that lipid interactions can indeed be both structural and functional in nature and are probably not restricted to individual proteins, but instead constitute a wide-spread phenomenon.
Implications for the functional organization of cell membranes
But what are the consequences of specific lipid interactions for membrane protein physiology? The plasma membrane is a complex lipid mixture whose components are not distributed randomly, but rather cluster into specific microenvironments (Simons & van Meer, 1988; Simons & Ikonen, 1997). Protein recruitment occurs predominantly through covalently attached lipid anchors. However, specific lipid interactions of membrane proteins have been suggested to contribute to the formation of lipid rafts, and lipid binding was found to affect the activity of some transmembrane receptors (Ernst & Brugger, 2014). Yet, elucidating the relationship between localization and activity of membrane proteins remains challenging, as it requires detailed knowledge about the molecular architecture as well as its modulation by the natural environment.
The synergistic binding of ligands and lipids can control membrane protein activity and their concerted interactions are of importance for physiological function in the plasma membrane (Fig.2). As many lipids are not uniformly distributed, they may only exert regulatory effects in a spatially constrained manner. If interactions with a substrate molecule increase a protein's affinity for a specific raft lipid, the formation of the active ligand–protein–lipid complex would only occur in an appropriate compartment of the plasma membrane (Fig.2). Alternatively, a protein could diffuse laterally until a high local concentration of the preferred lipid is found, which then increases ligand affinity or activity. At the same time, the stabilizing or energetically favourable protein–lipid interactions could help to tether the protein to its specific membrane section and serve as a more dynamic alternative to covalently attached lipid anchors. Such a concerted process amounts to a dynamic lateral organization of membrane protein functionality, as it can be coupled to regulatory factors on the inside and the outside of the membrane. Several observations are compatible with this concept, such as the modulation of G-protein-coupled receptor activity by lipid environments (Mondal et al. 2014), or how mechanosensitive channels are affected by the physical properties of their membrane surroundings (Bavi et al. 2014).
Figure 2.
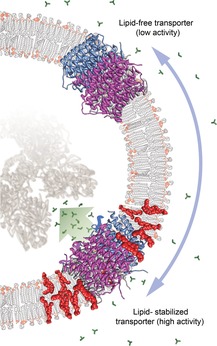
Possible mode for spatial regulation of membrane protein activity by specific lipids
Several multimeric transporter proteins (such as AmtB, shown here) are activated by specific lipids. The lipid-free form exhibits only low transport activity (top). If the transporter is relocated to a membrane raft (bottom) with a high local content of the preferred lipid (red), the complex is stabilized, which promotes the transport of its preferred substrate (green). Such a regulatory mode would result in a strictly localized influx of the substrate, e.g. to facilitate the direct delivery of the substrate to its target proteins (grey).
Up to 30% of the raft proteins associate via unknown mechanisms (Simons & Sampaio, 2011), and it is tempting to speculate that the specific protein–lipid interactions observed by MS will contribute to the organization of the plasma membrane.
Outlook and concluding remarks
In the light of these encouraging results, it appears possible that MS can be used to study other aspects of membrane proteins and lipid interactions, such as folding mechanisms. It has been demonstrated that specific interactions with phosphatidylethanolamine (PE) are crucial for the proper folding of the integral membrane protein lactose permase (LacY) from E. coli (Bogdanov et al. 1996). Mapping the orientation of the transmembrane helices of LacY upon insertion into membranes with different lipid compositions revealed that the entire N-terminal 6-helix bundle was inverted in the absence of PE (Bogdanov et al. 2014). The effects of PE are probably mediated by interactions with LacY folding intermediates and in this manner resemble the mechanisms used by conventional chaperones (Bogdanov & Dowhan, 1999). Given the ability of ‘native’ MS to correlate protein folding states and lipid binding, as well as the availability of complementary techniques to monitor folding processes in the membrane (Khanal et al. 2012), it appears likely that MS could prove useful in this area of membrane protein physiology.
With the advent of intact protein ionization, MS rapidly moved into structural biology, and from there it now extends towards physiology. In this article, we have summarized recent developments in MS and shown how they provide insights not only into the structure but also the action of membrane proteins. Challenging questions remain at the intersection of membrane physiology and structural biology, such as the complex protein–lipid interactions that facilitate hepatitis C virus replication (Liefhebber et al. 2009), the roles of lipids in neurotransmitter transport (Erkens et al. 2013), and the toxic membrane disruption events in protein aggregation diseases (Landreh et al. 2014b). It is, however, reasonable to believe that MS will contribute to solving some of the fascinating questions that exist within the molecular physiology of membrane proteins in health and disease.
Glossary
- ESI
electrospray ionization
- IM
ion mobility
- MS
mass spectrometry
- PE
phosphatidylethanolamine
- P-gp
P-glycoprotein
Biography
Michael Landreh is a Marie Curie Fellow in the group of Carol Robinson at the Department of Chemistry, and a Junior Research Fellow at St Cross College at the University of Oxford. He obtained his PhD at the Karolinska Institutet, where he studied the biophysical regulation of amyloid formation. His main research interest is the use of mass spectrometry to elucidate molecular under pinnings of membrane transport as well as protein assembly mechanisms. Carol V. Robinson is a Royal Society Research Professor and Doctor Lee s Professor of Chemistry Elect at the University of Oxford. She is recognized for pioneering the use ofmass spectrometry for her research into the 3D structure of proteins. Her most recent work is concerned with examining how small molecules, specifically lipids, impact on the structure and function of membrane assemblies.
Additional information
Competing interests
None declared.
Funding
C.V.R. acknowledges funding from the Royal Society, an ERC advanced investigator award (IMPRESS) as well as Wellcome Trust and MRC Programme Grants. M.L. holds a Marie Curie Fellowship and is a Junior Research Fellow at St. Cross College, Oxford.
Acknowledgements
We thank past and present members of the Robinson research group for their contributions to this research.
References
- Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL. Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RG. Jacobson K. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science. 2002;296:1821–1825. doi: 10.1126/science.1068886. [DOI] [PubMed] [Google Scholar]
- Barrera NP, Di Bartolo N, Booth PJ. Robinson CV. Micelles protect membrane complexes from solution to vacuum. Science. 2008;321:243–246. doi: 10.1126/science.1159292. [DOI] [PubMed] [Google Scholar]
- Barrera NP, Isaacson SC, Zhou M, Bavro VN, Welch A, Schaedler TA, Seeger MA, Miguel RN, Korkhov VM, van Veen HW, Venter H, Walmsley AR, Tate CG. Robinson CV. Mass spectrometry of membrane transporters reveals subunit stoichiometry and interactions. Nat Methods. 2009;6:585–587. doi: 10.1038/nmeth.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera NP. Robinson CV. Advances in the mass spectrometry of membrane proteins: from individual proteins to intact complexes. Annu Rev Biochem. 2011;80:247–271. doi: 10.1146/annurev-biochem-062309-093307. [DOI] [PubMed] [Google Scholar]
- Barrera NP, Zhou M. Robinson CV. The role of lipids in defining membrane protein interactions: insights from mass spectrometry. Trends Cell Biol. 2013;23:1–8. doi: 10.1016/j.tcb.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Bavi N, Nakayama Y, Bavi O, Cox CD, Qin QH. Martinac B. Biophysical implications of lipid bilayer rheometry for mechanosensitive channels. Proc Natl Acad Sci USA. 2014;111:13864–13869. doi: 10.1073/pnas.1409011111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M. Dowhan W. Lipid-assisted protein folding. J Biol Chem. 1999;274:36827–36830. doi: 10.1074/jbc.274.52.36827. [DOI] [PubMed] [Google Scholar]
- Bogdanov M, Dowhan W. Vitrac H. Lipids and topological rules governing membrane protein assembly. Biochim Biophys Acta. 2014;1843:1475–1488. doi: 10.1016/j.bbamcr.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Sun JZ, Kaback HR. Dowhan W. A phospholipid acts as a chaperone in assembly of a membrane transport protein. J Biol Chem. 1996;271:11615–11618. doi: 10.1074/jbc.271.20.11615. [DOI] [PubMed] [Google Scholar]
- Borysik AJ, Hewitt DJ. Robinson CV. Detergent release prolongs the lifetime of native-like membrane protein conformations in the gas-phase. J Am Chem Soc. 2013;135:6078–6083. doi: 10.1021/ja401736v. [DOI] [PubMed] [Google Scholar]
- Breuker K. McLafferty FW. Stepwise evolution of protein native structure with electrospray into the gas phase, 10–12 to 102 s. Proc Natl Acad Sci USA. 2008;105:18145–18152. doi: 10.1073/pnas.0807005105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay AT. Sharom FJ. Lipid bilayer properties control membrane partitioning, binding, and transport of P-glycoprotein substrates. Biochemistry. 2013;52:343–354. doi: 10.1021/bi301532c. [DOI] [PubMed] [Google Scholar]
- Contreras FX, Ernst AM, Haberkant P, Björkholm P, Lindahl E, Gönen B, Tischer C, Elofsson A, von Heijne G, Thiele C, Pepperkok R, Wieland F. Brügger B. Molecular recognition of a single sphingolipid species by a protein's transmembrane domain. Nature. 2012;481:525–529. doi: 10.1038/nature10742. [DOI] [PubMed] [Google Scholar]
- Contreras FX, Ernst AM, Wieland F. Brügger B. Specificity of intramembrane protein-lipid interactions. Cold Spring Harb Perspect Biol. 2011;3:a004705. doi: 10.1101/cshperspect.a004705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JM, Friess SD, Rajagopalan S, Wendt S. Zenobi R. Quantitative determination of noncovalent binding interactions using soft ionization mass spectrometry. Int J Mass Spectrom. 2002;216:1–27. [Google Scholar]
- Eichacker LA, Granvogl B, Mirus O, Müller BC, Miess C. Schleiff E. Hiding behind hydrophobicity. Transmembrane segments in mass spectrometry. J Biol Chem. 2004;279:50915–50922. doi: 10.1074/jbc.M405875200. [DOI] [PubMed] [Google Scholar]
- Erkens GB, Hanelt I, Goudsmits JM, Slotboom DJ. van Oijen AM. Unsynchronised subunit motion in single trimeric sodium-coupled aspartate transporters. Nature. 2013;502:119–123. doi: 10.1038/nature12538. [DOI] [PubMed] [Google Scholar]
- Ernst AM. Brugger B. Sphingolipids as modulators of membrane proteins. Biochim Biophys Acta. 2014;1841:665–670. doi: 10.1016/j.bbalip.2013.10.016. [DOI] [PubMed] [Google Scholar]
- Fagerberg L, Jonasson K, von Heijne G, Uhlen M. Berglund L. Prediction of the human membrane proteome. Proteomics. 2010;10:1141–1149. doi: 10.1002/pmic.200900258. [DOI] [PubMed] [Google Scholar]
- Fenn JB, Mann M, Meng CK, Wong SF. Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- Hall Z. Robinson CV. Do charge state signatures guarantee protein conformations? J Am Soc Mass Spectrom. 2012;23:1161–1168. doi: 10.1007/s13361-012-0393-z. [DOI] [PubMed] [Google Scholar]
- Hopkins AL. Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Hopper JT, Yu YT, Li D, Raymond A, Bostock M, Liko I, Mikhailov V, Laganowsky A, Benesch JL, Caffrey M, Nietlispach D. Robinson CV. Detergent-free mass spectrometry of membrane protein complexes. Nat Methods. 2013;10:1206–1208. doi: 10.1038/nmeth.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilag LL, Ubarretxena-Belandia I, Tate CG. Robinson CV. Drug binding revealed by tandem mass spectrometry of a protein-micelle complex. J Am Chem Soc. 2004;126:14362–14363. doi: 10.1021/ja0450307. [DOI] [PubMed] [Google Scholar]
- Jarrold MF. Helices and sheets in vacuo. Phys Chem Chem Phys. 2007;9:1659–1671. doi: 10.1039/b612615d. [DOI] [PubMed] [Google Scholar]
- Joh NH, Min A, Faham S, Whitelegge JP, Yang D, Woods VL. Bowie JU. Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature. 2008;453:1266–1270. doi: 10.1038/nature06977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas M. Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- Katta V. Chait BT. Observation of the heme globin complex in native myoglobin by electrospray-ionization mass-spectrometry. J Am Chem Soc. 1991;113:8534–8535. [Google Scholar]
- Khanal A, Pan Y, Brown LS. Konermann L. Pulsed hydrogen/deuterium exchange mass spectrometry for time-resolved membrane protein folding studies. J Mass Spectrom. 2012;47:1620–1626. doi: 10.1002/jms.3127. [DOI] [PubMed] [Google Scholar]
- Laganowsky A, Reading E, Allison TM, Ulmschneider MB, Degiacomi MT, Baldwin AJ. Robinson CV. Membrane proteins bind lipids selectively to modulate their structure and function. Nature. 2014;510:172–175. doi: 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreh M, Alvelius G, Johansson J. Jornvall H. Protective effects of dimethyl sulfoxide on labile protein interactions during electrospray ionization. Anal Chem. 2014a;86:4135–4139. doi: 10.1021/ac500879c. [DOI] [PubMed] [Google Scholar]
- Landreh M, Astorga-Wells J, Johansson J, Bergman T. Jornvall H. New developments in protein structure-function analysis by MS and use of hydrogen-deuterium exchange microfluidics. FEBS J. 2011;278:3815–3821. doi: 10.1111/j.1742-4658.2011.08215.x. [DOI] [PubMed] [Google Scholar]
- Landreh M, Johansson J. Jornvall H. Separate molecular determinants in amyloidogenic and antimicrobial peptides. J Mol Biol. 2014b;426:2159–2166. doi: 10.1016/j.jmb.2014.03.005. [DOI] [PubMed] [Google Scholar]
- Lanucara F, Holman SW, Gray CJ. Eyers CE. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat Chem. 2014;6:281–294. doi: 10.1038/nchem.1889. [DOI] [PubMed] [Google Scholar]
- Liefhebber JM, Brandt BW, Broer R, Spaan WJ. van Leeuwen HC. Hepatitis C virus NS4B carboxy terminal domain is a membrane binding domain. Virol J. 2009;6:62. doi: 10.1186/1743-422X-6-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HT, Bavro VN, Barrera NP, Frankish HM, Velamakanni S, van Veen HW, Robinson CV, Borges-Walmsley MI. Walmsley AR. MacB ABC transporter is a dimer whose ATPase activity and macrolide-binding capacity are regulated by the membrane fusion protein MacA. J Biol Chem. 2009;284:1145–1154. doi: 10.1074/jbc.M806964200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo JA. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom Rev. 1997;16:1–23. doi: 10.1002/(SICI)1098-2787(1997)16:1<1::AID-MAS1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Marcoux J. Robinson CV. Twenty years of gas phase structural biology. Structure. 2013;21:1541–1550. doi: 10.1016/j.str.2013.08.002. [DOI] [PubMed] [Google Scholar]
- Marcoux J, Wang SC, Politis A, Reading E, Ma J, Biggin PC, Zhou M, Tao H, Zhang Q, Chang G, Morgner N. Robinson CV. Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc Natl Acad Sci USA. 2013;110:9704–9709. doi: 10.1073/pnas.1303888110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez L, Arnaud O, Henin E, Tao H, Chaptal V, Doshi R, Andrieu T, Dussurgey S, Tod M, Di Pietro A, Zhang Q, Chang G. Falson P. Understanding polyspecificity within the substrate-binding cavity of the human multidrug resistance P-glycoprotein. FEBS J. 2014;281:673–682. doi: 10.1111/febs.12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S, Khelashvili G, Johner N. Weinstein H. How the dynamic properties and functional mechanisms of GPCRs are modulated by their coupling to the membrane environment. Adv Exp Med Biol. 2014;796:55–74. doi: 10.1007/978-94-007-7423-0_4. [DOI] [PubMed] [Google Scholar]
- Murata T, Yamato I, Kakinuma Y, Leslie AG. Walker JE. Structure of the rotor of the V-type Na+-ATPase from Enterococcus hirae. Science. 2005;308:654–659. doi: 10.1126/science.1110064. [DOI] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B. Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Pagel K, Natan E, Hall Z, Fersht AR. Robinson CV. Intrinsically disordered p53 and its complexes populate compact conformations in the gas phase. Angew Chem Int Ed Engl. 2013;52:361–365. doi: 10.1002/anie.201203047. [DOI] [PubMed] [Google Scholar]
- Pieper U, Schlessinger A, Kloppmann E, Chang GA, Chou JJ, Dumont ME, Fox BG, Fromme P, Hendrickson WA, Malkowski MG, Rees DC, Stokes DL, Stowell MH, Wiener MC, Rost B, Stroud RM, Stevens RC. Sali A. Coordinating the impact of structural genomics on the human α-helical transmembrane proteome. Nat Struct Mol Biol. 2013;20:135–138. doi: 10.1038/nsmb.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politis A, Stengel F, Hall Z, Hernandez H, Leitner A, Walzthoeni T, Robinson CV. Aebersold R. A mass spectrometry-based hybrid method for structural modeling of protein complexes. Nat Methods. 2014;11:403–406. doi: 10.1038/nmeth.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romantsov T, Battle AR, Hendel JL, Martinac B. Wood JM. Protein localization in Escherichia coli cells: comparison of the cytoplasmic membrane proteins ProP, LacY, ProW, AqpZ, MscS, and MscL. J Bacteriol. 2010;192:912–924. doi: 10.1128/JB.00967-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savas JN, Stein BD, Wu CC. Yates JR., 3rd Mass spectrometry accelerates membrane protein analysis. Trends Biochem Sci. 2011;36:388–396. doi: 10.1016/j.tibs.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schennach M. Breuker K. Proteins with highly similar native folds can show vastly dissimilar folding behavior when desolvated. Angew Chem Int Ed Engl. 2014;53:164–168. doi: 10.1002/anie.201306838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Zhou M, Marriott H, Morgner N, Politis A. Robinson CV. Comparative cross-linking and mass spectrometry of an intact F-type ATPase suggest a role for phosphorylation. Nat Commun. 2013;4:1985. doi: 10.1038/ncomms2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon M, Ilag LL. Robinson CV. Evidence for micellar structure in the gas phase. J Am Chem Soc. 2007;129:8740–8746. doi: 10.1021/ja067820h. [DOI] [PubMed] [Google Scholar]
- Sharon M. Robinson CV. The role of mass spectrometry in structure elucidation of dynamic protein complexes. Annu Rev Biochem. 2007;76:167–193. doi: 10.1146/annurev.biochem.76.061005.090816. [DOI] [PubMed] [Google Scholar]
- Simons K. Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Simons K. Sampaio JL. Membrane organization and lipid rafts. Cold Spring Harb Perspect Biol. 2011;3:a004697. doi: 10.1101/cshperspect.a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K. van Meer G. Lipid sorting in epithelial cells. Biochemistry. 1988;27:6197–6202. doi: 10.1021/bi00417a001. [DOI] [PubMed] [Google Scholar]
- Singer SJ. Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- Sonoda Y, Newstead S, Hu NJ, Alguel Y, Nji E, Beis K, Yashiro S, Lee C, Leung J, Cameron AD, Byrne B, Iwata S. Drew D. Benchmarking membrane protein detergent stability for improving throughput of high-resolution X-ray structures. Structure. 2011;19:17–25. doi: 10.1016/j.str.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundqvist B, Roepstorff P, Fohlman J, Hedin A, Hakansson P, Kamensky I, Lindberg M, Salehpour M. Sawe G. Molecular weight determinations of proteins by californium plasma desorption mass spectrometry. Science. 1984;226:696–698. doi: 10.1126/science.6387912. [DOI] [PubMed] [Google Scholar]
- Velamakanni S, Lau CH, Gutmann DA, Venter H, Barrera NP, Seeger MA, Woebking B, Matak-Vinkovic D, Balakrishnan L, Yao Y, U EC, Shilling RA, Robinson CV, Thorn P. van Veen HW. A multidrug ABC transporter with a taste for salt. PLoS One. 2009;4:e6137. doi: 10.1371/journal.pone.0006137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SC, Politis A, Di Bartolo N, Bavro VN, Tucker SJ, Booth PJ, Barrera NP. Robinson CV. Ion mobility mass spectrometry of two tetrameric membrane protein complexes reveals compact structures and differences in stability and packing. J Am Chem Soc. 2010;132:15468–15470. doi: 10.1021/ja104312e. [DOI] [PubMed] [Google Scholar]
- Warnke S, von Helden G. Pagel K. Protein structure in the gas phase: the influence of side-chain microsolvation. J Am Chem Soc. 2013;135:1177–1180. doi: 10.1021/ja308528d. [DOI] [PubMed] [Google Scholar]
- Weinglass AB, Whitelegge JP, Hu Y, Verner GE, Faull KF. Kaback HR. Elucidation of substrate binding interactions in a membrane transport protein by mass spectrometry. EMBO J. 2003;22:1467–1477. doi: 10.1093/emboj/cdg145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitelegge JP( Integral membrane proteins and bilayer proteomics. Anal Chem. 2013;85:2558–2568. doi: 10.1021/ac303064a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. Introduction to theme ‘membrane protein folding and insertion’. Annu Rev Biochem. 2011;80:157–160. doi: 10.1146/annurev-biochem-111910-091345. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cui W. Gross ML. Native electrospray ionization and electron-capture dissociation for comparison of protein structure in solution and the gas phase. Int J Mass Spectrom. 2013;354:288–291. doi: 10.1016/j.ijms.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong D. Blount P. Phosphatidylinositol is crucial for the mechanosensitivity of Mycobacterium tuberculosis MscL. Biochemistry. 2013;52:5415–5420. doi: 10.1021/bi400790j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Morgner N, Barrera NP, Politis A, Isaacson SC, Matak-Vinkovic D, Murata T, Bernal RA, Stock D. Robinson CV. Mass spectrometry of intact V-type ATPases reveals bound lipids and the effects of nucleotide binding. Science. 2011;334:380–385. doi: 10.1126/science.1210148. [DOI] [PMC free article] [PubMed] [Google Scholar]