

Abstract
The contractile performance of skeletal muscle declines during intense activities, i.e. fatigue develops. Fatigued muscle can enter a state of prolonged low-frequency force depression (PLFFD). PLFFD can be due to decreased tetanic free cytosolic [Ca2+] ([Ca2+]i) and/or decreased myofibrillar Ca2+ sensitivity. Increases in reactive oxygen and nitrogen species (ROS/RNS) may contribute to fatigue-induced force reductions. We studied whether pharmacological ROS/RNS inhibition delays fatigue and/or counteracts the development of PLFFD. Mechanically isolated mouse fast-twitch fibres were fatigued by sixty 150 ms, 70 Hz tetani given every 1 s. Experiments were performed in standard Tyrode solution (control) or in the presence of: NADPH oxidase (NOX) 2 inhibitor (gp91ds-tat); NOX4 inhibitor (GKT137831); mitochondria-targeted antioxidant (SS-31); nitric oxide synthase (NOS) inhibitor (l-NAME); the general antioxidant N-acetylcysteine (NAC); a cocktail of SS-31, l-NAME and NAC. Spatially and temporally averaged [Ca2+]i and peak force were reduced by ∼20% and ∼70% at the end of fatiguing stimulation, respectively, with no marked differences between groups. PLFFD was similar in all groups, with 30 Hz force being decreased by ∼60% at 30 min of recovery. PLFFD was mostly due to decreased tetanic [Ca2+]i in control fibres and in the presence of NOX2 or NOX4 inhibitors. Conversely, in fibres exposed to SS-31 or the anti ROS/RNS cocktail, tetanic [Ca2+]i was not decreased during recovery so PLFFD was only caused by decreased myofibrillar Ca2+ sensitivity. The cocktail also increased resting [Ca2+]i and ultimately caused cell death. In conclusion, ROS/RNS-neutralizing compounds did not counteract the force decline during or after induction of fatigue.
Key points.
Increased free radical production may contribute to decreased muscle force production during fatiguing exercise, and might delay recovery from fatigue.
We exposed mouse fast-twitch single fibres to antioxidants targeting specific cellular sites to determine whether these compounds delay fatigue development and/or improve the recovery from fatigue.
Antioxidants had no effect on the fatigue-induced decrease in contractile force.
During recovery from fatigue, a mitochondria-targeted antioxidant, SS-31, restored the fatigue-induced decrease in sarcoplasmic reticulum Ca2+ release, but did not improve force recovery.
We conclude that antioxidants cannot counteract the force decline during or after induction of muscle fatigue, although they may affect the underlying mechanisms.
Introduction
The production of reactive oxygen and nitrogen species (ROS/RNS) in skeletal muscle fibres increases with intense and prolonged contractions (for recent review see Sakellariou et al. 2014). Classically, increased ROS/RNS production is associated with deleterious effects on cell function and integrity. However, it is becoming increasingly clear that ROS/RNS also affect important physiological functions. In skeletal muscle these physiological functions include both acute effects, e.g. altered force production (Andrade et al. 2001; Mollica et al. 2012) and increased contraction-mediated glucose uptake (Balon & Nadler, 1996; Sandström et al. 2006; Kang et al. 2012), and prolonged effects, e.g. control of gene expression and adaptation to physical exercise (Ristow et al. 2009; Powers et al. 2011; Paulsen et al. 2014).
The superoxide anion (O2−) is the primary ROS species produced by skeletal muscle. Mitochondria have classically been considered as the major source of ROS during skeletal muscle contractile activities, where complexes I and III of the electron transport chain are identified as the primary locations of O2− production (Powers & Jackson, 2008). However, the results of recent studies indicate NADPH oxidases (NOX) to be the primary O2− producers during muscle contraction (Michaelson et al. 2010; Pal et al. 2013; Sakellariou et al. 2013). Skeletal muscles express two NOX isoforms, NOX2 and NOX4, and of these NOX2 is suggested to be of greatest importance during physical exercise (Sakellariou et al. 2014). Nitric oxide (NO) is the primary RNS. Nitric oxide is mainly synthesized from the amino acid l-arginine by nitric oxide synthases (NOS), but it can also be produced from nitrate and nitrite anions (Lundberg & Weitzberg, 2010). Adult skeletal muscle normally express the neuronal and the endothelial NOS isoforms and the production of nitric oxide increases during muscle contractions, mainly by increased neuronal NOS activity (Balon & Nadler, 1994; Kobzik et al. 1994; Hirschfield et al. 2000).
Intense muscle activity leads to fatigue development with decreased force production and slower contractions (Allen et al. 2008). Increased ROS/RNS production has been implicated in fatigue, but the role of ROS/RNS in this context is somewhat ambiguous: numerous studies have shown that ROS/RNS scavengers increase fatigue resistance, whereas others did not observe any endurance enhancing effects (Powers & Jackson, 2008; Powers et al. 2011). Generally, positive effects of ROS/RNS scavengers during exercise are more marked with submaximal than with near-maximal contractions (Reid et al. 1994). At the muscle fibre level, this indicates that ROS/RNS mainly affect sarcoplasmic reticulum (SR) Ca2+ release and/or myofibrillar Ca2+ sensitivity, because changes in these have large effects on submaximal contractions, which occur on the steep part of the force–Ca2+ relationship (see Fig. 6 in Allen et al. 2008). It is worth noting that everyday activities generally require low to moderate forces and the firing frequencies of motor units are therefore set to produce submaximal contractions (Marsden et al. 1971; Grimby & Hannerz, 1977).
Figure 6.
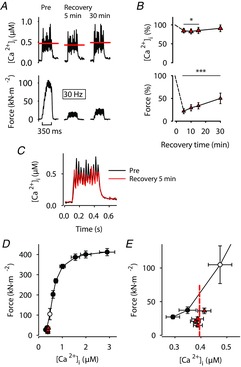
PLFFD in fibres exposed to the NOS inhibitor l-NAME is caused by a combination of decreased SR Ca2+ release and reduced myofibrillar Ca2+ sensitivity
A, representative records of [Ca2+]i (upper row) and force (lower row) obtained in one fibre stimulated with 30 Hz, 350 ms tetani and superfused by standard Tyrode before fatiguing stimulation (Pre) and subsequently in the presence of l-NAME (200 μm) during fatigue and recovery. Red lines show averaged [Ca2+]i during the stimulation periods. B, mean data (± SEM; n = 7) of [Ca2+]i (upper panel) and force (lower panel) in 30 Hz contractions produced 5–30 min after fatiguing stimulation. Data are expressed relative to pre-fatigue values, which were set to 100% in each fibre. *P < 0.05 and***P < 0.001 vs. pre-fatigue values with one-way repeated measures ANOVA. C, average [Ca2+]i records of all l-NAME-exposed fibres before (black trace) and 5 min (red trace) after induction of fatigue. D, mean (± SEM) force vs. [Ca2+]i data obtained in 15 to 150 Hz contractions produced at the start of each experiment. Data from 30 Hz contractions produced during the recovery period are also shown (red triangles) and compared to 30 Hz before fatigue (open circle). E, expanded view of data in D at low stimulation frequencies (15–30 Hz). Dashed red line indicates mean [Ca2+]i during the recovery period and its point of crossing of the force–[Ca2+]i relationship reflects the force expected from a decrease only in [Ca2+]i.
Increased ROS/RNS production has been implicated in the long-lasting depression in submaximal force following fatiguing exercise (Bruton et al. 2008; Lamb & Westerblad, 2011), which has been described as prolonged low-frequency force depression (PLFFD) and can persist for up to days after fatigue induction (Edwards et al. 1977; Westerblad et al. 1993; Chin & Allen, 1996; Allman & Rice, 2001; Hill et al. 2001; Allen et al. 2008). The PLFFD observed in human muscle in vivo can be replicated in isolated muscle fibres (e.g. Edwards et al. 1977; Westerblad et al. 1993), which provides strong evidence for PLFFD being caused by disturbances within the muscle fibres. At the cellular level, PLFFD can in principle be caused by reduced free cytosolic [Ca2+] ([Ca2+]i) during contractions and/or reduced myofibrillar Ca2+ sensitivity (Allen et al. 2008). The depressive effect of these two factors is additive and a reduction of both consequently results in severely decreased force production.
The aim of the present study was to answer the general question: Do pharmacological agents that decrease ROS/RNS improve muscle function during fatigue and recovery, and if so, via which mechanisms? Single muscle fibres were fatigued under control conditions (i.e. in standard Tyrode solutions) and in the presence of NOX inhibitors, a mitochondrial ROS scavenger, a NOS inhibitor, or a cocktail of antioxidants and NOS inhibitor. The results show no positive effect of any of the ROS/RNS-neutralizing compounds on force production during induction of fatigue or in the subsequent recovery period. Nevertheless, our results support an important, but highly complex, role of ROS/RNS in the development of PLFFD. For instance, the dominating mechanism underlying PLFFD was decreased tetanic [Ca2+]i in control fibres and reduced myofibrillar Ca2+ sensitivity in fibres exposed to mitochondrial ROS scavenging. Moreover, the reducing agent dithiothreitol (DTT) enhanced myofibrillar force production during PLFFD, whereas it decreased force in the unfatigued state.
Methods
Ethical approval
All experiments complied with the Swedish Animal Welfare Act, the Swedish Welfare Ordinance, and applicable regulations and recommendations from Swedish authorities. The study was approved by the Stockholm North Ethical Committee on Animal Experiments. The experiments comply with the policies of The Journal of Physiology (Drummond, 2009). Female C57BL/6 mice (n = 58) were killed by rapid neck disarticulation.
Force and [Ca2+]i measurements
Intact, single muscle fibres were mechanically dissected from the flexor digitorum brevis (FDB) muscle. This muscle contains mainly fast-twitch type IIa/x (Bruton et al. 2010) and the few type I fibres can be distinguished by their force–[Ca2+]i relationship and were not used in the present study. Aluminium clips were attached to the tendons and the fibre was mounted in a chamber between an Akers 801 force transducer (Kronex Technologies, Oakland, CA, USA) and an adjustable holder. Fibre length was adjusted to give maximum tetanic force. The diameter of the fibre at this length was measured at the widest and narrowest parts and these measurements were used to calculate the cross-sectional area. The fibre was electrically stimulated with supramaximal current pulses (0.5 ms duration) given via platinum electrodes placed along the long axis of the fibre.
Fibres were microinjected with the fluorescent Ca2+ indicator indo-1. We used indo-1, which is a high-affinity Ca2+ indicator, because it can detect changes in [Ca2+]i both at rest and during tetanic contractions (Westerblad & Allen, 1996). A disadvantage of high-affinity Ca2+ indicators is that they might significantly buffer [Ca2+]i resulting in decreased [Ca2+]i transient amplitudes and hence decreased force. Therefore we aimed at injecting the minimum amount of indo-1 required to allow reliable [Ca2+]i measurements. The calculated indo-1 concentration after injection was ≤100 μm and fibres were not used when the injection noticeably affected force production, indicating that indo-1 caused a physiologically significant buffering. Another disadvantage of high-affinity Ca2+ indicators, such as indo-1, is their relatively slow Ca2+ kinetics, which limits their ability to accurately follow rapid [Ca2+]i transient. Under the present experimental conditions, this will prevent accurate measurements of the rate of [Ca2+]i rise at the onset of contractions. However, the slow kinetics of indo-1 has no or little effect on measurements of time-averaged [Ca2+]i during complete tetanic stimulation periods (Westerblad & Allen, 1996), i.e. the type of measurement used in the present study.
The indo-1 fluorescence was measured with a system consisting of a xenon lamp, a monochromator, and two photomultiplier tubes (Photon Technology International, Wedel, Germany). About 1/3 of the muscle fibre length was excited with light at 360 nm, and the spatially averaged light emitted at 405 ± 5 and 495 ± 5 nm was measured. The ratio of the light emitted at 405 nm to that at 495 nm was converted to [Ca2+]i using an in vivo calibration as previously described (Andrade et al. 1998; Bruton et al. 2012). Force and fluorescence signals were sampled online at 100 Hz and stored on a computer for subsequent data analysis. We measured peak force (in kN m−2) and average [Ca2+]i during contractions.
The force–[Ca2+]i relationship for each fibre was first determined by producing 350 ms tetani at 1 min intervals with frequencies ranging between 15 and 150 Hz. Fibres were then allowed to rest for 20 min before being fatigued with repeated 70 Hz contractions of 150 ms duration given every 1 s for a total of 60 contractions. Force and [Ca2+]i during 30 and 120 Hz contractions of 350 ms duration were determined for each fibre immediately before the start of fatiguing stimulation and at regular intervals after fatiguing stimulation. In a subset of experiments, fibres were exposed to a reducing (DTT) or an oxidizing (tert-butyl hydroperoxide; t-BOOH) agent at 30 min after induction of fatigue and then followed for another 20 min with 30 Hz contractions produced at 2 min interval.
Solutions
Fibres were superfused by a standard Tyrode solution containing (in mm): 121 NaCl, 5.0 KCl, 1.8 CaCl2, 0.5 MgCl2, 0.4 NaH2PO4, 24.0 NaHCO3, 0.1 EDTA, and 5.5 glucose. The solution was bubbled with 95% O2–5% CO2, giving a bath pH of 7.4. Fetal calf serum (0.2%) was added to the solution. Experiments were performed at 32°C, i.e. an expected in vivo temperature of mouse FDB muscles during repeated contractions (Bruton et al. 1998). After establishing the force–[Ca2+]i relationship (see above) in standard Tyrode solution, fibres were superfused for 20 min before, during and after fatiguing stimulation with the standard Tyrode solution (control), or with Tyrode solution supplemented with: the NOX2 inhibitor gp91ds-tat (5 μm; Eurogentec, Seraing, Belgium; IC50 ∼1 μm; Csányi et al. 2011; Sakellariou et al. 2013); the NOX4 inhibitor GKT137831 (4 μm; Genkyotex, Geneva, Switzerland; IC50 ∼0.1 μm; Jiang et al. 2012); the mitochondria-targeted peptide SS-31 (200 nm; Andersson et al. 2011; Szeto & Schiller, 2011; Siegel et al. 2013); the nitric oxide synthase inhibitor NG-nitro-l-arginine methyl ester (l-NAME, 200 μm; TOCRIS Bioscience, Minneapolis, MN, USA); an antioxidant–nitric oxide synthase inhibitor cocktail consisting of SS-31 (200 nm), l-NAME (200 μm) and the general antioxidant N-acetylcysteine (NAC, 1 mm).
After 30 min of recovery, some fibres were superfused for 20 min with either the reducing agent DTT (1 mm) or the oxidizing agent t-BOOH (10 μm). Other fibres were exposed to either DTT or t-BOOH for 20 min in the unfatigued state.
ROS/RNS measurements
Mitochondrial ROS formation from fatiguing stimulation was measured by loading single fibres in Tyrode solution containing 5 μm of the membrane permeable fluorescent indicator MitoSOX Red (Invitrogen/Molecular Probes, Stockholm, Sweden) for 20 min (Aydin et al. 2009; Sakellariou et al. 2014); this indicator selectively targets mitochondria and its fluorescence increases when oxidized by O2− but not by H2O2 or NO (Sakellariou et al. 2014). Fibres were then washed for 35 min in normal Tyrode. Measurements were performed with a Bio-Rad MRC 1024 confocal unit attached to a Nikon Diaphot 200 inverted microscope with a ×20 objective lens (NA 0.75). Excitation and emission wavelengths were set at 531 nm and 605 nm, respectively. Confocal images were obtained before and after fatiguing stimulation. To assess the dynamic range of the MitoSOX Red fluorescence, experiments ended with application of 1 mm H2O2 and an image was obtained after 5 min exposure.
Fatigue-induced changes in NO were assessed by loading single fibres with the fluorescent NO indicator 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) diacetate (Invitrogen/Molecular Probes) as described above for MitoSOX Red. For DAF-FM we used excitation at 491 nm and measured the emitted light at 515 nm and the dynamic range was assessed by finally exposing fibres to 1 mm of the NO donor S-nitroso-N-acetyl-d,l-penicillamine (SNAP; Cayman Chemical, Ann Arbor, MI, USA).
Western blotting
Individual FDB fibres were placed in 5 μl Laemmli buffer before (Pre) and 5 min after fatiguing stimulation (Post). Protein separation was performed by loading one fibre per well on an 8% Tris–glycine gel with 30% glycerol content. Proteins were transferred onto PVDF membranes and following blocking (Odyssey buffer; LI-COR Biotechnology, Cambridge, UK), they were incubated with the primary antibody, anti-malondialdehyde (MDA; 1:1000; Abcam (ab27642)). Membranes were then incubated with infrared-labelled secondary antibody (IRDye 800, 1:15000, LI-COR (926-32213)) and immunoreactive bands were visualized using the Odyssey Infrared Imaging System (LI-COR). Finally membranes were stained with Coomassie to assess the total protein content. Image J was used to normalize the anti-MDA myosin band staining to the total myosin protein content, which was obtained from the Coomassie staining. In control experiments, enzymatically dissociated FDB fibres (Zong et al. 2012) were incubated with or without 10 mm H2O2 for 10 min and then assessed for myosin MDA adducts as described above.
Statistics
Data are presented as means ± SEM. Student's paired and unpaired t tests, as well as one-way ANOVA and one-way repeated measures ANOVA were used to determine statistically significant differences as appropriate (Sigmaplot, Systat Software Inc, San Jose, CA, USA). The Holm–Sidak method was used for post hoc analyses when significant differences were determined using ANOVA. The level of significance was set at P < 0.05.
Results
ROS/RNS measurements
Initial experiments were performed to establish whether our fatiguing stimulation protocol actually induced detectable increases in ROS/RNS. We used the superoxide sensitive fluorescent indicator MitoSOX Red to measure fatigue-induced changes in mitochondrial ROS production. The MitoSOX Red fluorescence was stable before induction of fatigue. Conversely, it increased during the 1 min of fatiguing stimulation and remained elevated during 20 min of recovery, reaching a maximum of 19.1 ± 3.4% above the pre-fatigue level (n = 11, P < 0.001). Experiments were also performed in the presence of the mitochondria-targeted ROS-scavenging peptide SS-31 (Szeto & Schiller, 2011; Siegel et al. 2013) and the fatigue-induced increase in MitoSOX Red fluorescence was then smaller (11.9 ± 3.1%, n = 14) than under control conditions, but still statistically significant (P < 0.01). MitoSOX Red is a derivative of dihydroethidium and not directly oxidized by H2O2 (Sakellariou et al. 2014). Nevertheless, application of H2O2 results in markedly increased MitoSOX Red fluorescence, which depends on mitochondrial respiration and is likely to be caused by H2O2-induced inhibition of superoxide dismutase 2 (SOD2) and hence accumulation of O2− produced in the mitochondrial electron transport chain (Hearn et al. 1999; Aydin et al. 2009). Fibres were exposed to 1 mm H2O2 at the end of each experiment and within 5 min this increased the MitoSOX Red fluorescence by 278 ± 40% (P < 0.001) under control conditions and by 179 ± 51% (P < 0.01) in the presence of SS-31, respectively. Thus, the MitoSOX Red experiments revealed a modest fatigue-induced increase in ROS, which was partly inhibited by SS-31.
The fluorescence of the NO indicator DAF-FM increased during fatiguing stimulation reaching a maximum within 20 min after the end of stimulation of 9.1 ± 2.1% above the pre-fatigue value (n = 7, P < 0.01). To get an estimate of the dynamic range of DAF, fibres were subsequently exposed to the NO donor SNAP and this increased fluorescence to 47.8 ± 6.9% above the basal.
We next searched for signs of permanent protein modifications caused by a fatigue-induced increase in ROS/RNS. Single FDB fibres were then fixed for Western blotting before or 5 min after fatiguing stimulation and we measured MDA protein adducts, which are permanent protein modifications reflecting the extent of lipid peroxidation Since these analyses were performed on single fibres, we focused on myosin, which is the most abundant protein in skeletal muscle. Five minutes after fatiguing stimulation we observed an increased anti-MDA myosin band (Fig. 1A), whereas the total myosin protein content was not altered (Fig. 1B). Mean data showed an ∼30% increase in the relative amount of MDA adducts on myosin (n = 4, P < 0.01; Fig. 1C). A set of additional experiments was performed to assess the magnitude of MDA adducts that can be achieved by supraphysiological ROS exposure. FDB fibres were exposed to 10 mm H2O2 for 10 min and this resulted in an ∼200% increase in the relative amount of MDA adducts on myosin (n = 5, P < 0.01; Fig. 1A and D). To sum up, the results of the MitoSOX Red, DAF-FM and MDA–myosin-adduct experiments give a coherent picture of a modest increase in ROS/RNS production during fatiguing stimulation.
Figure 1.
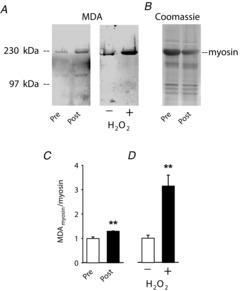
Fatiguing stimulation induces lipid peroxidation resulting in increased malondialdehyde (MDA) binding to myosin
A, Western blots with anti-MDA staining from one FDB fibre obtained before (Pre) and one fibre obtained 5 min after fatiguing stimulation (Post), and from fibres incubated without (−) or with (+) 10 mm H2O2 for 10 min. B, Coomassie-stained membrane showing total protein content in one FDB fibre obtained before and one fibre obtained 5 min after fatiguing stimulation. C and D, mean data (± SEM) of MDA bound to myosin before and after fatiguing stimulation (C, n = 4) and after incubation without or with 10 mm H2O2 (D, n = 5). **P < 0.01.
[Ca2+]i and force during fatigue
The force–[Ca2+]i relationship was studied at the start of experiments. Force (P) and [Ca2+]i measurements from 350 ms contractions at 15–150 Hz were then used to establish the maximum force (Pmax), the [Ca2+]i at 50% of Pmax (Ca50), and the steepness of the relation (N) according to the following equation:
This analysis gave mean (± SEM) values in all fibres of: Pmax = 391 ± 6 kN m−2; Ca50 = 673 ± 23 nm; N = 4.29 ± 0.17 (n = 69). No consistent differences were observed between fibres subsequently exposed only to standard Tyrode solution (control fibres) and those exposed to different ROS/RNS modifying compounds. Resting [Ca2+]i at the start of experiments was 61 ± 3 nm (n = 69) and again there were no consistent difference between fibres that subsequently were assigned to the different groups.
The present fatiguing stimulation protocol (150 ms, 70 Hz contractions produced at 1 s interval for 1 min) was designed to severely stress the fibres’ energy metabolism and decrease force by at least 50% in control fibres. The force produced in the first fatiguing contraction was 273 ± 8 kN m−2 (n = 69), i.e. ∼70% of Pmax. Figure 2A and B show representative [Ca2+]i and force records from fatiguing stimulation of a control fibre: tetanic [Ca2+]i increased over the first ten contractions and then decreased gradually until the end of fatigue, while tetanic force decreased monotonically. A similar pattern was observed in fibres exposed to the different ROS/RNS-modulating compounds (not shown) and the decrease in tetanic [Ca2+]i (Fig. 2C) and force (Fig. 2D) at the end of fatiguing stimulation did not differ between the groups (P > 0.5).
Figure 2.
The decrease in tetanic [Ca2+]i and force during fatiguing stimulation was not affected by the presence of ROS/RNS-modulating compounds
Typical fatigue profiles of [Ca2+]i (A) and force (B) from an intact single FDB fibre stimulated with 60 tetani (70 Hz, 150 ms duration given at 1 s intervals). Relative tetanic [Ca2+]i (C) and force (D) at the end vs. the start of fatiguing stimulation in standard Tyrode solution (control; n = 26) compared with fibres exposed to: the NOX2 inhibitor gp91ds-tat (5 μm, n = 8); the NOX4 inhibitor GKT137831 (4 μm, n = 8); the mitochondria-targeting antioxidant SS-31 (200 nm, n = 9); the NOS inhibitor l-NAME (200 μm, n = 7); an antioxidant–NOS inhibitor cocktail (SS-31, l-NAME and NAC, n = 5). Data are means ± SEM. One-way ANOVA showed no difference between groups for either [Ca2+]i (P = 0.70) or force (P = 0.52).
[Ca2+]i and force during recovery
Control fibres
Representative records obtained from 30 Hz stimulations of a control fibre (i.e. not exposed to a pharmacological agent) show markedly lower [Ca2+]i and force at 5 and 30 min of recovery than before fatiguing stimulation (Fig. 3A). Mean data from the recovery of control fibres (n = 26) show that [Ca2+]i during 30 Hz contractions was decreased to ∼80% of the pre-fatigue value, whereas force was decreased to ∼20% at 5 min and to ∼45% at 30 min (Fig. 3B). Superimposed average [Ca2+]i records from 30 Hz contractions produced before fatigue and at 5 min recovery show lower peaks for each stimulation pulse during recovery, but otherwise the general shape of the records is similar (Fig. 3C). Figure 3D and E shows mean force–[Ca2+]i data obtained before fatiguing stimulation and from 30 Hz contractions produced at 5–30 min of recovery. In addition to displaying decreased tetanic [Ca2+]i (see Fig. 3B), data points obtained from 30 Hz contractions during recovery (red triangles) lie to the right of control values, which indicates decreased myofibrillar Ca2+ sensitivity. To get an estimate of the relative contribution of these two force-depressing mechanisms, a straight vertical line was drawn from the mean [Ca2+]i of the recovery values (red dashed line in Fig. 3E). The force where this line crosses the pre-fatigue force–[Ca2+]i relationship (∼50 kN m−2) provides an estimate of the relative force decrease that can be attributed to the reduction in [Ca2+]i. Since the total reduction in force at 30 Hz was ∼75 kN m−2 (from ∼100 kN m−2 before to ∼25 kN m−2 after fatigue), this indicates that ∼2/3 of the decrease in 30 Hz force after fatigue is due to the decreased [Ca2+]i and the remaining ∼1/3 would then be due to decreased myofibrillar Ca2+ sensitivity.
Figure 3.
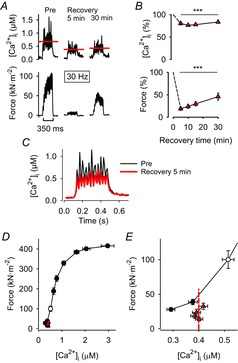
Prolonged decrease in SR Ca2+ release is the main cause of PLFFD in control fibres
A, representative records of [Ca2+]i (upper row) and force (lower row) obtained in one control fibre (i.e. superfused only with standard Tyrode solution) and stimulated with 30 Hz, 350 ms tetani before (Pre) and 5 and 30 min after fatiguing stimulation (Recovery 5 min and 30 min). Red lines show averaged [Ca2+]i during the stimulation periods. B, mean data (± SEM; n = 26) of [Ca2+]i (upper panel) and force (lower panel) in 30 Hz contractions produced 5–30 min after fatiguing stimulation. Data are expressed relative to pre-fatigue values, which were set to 100% in each fibre. ***P < 0.001 vs. pre-fatigue values with one-way repeated measures ANOVA. C, average [Ca2+]i records of all control fibres; note the similar resting [Ca2+]i and time course of the [Ca2+]i transients before (black trace) and 5 min (red trace) after induction of fatigue. D, mean (± SEM) force vs. [Ca2+]i data obtained in 15 to 150 Hz contractions produced in all control fibres at the start of each experiment. Data from 30 Hz contractions induced during the recovery period are also shown (red triangles) and compared to 30 Hz before fatigue (open circle). E, expanded view of data in D at low stimulation frequencies (15–30 Hz). Dashed red line indicates mean [Ca2+]i during the recovery period and its point of crossing of the force–[Ca2+]i relationship reflects the force expected from a decrease only in [Ca2+]i.
It might be noted that a decrease in Pmax would also tend to move data points to the right of the control force–[Ca2+]i relationship by inducing a proportional force decrease at all stimulation frequencies. However, in the present study this tentative effect of decreased Pmax would be small at the low forces produced for 30 Hz contractions. Moreover, the sustained force depression after fatigue is much larger at 30 Hz than at 120 Hz stimulation (see Fig. 9); the force for 120 Hz contractions is close to Pmax, which then indicates little change in Pmax.
Figure 9.
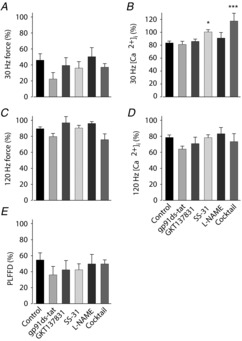
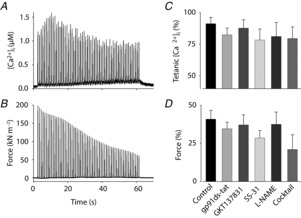
Markedly increased [Ca2+]i during 30 Hz contractions in fibres exposed to the antioxidant–NOS inhibitor cocktail
Relative force and [Ca2+]i during 30 Hz (A and B) and 120 Hz (C and D) contractions produced 30 min after fatiguing stimulation; data expressed relative to pre-fatigue value in each fibre. E, PLFFD calculated as the 30/120 Hz force ratio at 30 min recovery divided by the pre-fatigue ratio. Groups defined below panels D and E. Data are means ± SEM.*P < 0.05 and***P < 0.001 vs. control fibres (one-way ANOVA).
Fibres exposed to NOX2 or NOX4 inhibitors
Fibres exposed to either the NOX2 inhibitor gp91ds-tat (n = 8) or the NOX4 inhibitor GKT137831 (n = 8) during fatigue and recovery showed a pattern similar to that observed in control fibres (Fig. 4); that is, [Ca2+]i was somewhat decreased and force was markedly depressed (Fig. 4D) during 30 Hz contractions throughout the 30 min recovery period. Thus, PLFFD in fibres exposed to either gp91ds-tat or GKT137831 was mainly due to decreased [Ca2+]i but also reduced myofibrillar Ca2+ sensitivity, i.e. results similar to those in control fibres.
Figure 4.
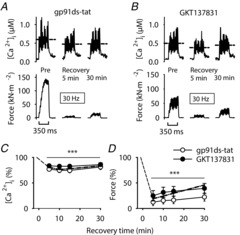
The decreases in SR Ca2+ release and force during PLFFD in the presence of NOX2 or NOX4 inhibitors are similar to those in control
Representative records of [Ca2+]i (upper row) and force (lower row) from muscle fibres stimulated with 30 Hz, 350 ms tetani while being superfused by standard Tyrode before fatiguing stimulation (Pre) and subsequently exposed to the NOX2 inhibitor gp91ds-tat (5 μm; A) or the NOX4 inhibitor GKT137831 (4 μm; B) during fatiguing stimulation and recovery. Dashed horizontal lines show averaged [Ca2+]i during stimulation periods. C and D show mean data (± SEM) of 30 Hz [Ca2+]i and force, respectively, at 5 to 30 min after fatigue in the presence of gp91ds-tat (○, n = 8) or GKT137831 (•, n = 8); for comparison, mean values from control fibres are also shown (dashed–dotted line; data from Fig. 3). Data are expressed relative to pre-fatigue values, which were set to 100%. ***P < 0.001 vs. pre-fatigue values with repeated measures ANOVA.
Fibres exposed to the mitochondria-targeted peptide SS-31
Fibres exposed to SS-31 during fatigue and recovery (n = 9) showed a prolonged force decrease similar to that seen in control fibres (Fig. 5). However, the force decrease in SS-31-exposed fibres was not accompanied by any decrease in [Ca2+]i during contractions. Thus, in the presence of SS-31 the force decrease was due to a fatigue-induced reduction in myofibrillar Ca2+ sensitivity, i.e. a rightward shift of the force–[Ca2+]i relationship (Fig. 5E).
Figure 5.
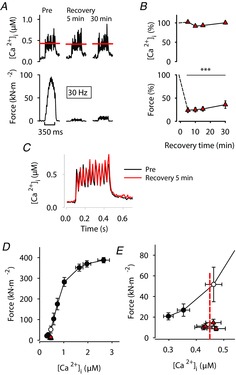
Decreased myofibrillar Ca2+ sensitivity is the main cause of PLFFD in fibres exposed to the mitochondrial targeted antioxidant SS-31
A, representative records of [Ca2+]i (upper row) and force (lower row) obtained in one fibre stimulated with 30 Hz, 350 ms tetani and superfused by standard Tyrode before fatiguing stimulation (Pre) and subsequently in the presence of SS-31 (200 nm) during fatigue and recovery. Red lines show averaged [Ca2+]i during the stimulation periods. B, mean data (± SEM; n = 9) of [Ca2+]i (upper panel) and force (lower panel) in 30 Hz contractions produced 5–30 min after fatiguing stimulation. Data are expressed relative to pre-fatigue values, which were set to 100% in each fibre. ***P < 0.001 vs. pre-fatigue values with one-way repeated measures ANOVA. C, average [Ca2+]i records of all SS-31-exposed fibres; note the similarity in amplitude and time course before (black trace) and 5 min (red trace) after induction of fatigue. D, mean (± SEM) force vs. [Ca2+]i data for 15–150 Hz contractions produced at the start of each experiment. Data from 30 Hz contractions induced during the recovery period are also shown (red triangles) and compared to 30 Hz before fatigue (open circle). E, expanded view of data in D at low stimulation frequencies (15–30 Hz). Dashed red line indicates mean [Ca2+]i during the recovery period and its point of crossing of the force–[Ca2+]i relationship reflects the force expected from a decrease only in [Ca2+]i.
Fibres exposed to the NOS inhibitor l-NAME
l-NAME is a well characterized inhibitor of NOS, which has been shown to be effective in skeletal muscle (Thomas & Victor, 1998). The extent of force decrease during 30 Hz contractions in fibres exposed to l-NAME (n = 7) was similar to that in control fibres during and after fatigue (Fig. 6). [Ca2+]i during contractions was ∼85% of the pre-fatigue value 5–15 min after fatigue and the decrease was no longer statistically significant after 30 min. Thus, the decrease in [Ca2+]i appeared smaller in l-NAME-exposed fibres than in control fibres. Accordingly, a larger proportion of the force decrease can be explained by decreased myofibrillar Ca2+ sensitivity in l-NAME-exposed fibres (∼50%) than in control fibres (Fig. 6E vs. Fig. 3E).
Fibres exposed to a cocktail of antioxidants and l-NAME
In contrast to the other groups, mean [Ca2+]i during 30 Hz stimulation was actually increased throughout the recovery period in fibres exposed to SS-31, l-NAME and NAC, albeit the increase did not reach statistical significance (P = 0.053; Fig. 7). The extent of force decrease was, nevertheless, similar to the other groups, which indicates severely impaired myofibrillar function. Accordingly, data points during recovery lie markedly to the right of the pre-fatigue force–[Ca2+]i relationship (Fig. 7E).
Figure 7.
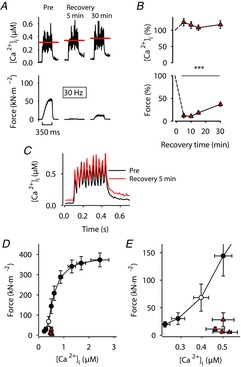
PLFFD in fibres exposed to a cocktail of antioxidants and NOS inhibitor is caused by a marked decrease in myofibrillar Ca2+ sensitivity
A, representative records of [Ca2+]i (upper row) and force (lower row) obtained in one fibre stimulated with 30 Hz, 350 ms tetani and superfused by standard Tyrode before fatiguing stimulation (Pre) and subsequently in the presence of SS-31 (200 nm), l-NAME (200 μm) and NAC (1 mm) during fatigue and recovery. Red lines show averaged [Ca2+]i during the stimulation periods. B, mean data (± SEM; n = 5) of [Ca2+]i (upper panel) and force (lower panel) in 30 Hz contractions produced 5–30 min after fatiguing stimulation. Data are expressed relative to pre-fatigue values, which were set to 100% in each fibre.***P < 0.001 vs. pre-fatigue values with one-way repeated measures ANOVA. C, average [Ca2+]i records of all fibres before (black trace) and 5 min (red trace) after induction of fatigue; note the major increase in resting [Ca2+]i during recovery. D, mean (± SEM) force vs. [Ca2+]i data for 15–150 Hz contractions produced at the start of each experiment. Data from 30 Hz contractions induced during the recovery period are also shown (red triangles) and compared to 30 Hz before fatigue (open circle). E, expanded view of data in D at low stimulation frequencies (15–40 Hz).
There was a marked increase in resting [Ca2+]i in fibres exposed to the antioxidant–NOS inhibitor cocktail (see Fig. 7C) and this increase had already started in the initial 20 min exposure period at rest (Fig. 8A) and continued during and after fatiguing stimulation (Fig. 8B). Thus, the increase in resting [Ca2+]i 30 min after the end of fatiguing stimulation was ∼5 times larger in fibres exposed to the antioxidant–NOS inhibitor cocktail than in control fibres. Severely increased resting [Ca2+]i, is an important sign of defective cellular integrity. Accordingly, resting [Ca2+]i continued to increase in antioxidant–NOS inhibitor cocktail-exposed fibres and they stopped contracting or developed an irreversible contracture 45–105 min after the end of fatiguing stimulation.
Figure 8.
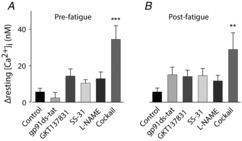
Markedly increased resting [Ca2+]i in fibres exposed to the antioxidant–NOS inhibitor cocktail
Mean data (± SEM) of the increase in resting [Ca2+]i during 20 min before fatiguing stimulation (A) and the additional increase observed 30 min after fatiguing stimulation (B). Groups defined below panels.**P < 0.01 and***P < 0.001 vs. control fibres (one-way ANOVA).
Control experiments were performed with fibres exposed only to NAC (1 mm; n = 4) to make sure that the deleterious effects of the antioxidant–NOS inhibitor cocktail were not due to the addition of the general antioxidant NAC itself. The results from NAC-exposed fibres were similar to those observed in control fibres, which is consistent with previously published data (Bruton et al. 2008). For instance, at 30 min of recovery [Ca2+]i and force during 30 Hz stimulation in NAC-exposed fibres (n = 4) were decreased to 80 ± 5% (P < 0.05) and 33 ± 6% (P < 0.01) of the pre-fatigue value, respectively, which compares to mean decreases to 84 and 46% of the pre-fatigue value in control fibres (data from Fig. 3B). Furthermore, the mean increase in resting [Ca2+]i at 30 min recovery was small (16 ± 1 nm) and not statistically significant (P = 0.20). Thus, the deleterious effects of the antioxidant–NOS inhibitor cocktail cannot be explained by effects of NAC on its own.
Force and [Ca2+]i at 30 min of recovery
Figure 9A–E summarizes changes of force and [Ca2+]i at 30 min of recovery vs. pre-fatigue values. Pre-fatigue measurements were performed before exposure to any ROS/RNS modifying compound and mean values of force and [Ca2+]i were 97 ± 8 kN m−2 and 0.50 ± 0.01 μm at 30 Hz stimulation and 401 ± 8 kN m−2 and 2.13 ± 0.08 μm at 120 Hz stimulation (n = 69); there were no consistent differences between fibres that subsequently were exposed to the different compounds (P > 0.05, one-way ANOVA). At 30 min of recovery, there were no statistically significant differences between control fibres and the other groups regarding the relative 30 Hz force (Fig. 9A), 120 Hz force (Fig. 9C), 120 Hz [Ca2+]i (Fig. 9D), or the extent of PLFFD (measured as the relative change in 30/120 Hz force ratio between 30 min recovery and pre-fatigue; Fig. 9E). On the other hand, [Ca2+]i during 30 Hz contractions was significantly higher in fibres exposed to the antioxidant–NOS inhibitor cocktail (∼120% of pre-fatigue; P < 0.001) and to SS-31 (∼100% of pre-fatigue; P < 0.05) than in control fibres (∼80% of pre-fatigue; Fig. 9B).
Fibres exposed to the reducing agent DTT or the oxidizing agent t-BOOH after 30 min of recovery
In the last set of experiments we studied how a reducing or an oxidizing compound affects contractile function in control fibres that were in a stable PLFFD state both regarding [Ca2+]i and force. The reducing agent DTT was applied 30 min after the end of fatiguing stimulation and this had no obvious effect on [Ca2+]i during 30 Hz contractions, whereas there was a marked increase in force (P < 0.001 at 4–12 min after application), which reached a peak after 6 min and then declined towards the pre-exposure level (Fig. 10A). This force increase with DTT is distinctly different from the effect in unfatigued fibres, where application of DTT resulted in decreased force production (open circles in Fig. 10A). These results are consistent with the concept that fatigue causes ‘oxidative stress’, which induces prolonged impairments in muscle function that can be temporarily reversed by a reducing agent. Unexpectedly, application of the oxidizing agent t-BOOH 30 min after the end of fatiguing stimulation gave a similar pattern with little effect on [Ca2+]i during 30 Hz contractions combined with a marked and transitory force increase (P < 0.05 at 4–10 min after application; Fig. 10B). Moreover, the response to t-BOOH was similar in fatigued and unfatigued fibres.
Figure 10.
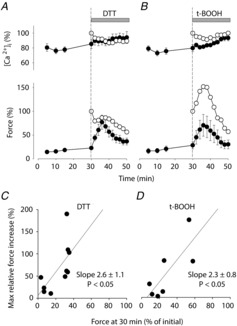
Both the reducing agent DTT and the oxidizing agent t-BOOH temporarily improve force production in fibres displaying PLFFD
Mean data (± SEM; •) of Ca2+]i (upper panel) and force (lower panel) during 30 Hz contractions produced during recovery from fatigue initially in standard Tyrode, and then with addition of DTT (1 mm, n = 10; A) or t-BOOH (10 μm; n = 7; B) at 30 min of recovery. Mean data from experiments where unfatigued fibres were exposed to DTT (n = 5) or t-BOOH (n = 2) are also shown (○). C, the maximum increase in 30 Hz force in individual fibres exposed to DTT (% of force at 30 min of recovery) plotted against their force at 30 min of recovery (percentage of force before fatiguing stimulation). D, same as in C but for fibres exposed to t-BOOH. Linear regression analyses (lines in C and D) show a significant positive correlation (P < 0.05) between the maximum force increase with DTT or t-BOOH and the force at the start of exposures.
The effect of ROS/RNS might be mediated both by reversible alterations (e.g. S-nitrosylation of thiol groups or oxidation of thiol groups to form disulphide bonds) and irreversible modifications (e.g. lipid peroxidation leading to formation of MDA–protein adducts (see Fig. 1) or nitration of tyrosine; for review see Powers & Jackson, 2008). In an attempt to distinguish between these two possibilities we took advantage of the fact that fatigued fibres showed relatively large differences in the force response to DTT or t-BOOH. We then observed a positive correlation between the maximum force increase with DTT and t-BOOH and the force before application of these compounds (P < 0.05; Fig. 10C and D). These analyses indicate a positive association between low force produced at 30 min after fatiguing stimulation and stable covalent modifications that are not affected by application of strong reducing or oxidizing agents.
Discussion
The present study focuses on the effect of ROS/RNS on the prolonged force depression, PLFFD, which is frequently observed after induction of fatigue (Keeton & Binder-Macleod, 2006). The results imply multifaceted mechanisms underlying PLFFD with disparate detrimental effects of ROS/RNS on SR Ca2+ release and myofibrillar Ca2+ sensitivity playing a central role.
Fatiguing stimulation induced increases in MitoSOX Red (∼19%) and DAF-FM (∼9%) fluorescence. We also observed ∼30% increased MDA content on myosin in fatigued fibres (see Fig. 1). Compared to the fatigue-induced changes, markedly larger increases in MitoSOX Red fluorescence and MDA–myosin adducts were obtained with direct exposure to the oxidant H2O2, and DAF-FM fluorescence increased more with application of the NO donor SNAP. Thus, our results show modest fatigue-induced increases in ROS/RNS both in the cytosol and mitochondria.
ROS/RNS have larger effects after than during fatiguing stimulation
We employed a high-intensity fatiguing stimulation protocol, where severe fatigue occurred within 1 min (i.e. tetanic force decreased by more than 50%). During fatiguing stimulation, neither tetanic [Ca2+]i nor force was affected by any of the ROS/RNS-neutralizing agents tested (see Fig. 2). This result is in general agreement with previous studies, which show a small or absent effect of decreased ROS/RNS on endurance when fatigue is induced with maximal contractions, whereas a clear positive effect on endurance is observed with submaximal contractions (Reid et al. 1994; Powers et al. 2011). Thus, other fatigue mechanisms dominate during the type of high-intensity fatiguing stimulation used in the present study, e.g. accumulation of inorganic phosphate ions due breakdown of creatine phosphate and depletion of inter-and intramyofibrillar glycogen (Dahlstedt et al. 2003; Allen et al. 2008; Nielsen et al. 2014).
Intriguingly, ROS/RNS produced during fatiguing stimulation seem to have markedly larger effects on the recovery process than on the actual fatigue development (Westerblad & Allen, 2011). A striking example is the finding that isolated mouse soleus fibres did not fatigue prematurely when exposed to severe oxidative stress (stimulated at 43°C in the presence of 10 μm H2O2 or t-BOOH), but they went into contracture and died ∼10 min after cessation of fatiguing stimulation (Place et al. 2009). The present study shows a marked PLFFD after fatiguing stimulation in control fibres not exposed to any exogenous ROS/RNS-neutralizing agents (see Fig. 3). This force depression was mainly due to decreased SR Ca2+ release, which is in agreement with previous studies on mouse FDB fibres fatigue by repeated tetani (Westerblad et al. 1993; Bruton et al. 2008), although we also observed some decrease in myofibrillar Ca2+ sensitivity.
The decreased [Ca2+]i during contractions observed at the end of fatiguing stimulation and during recovery in control fibres might involve impaired sarcolemmal excitability and defective action potential activation of SR Ca2+ release. A complete excitation failure in response to a stimulation pulse will result in a transitory decrease in [Ca2+]i. Such transient decreases were neither observed during fatiguing stimulation (data not shown) nor during recovery. Rather, the control fibres showed a clear increase in [Ca2+]i for each stimulation pulse (see Fig. 3C). Furthermore, transient decreases in [Ca2+]i during contractions were not observed in any of the groups of fibres exposed to ROS/RNS-neutralizing agents (see Figs. 7). However, this does not exclude the occurrence of a partial excitation failure with decreased action potential amplitude and hence less effective triggering of SR Ca2+ release.
Pharmacological inhibition of ROS/RNS has no beneficial effects on PLFFD
The results of recent studies indicate that NAD(P)H oxidases, rather than mitochondria, are the main producers of ROS in contracting skeletal muscles (Sakellariou et al. 2014). In particular, NOX2 has been proposed to be the main controller of cytosolic ROS production during contractions, because the contraction-mediated increase in cytosolic ROS was prevented by pharmacological inhibition or genetic knockdown of NOX2 (Michaelson et al. 2010; Pal et al. 2013; Sakellariou et al. 2013). In the present study muscle fibres were exposed to either the NOX2 inhibitor gp91ds-tat or the NOX4 inhibitor GKT137831. The gp91ds-tat peptide has been shown to selectively inhibit NOX2 (Csányi et al. 2011) and it was recently used in experiments on isolated mouse FDB fibres, where it abolished the contraction-mediated increase in cytosolic superoxide, as measured with the fluorescent indicator dihydroethidium (Sakellariou et al. 2013). GKT137831 is a well-characterized NOX inhibitor that potently inhibits NOX4 (IC50 value <1 μm), whereas it has little effect on NOX2 (Jiang et al. 2012; Altenhöfer et al. 2014); GKT137831 also effectively inhibits NOX1 and NOX5 (Altenhöfer et al. 2014), but these are of little importance in skeletal muscle (Sakellariou et al. 2014). Neither gp91ds-tat nor GKT137831 had any obvious effect on the fatigue-induced decrease in 30 Hz force or [Ca2+]i, or the extent of PLFFD (see Figs 4 and 9). Thus, pharmacological inhibition of NOX2 or NOX4 did not affect the development of PLFFD under our experimental conditions. This might suggest that ROS produced by NOX2 or NOX4 do not have any major effects on SR Ca2+ handling or myofibrillar force production, but additional experiments (e.g. genetic manipulation of NOX2 and NOX4 expression and function) are clearly required to verify this suggestion.
The mitochondria-targeted peptide SS-31 has been shown to decrease mitochondrial ROS associated with, for instance, ischaemic kidney injury (Szeto et al. 2011), skeletal muscle ageing (Siegel et al. 2013), and β-adrenergic stimulation of cardiomyocytes (Andersson et al. 2011). Our results show ∼40% less fatigue-induced increase in MitoSOX Red fluorescence in the presence of SS-31, which implies that the increased MitoSOX Red signal genuinely reflects augmented mitochondrial ROS production and that SS-31 counteracted this ROS increase. SS-31 prevented the prolonged decrease in SR Ca2+ release after fatiguing stimulation (see Fig. 5). Nevertheless, the magnitude of PLFFD was similar in SS-31-exposed and control fibres. Thus, PLFFD can be fully explained by decreased myofibrillar Ca2+ sensitivity in SS-31-exposed fibres, whereas it was due to the combined effect of decreased tetanic [Ca2+]i and reduced myofibrillar Ca2+ sensitivity in control fibres (compare Figs 3E and 5E). This is consistent with our previous results showing that PLFFD is caused by decreased myofibrillar Ca2+ sensitivity in FDB fibres of rats and SOD2 overexpressing mice, which have a higher SOD2 content and hence convert mitochondrial O2− to H2O2 more effectively than wild-type mouse FDB fibres (Bruton et al. 2008). A recent study using mouse FDB fibres transfected with a novel mitochondrial-targeted superoxide biosensor (mt-cpYFP) shows strictly localized mitochondrial O2− production during repetitive contractions (Wei et al. 2011). Electron microscopy has revealed small bridges, or tethers, that link mitochondria to adjacent SR in adult mouse FDB fibres (Boncompagni et al. 2009), which means that local mitochondrial ROS/RNS production may directly affect proteins involved in SR Ca2+ release without increasing global cytoplasmic ROS/RNS concentration. Thus, a model can be envisaged where: (i) the mitochondrial O2− concentration increases during repeated contractions in wild-type mouse FDB fibres, which have limited SOD2 capacity, and this increase has a detrimental effect on Ca2+ release in the adjacent SR; (ii) SOD2 overexpressing and SS-31-treated FDB fibres rapidly eliminate mitochondrial O2−, but the ROS/RNS challenge then occurs in the cytosol resulting in decreased myofibrillar Ca2+ sensitivity. This cytosolic ROS/RNS challenge would be related to an increased H2O2 concentration in SOD2 overexpressing fibres. An exaggerated increase in cytosolic H2O2 concentration is unlikely to occur in the SS-31-treated fibres (Szeto & Schiller, 2011) and the myofibrillar modification could instead be related to a ROS/RNS challenge caused by some product of SS-31 mitochondrial ROS scavenging.
In an attempt to counteract the development of PLFFD by extensive inhibition of fatigue-induced increases in mitochondrial and cytosolic ROS/RNS, some muscle fibres were exposed to a cocktail of antioxidants (SS-31 and NAC) and the NOS inhibitor l-NAME. This attempt turned out to be unsuccessful as the extent of PLFFD in fibres exposed to the cocktail was similar to that in the other groups. The cocktail preserved, or even increased, [Ca2+]i during contractions and hence the force depression was due to a very marked reduction of myofibrillar Ca2+ sensitivity (see Fig. 7). Moreover, exposure to the cocktail resulted in a marked increase in [Ca2+]i at rest, prior to fatiguing stimulation, and this increase continued after the stimulation period (see Fig. 8), which indicates deleterious changes in cell function. Accordingly, fibres exposed to the cocktail eventually stopped contracting or developed an irreversible contracture. Thus, rather than preventing deleterious effects induced by fatiguing stimulation, exposure to the antioxidant–NOS inhibitor cocktail resulted in additional harmful effects. This means that effective inhibition of ROS/RNS-mediated changes during fatiguing stimulation might have highly detrimental effects on muscle function. However, we cannot rule out the possibility that the deleterious effects were due to unexpected interactions between the potent antioxidants SS-31and NAC and the effective NOS inhibitor l-NAME.
Reducing and oxidizing agents exert complex effects on myofibrillar function in fibres displaying PLFFD
Some fibres were exposed to either the reducing agent DTT or the non-metabolized H2O2 analogue t-BOOH (see Fig.10). In agreement with previous results from unfatigued mouse FDB fibres (Andrade et al. 1998), neither DTT nor t-BOOH had any major effect on tetanic [Ca2+]i in fibres displaying PLFFD. These results show that increased H2O2 has little impact on tetanic [Ca2+]i and is therefore unlikely to cause the prolonged decease in SR Ca2+ release after fatiguing stimulation. Furthermore, the finding that the prolonged decrease in [Ca2+]i during PLFFD was not reversed by DTT implies that the underlying functional defect is not caused by any molecular modifications accessible to this strong reducing agent; e.g. it is unlikely to be caused by formation of disulphide bonds between amino acids.
In the rested state, DTT, H2O2 and t-BOOH act mainly on the myofibrils and DTT decreases force production whereas H2O2 and t-BOOH induce an initial force increase followed by a decline (Andrade et al. 1998, 2001). Thus, the myofibrils of rested fibres can be considered to be in an excessively reduced state (Andrade et al. 1998; Lamb & Westerblad, 2011; Powers et al. 2011). Application of DTT during PLFFD resulted in a major, but transient, increase in myofibrillar force production, which is markedly different to the decrease observed in the unfatigued state (see Fig. 10A). The diverging effects when adding DTT can be explained by a model where sites accessible to reduction by DTT are in a suboptimally reduced state under resting conditions and become overly oxidized during fatigue. This fatigue-induced oxidation is reversed by DTT, resulting in an initial increase in force production. Continued exposure to DTT results in a suboptimally reduced environment both after fatigue and at rest, decreasing force. Application of t-BOOH, on the other hand, resulted in transiently improved myofibrillar force production both in the unfatigued state and during PLFFD (see Fig. 10B). The latter finding was highly unexpected and indicates that some myofibrillar proteins were still in a suboptimally reduced state after fatigue and the function of these was temporarily improved by exogenous hydroperoxides.
Our results show statistically significant positive correlations between the force increase in response to DTT or t-BOOH and the extent of PLFFD (see Fig. 10C and D). In particular, fibres displaying severe PLFFD were little affected when exposed to DTT or t-BOOH. These results further illustrate a complex interplay between different molecular targets of oxidation/reduction and fatigue-induced reversible and irreversible modifications, which are DTT or t-BOOH accessible and inaccessible, respectively. In line with this, experiments on skinned muscle fibres show markedly different and fibre type-dependent effects on myofibrillar Ca2+ sensitivity of application of H2O2 in the presence or absence of myoglobin and glutathione, which are normally present in skeletal muscle fibres (Murphy et al. 2008; Lamb & Westerblad, 2011). For instance, application of H2O2 on its own has little effect in fast-twitch fibres, whereas it results in a marked decrease in myofibrillar Ca2+ sensitivity in the presence of myoglobin. This H2O2–myoglobin-induced decrease can be reversed by DTT, but only if DTT is applied before any activation of the contractile machinery in the presence of H2O2 and myoglobin. Moreover, application of H2O2 and myoglobin in the presence of glutathione results in an initial increase in myofibrillar Ca2+ sensitivity followed by a decrease (Murphy et al. 2008), i.e. a pattern very similar to that observed with exposure to t-BOOH in the present study.
Conclusions
It appears irrelevant to discuss mechanisms underlying PLFFD in terms of one specific ROS/RNS acting on one specific molecular site. Instead our data support complex interactions between several ROS/RNS affecting both SR Ca2+ handling and myofibrillar contractile function (Fig.11). Additional intake of antioxidants is often assumed to be beneficial and improve exercise performance, but there is little scientific support for this belief (Hernandez et al. 2012). In fact, beneficial adaptations to endurance training can be hampered by treatment with antioxidants (e.g. Ristow et al. 2009; Paulsen et al. 2014). The present results provide a tentative explanation for this undesirable effect: antioxidant treatment induces a shift from exercise-induced modifications in cellular Ca2+ handling, which can serve as an effective trigger of beneficial adaptations (Wright et al. 2007; Bruton et al. 2010), towards impaired myofibrillar function, which causes intrinsically less stress to the cell and is therefore unlikely to trigger major adaptations.
Figure 11.
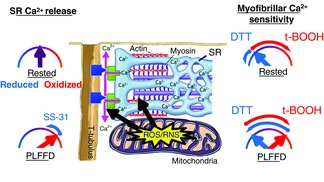
Cartoon illustrating the intricate effects of ROS/RNS and redox states on SR Ca2+ release and myofibrillar Ca2+ sensitivity
In this simplified model ROS/RNS mainly affect SR Ca2+ release and myofibrillar Ca2+ sensitivity. Key proteins for SR Ca2+ release are the t-tubular voltage sensors, dihydropyridine receptors (blue boxes), and the SR Ca2+ release channels, ryanodine receptors (green boxes). In the rested state, proteins involved in SR Ca2+ release appear in an optimal redox state and become overly oxidized during fatiguing stimulation; the presence of the mitochondrial antioxidant SS-31 during induction of fatigue prevents the oxidation and Ca2+ release remains optimal. In the rested state, myofibrillar proteins are in a reduced state with decreased myofibrillar Ca2+ sensitivity. Some of these proteins become overly oxidized during fatiguing stimulation and this effect can be reversed by application of the reducing agent DTT. On the other hand, other myofibrillar proteins remain reduced during fatigue and application of the oxidizing agent t-BOOH temporarily improves their function.
Glossary
- [Ca2+]i
free cytosolic [Ca2+]
- Ca50
[Ca2+]i at 50% of maximum force
- DTT
dithiothreitol
- FDB
flexor digitorum brevis
- l-NAME
NG-nitro-l-arginine methyl ester
- MDA
malondialdehyde
- NAC
N-acetylcysteine
- NO
nitric oxide
- NOS
nitric oxide synthase
- NOX
NADPH oxidase
- P
force
- PLFFD
prolonged low-frequency force depression
- Pmax
maximum force
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SOD2
superoxide dismutase 2
- t-BOOH
tert-butyl hydroperoxide
- SR
sarcoplasmic reticulum
Additional information
Competing interests
There are no competing interests.
Author contributions
All authors contributed to the conception, design, data interpretation and manuscript preparation. A.J.C., J.D.B., and J.T.L. were responsible for experimental work and H.W. contributed to data collection and analysis. All authors approved the final version of the manuscript.
Funding
This work was supported by the Swedish Research Council, the Swedish National Center for Sports Research, Association Francaise Contre les Myopathies (AFM), Lars Hierta Minne Stiftelsen, and Jeanssons Stiftelser.
References
- Allen DG, Lamb GD. Westerblad H. Skeletal muscle fatigue: cellular mechanisms. Physiol Rev. 2008;88:287–332. doi: 10.1152/physrev.00015.2007. [DOI] [PubMed] [Google Scholar]
- Allman BL. Rice CL. Incomplete recovery of voluntary isometric force after fatigue is not affected by old age. Muscle Nerve. 2001;24:1156–1167. doi: 10.1002/mus.1127. [DOI] [PubMed] [Google Scholar]
- Altenhöfer S, Radermacher KA, Kleikers PW, Wingler K. Schmidt HH. Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid Redox Signal. 2014 doi: 10.1089/ars.2013.5814. doi: 10.1089/ars.2013.5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DC, Fauconnier J, Yamada T, Lacampagne A, Zhang SJ, Katz A. Westerblad H. Mitochondrial production of reactive oxygen species contributes to the β-adrenergic stimulation of mouse cardiomyocytes. J Physiol. 2011;589:1791–1801. doi: 10.1113/jphysiol.2010.202838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade FH, Reid MB, Allen DG. Westerblad H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J Physiol. 1998;509:565–575. doi: 10.1111/j.1469-7793.1998.565bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade FH, Reid MB. Westerblad H. Contractile response of skeletal muscle to low peroxide concentrations: myofibrillar calcium sensitivity as a likely target for redox-modulation. FASEB J. 2001;15:309–311. doi: 10.1096/fj.00-0507fje. [DOI] [PubMed] [Google Scholar]
- Aydin J, Andersson DC, Hänninen SL, Wredenberg A, Tavi P, Park CB, Larsson NG, Bruton JD. Westerblad H. Increased mitochondrial Ca2+ and decreased sarcoplasmic reticulum Ca2+ in mitochondrial myopathy. Human Mol Genet. 2009;18:278–288. doi: 10.1093/hmg/ddn355. [DOI] [PubMed] [Google Scholar]
- Balon TW. Nadler JL. Nitric oxide release is present from incubated skeletal muscle preparations. J Appl Physiol. 1994;77:2519–2521. doi: 10.1152/jappl.1994.77.6.2519. [DOI] [PubMed] [Google Scholar]
- Balon TW. Nadler JL. Nitric oxide mediates skeletal glucose transport. Am J Physiol Endocrinol Metab. 1996;270:E1058–E1059. doi: 10.1152/ajpendo.1996.270.6.E1058. [DOI] [PubMed] [Google Scholar]
- Boncompagni S, Rossi AE, Micaroni M, Beznoussenko GV, Polishchuk RS, Dirksen RT. Protasi F. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol Biol Cell. 2009;20:1058–1067. doi: 10.1091/mbc.E08-07-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruton J, Cheng A. Westerblad H. Methods to detect Ca2+ in living cells. Adv Exp Med Biol. 2012;740:27–43. doi: 10.1007/978-94-007-2888-2_2. [DOI] [PubMed] [Google Scholar]
- Bruton JD, Aydin J, Yamada T, Shabalina IG, Ivarsson N, Zhang SJ, Wada M, Tavi P, Nedergaard J, Katz A. Westerblad H. Increased fatigue resistance linked to Ca2+-stimulated mitochondrial biogenesis in muscle fibres of cold-acclimated mice. J Physiol. 2010;588:4275–4288. doi: 10.1113/jphysiol.2010.198598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruton JD, Lännergren J. Westerblad H. Effects of CO2-induced acidification on the fatigue resistance of single mouse muscle fibers at 28oC. J Appl Physiol. 1998;85:478–483. doi: 10.1152/jappl.1998.85.2.478. [DOI] [PubMed] [Google Scholar]
- Bruton JD, Place N, Yamada T, Silva JP, Andrade FH, Dahlstedt AJ, Zhang SJ, Katz A, Larsson NG. Westerblad H. Reactive oxygen species and fatigue-induced prolonged low-frequency force depression in skeletal muscle fibres of rats, mice and SOD2 overexpressing mice. J Physiol. 2008;586:175–184. doi: 10.1113/jphysiol.2007.147470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin ER. Allen DG. The role of elevations in intracellular [Ca2+] in the development of low frequency fatigue in mouse single muscle fibres. J Physiol. 1996;491:813–824. doi: 10.1113/jphysiol.1996.sp021259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csányi G, Cifuentes-Pagano E, Al Ghouleh I, Ranayhossaini D, Egaña L, Lopes L, Jackson H, Kelley E. Pagano P. Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Radic Biol Med. 2011;51:1116–1125. doi: 10.1016/j.freeradbiomed.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlstedt AJ, Katz A, Tavi P. Westerblad H. Creatine kinase injection restores contractile function in creatine-kinase-deficient mouse skeletal muscle fibres. J Physiol. 2003;547:395–403. doi: 10.1113/jphysiol.2002.034793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RH, Hill DK, Jones DA. Merton PA. Fatigue of long duration in human skeletal muscle after exercise. J Physiol. 1977;272:769–778. doi: 10.1113/jphysiol.1977.sp012072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimby L. Hannerz J. Firing rate and recruitment order of toe extensor motor units in different modes of voluntary conraction. J Physiol. 1977;264:865–879. doi: 10.1113/jphysiol.1977.sp011699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearn AS, Tu C, Nick HS. Silverman DN. Characterization of the product-inhibited complex in catalysis by human manganese superoxide dismutase. J Biol Chem. 1999;274:24457–24460. doi: 10.1074/jbc.274.35.24457. [DOI] [PubMed] [Google Scholar]
- Hernandez A, Cheng A. Westerblad H. Antioxidants and skeletal muscle performance: "Common knowledge" vs. experimental evidence. Front Physiol. 2012;3:46. doi: 10.3389/fphys.2012.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CA, Thompson MW, Ruell PA, Thom JM. White MJ. Sarcoplasmic reticulum function and muscle contractile character following fatiguing exercise in humans. J Physiol. 2001;531:871–878. doi: 10.1111/j.1469-7793.2001.0871h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschfield W, Moody MR, O'Brien WE, Gregg AR, Bryan RM., Jr Reid MB. Nitric oxide release and contractile properties of skeletal muscles from mice deficient in type III NOS. Am J Physiol Regul Integr Comp Physiol. 2000;278:R95–R100. doi: 10.1152/ajpregu.2000.278.1.R95. [DOI] [PubMed] [Google Scholar]
- Jiang J, Chen X, Serizawa N, Szyndralewiez C, Page P, Schröder K, Brandes R, Devaraj S. Török N. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med. 2012;53:289–296. doi: 10.1016/j.freeradbiomed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L, Lustig M, Bonner J, Lee-Young R, Mayes W, James F, Lin C-T, Perry C, Anderson E, Neufer P. Wasserman D. Mitochondrial antioxidative capacity regulates muscle glucose uptake in the conscious mouse: effect of exercise and diet. J Appl Physiol. 2012;113:1173–1183. doi: 10.1152/japplphysiol.01344.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeton RB. Binder-Macleod SA. Low-frequency fatigue. Phys Ther. 2006;86:1146–1150. [PubMed] [Google Scholar]
- Kobzik L, Reid MB, Bredt DS. Stamler JS. Nitric oxide in skeletal muscle. Nature. 1994;372:546–548. doi: 10.1038/372546a0. [DOI] [PubMed] [Google Scholar]
- Lamb GD. Westerblad H. Acute effects of reactive oxygen and nitrogen species on the contractile function of skeletal muscle. J Physiol. 2011;589:2119–2127. doi: 10.1113/jphysiol.2010.199059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg JO. Weitzberg E. NO-synthase independent NO generation in mammals. Biochem Biophys Res Commun. 2010;396:39–45. doi: 10.1016/j.bbrc.2010.02.136. [DOI] [PubMed] [Google Scholar]
- Marsden CD, Meadows JC. Merton PA. Isolated single motor units in human muscle and their rate of discharge during maximal voluntary effort. J Physiol. 1971;217:12P–13P. [PubMed] [Google Scholar]
- Michaelson LP, Shi G, Ward CW. Rodney GG. Mitochondrial redox potential during contraction in single intact muscle fibers. Muscle Nerve. 2010;42:522–529. doi: 10.1002/mus.21724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollica JP, Dutka TL, Merry TL, Lamboley CR, McConell GK, McKenna MJ, Murphy RM. Lamb GD. S-Glutathionylation of troponin I(fast) increases contractile apparatus Ca2+ sensitivity in fast-twitch muscle fibres of rats and humans. J Physiol. 2012;590:1443–1463. doi: 10.1113/jphysiol.2011.224535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy RM, Dutka TL. Lamb GD. Hydroxyl radical and glutathione interactions alter calcium sensitivity and maximum force of the contractile apparatus in rat skeletal muscle fibres. J Physiol. 2008;586:2203–2216. doi: 10.1113/jphysiol.2007.150516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J, Cheng AJ, Ørtenblad N. Westerblad H. Subcellular distribution of glycogen and decreased tetanic Ca2+ in fatigued single intact mouse muscle fibres. J Physiol. 2014;592:2003–2012. doi: 10.1113/jphysiol.2014.271528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal R, Basu Thakur P, Li S, Minard C. Rodney GG. Real-time imaging of NADPH oxidase activity in living cells using a novel fluorescent protein reporter. PLoS One. 2013;8:e63989. doi: 10.1371/journal.pone.0063989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen G, Cumming KT, Holden G, Hallen J, Ronnestad BR, Sveen O, Skaug A, Paur I, Bastani NE, Ostgaard HN, et al. Vitamin C and E supplementation hampers cellular adaptation to endurance training in humans: a double-blind, randomised, controlled trial. J Physiol. 2014;592:1887–1901. doi: 10.1113/jphysiol.2013.267419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Place N, Yamada T, Zhang SJ, Westerblad H. Bruton JD. High temperature does not alter fatigability in intact mouse skeletal muscle fibres. J Physiol. 2009;587:4717–4724. doi: 10.1113/jphysiol.2009.176883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK. Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Ji LL, Kavazis AN. Jackson MJ. Reactive oxygen species: impact on skeletal muscle. Compr Physiol. 2011;1:941–969. doi: 10.1002/cphy.c100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MB, Stokic DS, Koch SM, Khawli FA. Leis AA. N-acetylcysteine inhibits muscle fatigue in humans. J Clin Invest. 1994;94:2468–2474. doi: 10.1172/JCI117615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Klöting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR. Blüher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakellariou GK, Jackson MJ. Vasilaki A. Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD(P)H oxidases. Free Radic Res. 2014;48:12–29. doi: 10.3109/10715762.2013.830718. [DOI] [PubMed] [Google Scholar]
- Sakellariou GK, Vasilaki A, Palomero J, Kayani A, Zibrik L, McArdle A. Jackson MJ. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid Redox Signal. 2013;18:603–621. doi: 10.1089/ars.2012.4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandström ME, Zhang SJ, Bruton J, Silva JP, Reid MB, Westerblad H. Katz A. Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. J Physiol. 2006;575:251–262. doi: 10.1113/jphysiol.2006.110601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel MP, Kruse SE, Percival JM, Goh J, White CC, Hopkins HC, Kavanagh TJ, Szeto HH, Rabinovitch PS. Marcinek DJ. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell. 2013;12:763–771. doi: 10.1111/acel.12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, Zhao Z, Ganger M, Tow CY. Seshan SV. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. 2011;22:1041–1052. doi: 10.1681/ASN.2010080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto H. Schiller P. Novel therapies targeting inner mitochondrial membrane – from discovery to clinical development. Pharm Res. 2011;28:2669–2679. doi: 10.1007/s11095-011-0476-8. [DOI] [PubMed] [Google Scholar]
- Thomas GD. Victor RG. Nitric oxide mediates contraction-induced attenuation of sympathetic vasoconstriction in rat skeletal muscle. J Physiol. 1998;506:817–826. doi: 10.1111/j.1469-7793.1998.817bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Salahura G, Boncompagni S, Kasischke KA, Protasi F, Sheu SS. Dirksen RT. Mitochondrial superoxide flashes: metabolic biomarkers of skeletal muscle activity and disease. FASEB J. 2011;25:3068–3078. doi: 10.1096/fj.11-187252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerblad H. Allen DG. Mechanisms underlying changes of tetanic [Ca2+]i and force in skeletal muscle. Acta Physiol Scand. 1996;156:407–416. doi: 10.1046/j.1365-201X.1996.196000.x. [DOI] [PubMed] [Google Scholar]
- Westerblad H. Allen DG. Emerging roles of ROS/RNS in muscle function and fatigue. Antioxid Redox Signal. 2011;15:2487–2499. doi: 10.1089/ars.2011.3909. [DOI] [PubMed] [Google Scholar]
- Westerblad H, Duty S. Allen DG. Intracellular calcium concentration during low-frequency fatigue in isolated single fibers of mouse skeletal muscle. J Appl Physiol. 1993;75:382–388. doi: 10.1152/jappl.1993.75.1.382. [DOI] [PubMed] [Google Scholar]
- Wright DC, Geiger PC, Han DH, Jones TE. Holloszy JO. Calcium induces increases in peroxisome proliferator-activated receptor gamma coactivator-1α and mitochondrial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation. J Biol Chem. 2007;282:18793–18799. doi: 10.1074/jbc.M611252200. [DOI] [PubMed] [Google Scholar]
- Zong M, Bruton JD, Grundtman C, Yang H, Li JH, Alexanderson H, Palmblad K, Andersson U, Harris HE, Lundberg IE. Westerblad H. TLR4 as receptor for HMGB1 induced muscle dysfunction in myositis. Ann Rheum Dis. 2012;72:1390–1399. doi: 10.1136/annrheumdis-2012-202207. [DOI] [PubMed] [Google Scholar]