

Abstract
Lines of evidence coming from many branches of neuroscience indicate that anxiety disorders arise from a dysfunction in the modulation of brain circuits which regulate emotional responses to potentially threatening stimuli. The concept of anxiety disorders as a disturbance of emotional response regulation is a useful one as it allows anxiety to be explained in terms of a more general model of aberrant salience and also because it identifies avenues for developing psychological, behavioral, and pharmacological strategies for the treatment of anxiety disorder. These circuits involve bottom-up activity from the amygdala, indicating the presence of potentially threatening stimuli, and top-down control mechanisms originating in the prefrontal cortex, signaling the emotional salience of stimuli. Understanding the factors that control cortical mechanisms may open the way to identification of more effective cognitive behavioral strategies for managing anxiety disorders. The brain circuits in the amygdala are thought to comprise inhibitory networks of γ-aminobutyric acid-ergic (GABAergic) interneurons and this neurotransmitter thus plays a key role in the modulation of anxiety responses both in the normal and pathological state. The presence of allosteric sites on the GABAA receptor allows the level of inhibition of neurons in the amygdala to be regulated with exquisite precision, and these sites are the molecular targets of the principal classes of anxiolytic drugs. Changes in the levels of endogenous modulators of these allosteric sites as well as changes in the subunit composition of the GABAA receptor may represent mechanisms whereby the level of neuronal inhibition is downregulated in pathological anxiety states. Neurosteroids are synthesized in the brain and act as allosteric modulators of the GABAA receptor. Since their synthesis is itself regulated by stress and by anxiogenic stimuli, targeting the neurosteroid-GABAA receptor axis represents an attractive target for the modulation of anxiety.
Keywords: allosteric modulation, amygdala, salience, γ-aminobutyric acid, neurosteroids
Objective
Better understanding of the neurobiological networks underlying anxiety and how they are deregulated in anxiety disorders is crucial, not only to understand the observed clinical features but also to propose individualized treatments. Treatment should aim to “retrain” and normalize the various neurobiological circuits that are deregulated in anxiety disorders. This review aims to describe the molecular and neurobiological modulatory processes associated with anxiety disorders, in particular those involving γ-aminobutyric acid-ergic (GABAergic) neurotransmission.
Anxiety disorders: definitions, presentation, and classification
Anxiety is a frequent negative emotional state characterized by feelings of worry and apprehension, and accompanied by specific somatic, cognitive, and behavioral manifestations. Anxiety can be distinguished from fear in that it is a nonspecific state of heightened awareness and apprehension, whereas fear is directed at a specific identified threat. Anxiety is a part of the normal behavioral repertoire and is of value as a defense mechanism in raising awareness and responsiveness to deal with novel situations. Nonetheless, when it is excessively severe or frequent or appears in inappropriate contexts, it can interfere with normal functioning and can thus be considered as pathological. The boundary between “normal” anxiety and “pathological” anxiety is not easy to define and can vary between individuals as a function of personality traits or, notably, as a function of what has been described as an “anxiety-prone cognitive style.”1–3 The Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) and Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) diagnostic criteria state that anxiety should be considered pathological when “the anxiety, worry, or physical symptoms cause clinically significant distress or impairment in social, occupational, or other important areas of functioning.”4,5 It is useful to distinguish between state anxiety, which is episodic and transitory, and trait anxiety, which is persistent and may reflect an “anxiety-prone” personality. Moreover, anxiety symptoms may appear in response to specific environmental clues, as in phobias, or be aspecific and pervasive, as in generalized anxiety disorders. In addition, the expression of anxious states evolves over time as new behaviors are learned or new cognitive strategies are put in place, as well as in response to changes in the environmental context.
The classification of anxiety disorders proposed in DSM-55 proposes three high-level categories, namely anxiety disorders, obsessive–compulsive and related disorders, and trauma- and stressor-related disorders. The last two categories, which were classified within anxiety disorders in DSM-IV, are now categories in their own right. The new anxiety disorders category includes generalized anxiety disorder, panic disorder, agoraphobia, phobias, and social anxiety disorders, whereas the new category of trauma- and stressor-related disorders includes posttraumatic stress disorder, acute stress disorder, and adjustment disorders.
For the best part of 50 years, anxiety disorders were primarily treated with benzodiazepines. Although these drugs are generally well tolerated, they carry some risk of dependence during long-term use and have unwanted sedative effects.6 For these reasons, current practice guidelines7–10 recommend the use of antidepressants, in particular selective serotonin reuptake inhibitors (SSRIs) or serotonin–noradrenaline reuptake inhibitors, as the preferred first-line treatment for anxiety disorders. The use of benzodiazepines is now reserved for second-line treatment or for short-term adjunctive use with SSRIs at the beginning of treatment to cover the lag period of 4–6 weeks before SSRIs become fully effective. With regard to nonpharmacological treatment of anxiety disorders, cognitive behavioral therapy is the best established.11,12
Brain regions involved in the modulation of anxiety
Simplified view of the brain circuitry involved in anxiety
Many brain regions appear to be involved in the recognition and regulation of negative emotional stimuli and in the generation of cognitive, behavioral, or somatic responses to these stimuli. Nonetheless, a set of limbic structures appear to be critical for the regulation of negative emotion. In particular, the amygdala – nuclei situated in the median temporal lobes – appears to play a crucial role. The principal neural circuits thought to be related to anxiety are presented in Figure 1. It is important to bear in mind that this circuitry has been established from research on experimental animals. Although data from functional imaging are consistent with this model, it should be noted that these pathways have not all been demonstrated conclusively in the human brain.
Figure 1.
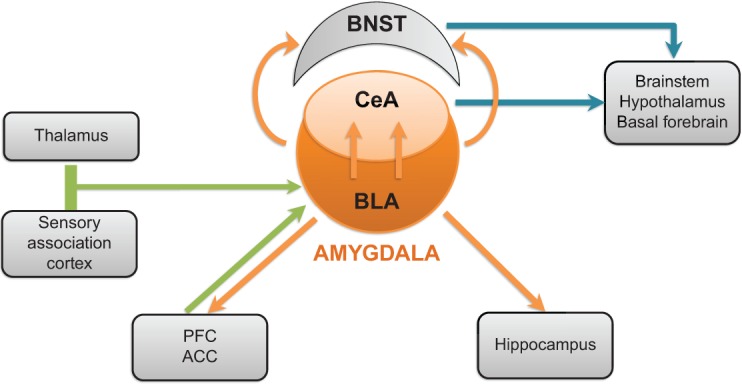
Neural circuits implicated in anxiety disorders.
Notes: Green arrows: principal inputs to the BLA; orange arrows: principal outputs of the BLA; blue arrows: principal outputs of the CeA and BNST.
Abbreviations: ACC, anterior cingulate cortex; BLA, basolateral amygdala; BNST, bed nucleus of the stria terminalis; CeA, central nucleus of the amygdala; PFC, prefrontal cortex.
In humans, bilateral lesions of the amygdala have been associated with deficits in recognizing facial expressions of fear and other negative emotions.13 In contrast, electrical stimulation of this structure leads to feelings of fear and anxiety.14 Since the development of functional imagery, a plethora of studies have evaluated the activation of different brain regions in response to negative emotional stimuli and in anxiety disorders (reviewed in detail by Etkin15 and Forster et al16). A consistent finding of these studies has been the activation of the amygdala.17–19 Moreover, patients with anxiety disorders appear to activate the amygdala in response to a given stimulus more than non-anxious controls.20 In addition, successful treatment of anxiety disorders with cognitive behavioral therapy leads to extinction of this hyperactivation of the amygdala.21
The amygdala is composed of a number of distinct nuclei but, for the purposes of a discussion of the brain circuitry relevant to anxiety, two groups of nuclei are of particular interest, namely the basolateral amygdala complex (BLA) and the centromedial amygdala complex, in particular the central nucleus (CeA).15,22 The BLA receives incoming information on potentially negative emotional signals from the thalamus and the sensory association cortex (Figure 1). The BLA activates the CeA directly through an excitatory glutamatergic pathway as well as activating a relay of inhibitory GABAergic interneurons – the intercalated neurons – that lie between the BLA and the CeA and exert an inhibitory influence upon the latter.23,24 The CeA is the principal output pathway from the amygdala. Inhibitory GABAergic neurons project from the CeA to the hypothalamus and brainstem; the activation of these neurons leads to the somatic manifestations of anxiety.25 Projections to other basal forebrain nuclei such as the ventrotegmental area and the locus ceruleus may be involved in the dysphoria associated with anxiety.16 In addition to activation of the CeA, neurons from the BLA also activate cells in the adjacent bed nucleus of the stria terminalis, which project to the same areas as the CeA and apparently play a similar role.16,22
In addition to the role of the amygdala in the regulation of anxiety, forebrain areas such as the medial prefrontal cortex (PFC) and anterior cingulate cortex also appear to play an important role.15 These cortical areas receive and send excitatory glutamatergic projections to and from the BLA, and are activated concomitantly with the amygdala during presentation of emotional stimuli.26
It has been suggested that the medial PFC regulates the experience or expression of anxiety through modulation of neuronal activity in the BLA, with more dorsal cortical areas being responsible for conscious, voluntary control of anxiety and more ventral areas responsible for implicit, subconscious control.15,27–29 This “top-down” control would lead to inhibition of output from the amygdala.
Again, neuroimaging studies have shown the medial PFC to be hypoactive in certain anxiety disorders, notably posttraumatic stress disorder20,30 and generalized anxiety disorder.15 Moreover, in subjects exerting voluntary effort to control their emotional reactivity31,32 to negative emotional stimuli, the lateral and medial PFC was strongly activated. Interestingly, anxious individuals needed to achieve higher levels of PFC activation than did non-anxious participants in order to reduce negative emotions successfully. Also of interest in this context is a neuroimaging study that demonstrated activation of the anterior cingulate cortex associated with an anxiolytic response to a placebo in subjects reacting to negatively charged cues, without any change in the activation of the amygdala.33 The strength of the activation of the anterior cingulate cortex was strongly correlated with the robustness of the placebo effect.33 Understanding how these cortical–subcortical regulatory mechanisms work and how they can be harnessed could be very important in developing more effective interventions to reduce anxiety.
Role of GABA in the amygdala
GABAergic neurotransmission in the amygdala is a promising candidate for modulation of anxiety-related responses. A number of lines of research in experimental animals have provided evidence for an important role of GABAergic neurotransmission in the amygdala in modulating anxiety-related behaviors. For example, infusions of GABA or GABA receptor agonists into the amygdala decrease measures of fear and anxiety in several animal species while infusions of GABA antagonists tend to have anxiogenic effects.34,35 Likewise, selective deactivation of expression of the GABA synthetic enzyme glutamic acid decarboxylase in the amygdala leads to loss of the anxiolytic response to benzodiazepines.36 In humans, administration of benzodiazepines attenuates the activation of the amygdala in the presence of negative emotional stimuli.37,38
As described above, the GABAergic intercalated neurons that lie entirely within the amygdala (Figure 2) regulate activation of the CeA by the BLA. In addition, these neurons are also a principal target of the excitatory glutamatergic projection from the PFC to the amygdala39,40 and make numerous monosynaptic contacts between different groups of intercalated neurons.41,42 These neurons possess a slowly deactivating potassium conductance, which confers on them the ability to modify their firing pattern in response to changes in the overall activity within the amygdala.40,43 These GABAergic neurons are thus well placed to play a key modulatory role in the gating of information related to anxiety in the amygdala.24,44,45
Figure 2.
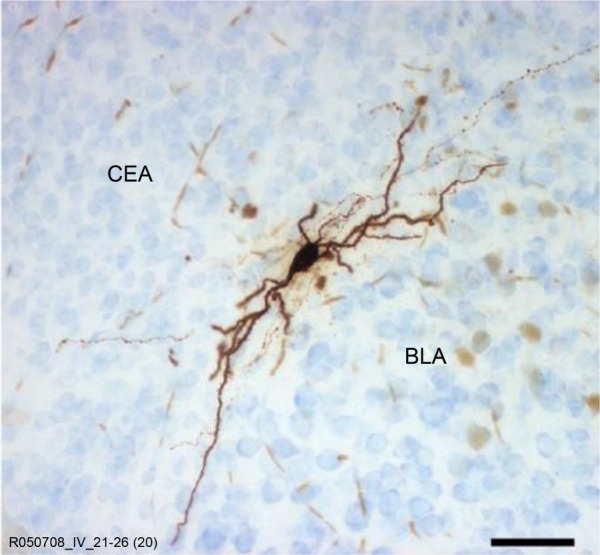
A GABAergic medial paracapsular intercalated cell in the mouse amygdala.
Notes: Scale bar: 50 μm. Republished with permission of the Society for Neuroscience, from Different fear states engage distinct networks within the intercalated cell clusters of the amygdala, Busti D, Geracitano R, Whittle N, et al, J Neurosci, 31(13), 2011;41 permission conveyed through Copyright Clearance Center, Inc.
Abbreviations: BLA, basolateral amygdala; CeA, central nucleus of the amygdala.
Neurotransmitters involved in the modulation of anxiety
The expression of anxiety involves a coordinated activity of numerous brain pathways involving different neurotransmitters, all of which interact and are modulated by local and distant synaptic relays. The role of the inhibitory neurotransmitter GABA has long been regarded as central to the regulation of anxiety and this neurotransmitter system is the target of benzodiazepines and related drugs used to treat anxiety disorders.46 It should nevertheless be recognized that GABA is not the only neurotransmitter important in the modulation of anxiety responses in the amygdala, and many other neurotransmitters have been implicated, including serotonin,47,48 opioid peptides,49 endocannabinoids,50,51 neuropeptide Y,52,53 oxytocin,54 and corticotropin-releasing hormone.53 These important neurotransmitter pathways will not be considered in detail in this review.
GABA, GABA receptors, and GABA-involved structures
GABA is the major inhibitory neurotransmitter in the central nervous system (CNS) and it has been estimated that at least one-third of all CNS neurons utilize GABA as their primary neurotransmitter.55 GABAergic inhibition is essential for maintaining a balance between neuronal excitation and inhibition, for tight temporal and spatial control of transsynaptic signaling, temporal modulation of neuronal excitability, and for maintaining oscillatory “pacemaker” activities in numerous brain regions.56 Most GABA-containing neurons are interneurons, which control the excitability of local circuits within a given brain region, although some major projection pathways – notably arising in the thalamus and cortex – are GABAergic.57
Neuronal inhibition by GABA is mediated by two distinct classes of GABA receptors. Ionotropic GABAA receptors are fast-acting ligand-gated chloride channels responsible for rapid inhibition,58 while metabotropic GABAB receptors are coupled indirectly via G-proteins to either calcium or potassium channels to produce slow and prolonged inhibitory responses.59 Their role in neurological and psychological function remains poorly characterized, although baclofen – a molecule that mimics the action of GABA at these GABAB receptors – has potent myorelaxant properties and has been proposed to be useful in the treatment of alcohol dependence.60
Activation of GABAA receptors causes an immediate and substantial rise in chloride conductance across the cell membrane, which renders the neuron unable to raise an action potential and leads to “phasic” inhibition of the neuron.61 In addition, low (submicromolar) concentrations of GABA in the extracellular space can persistently activate extrasynaptic GABAA receptors to generate a persistent or “tonic” inhibitory state, whereby the neuron will be rendered less responsive to excitatory stimuli.62
GABAA receptor structure and function
The GABAA receptor is a hetero-oligomer composed of five protein subunits which traverse the neuronal membrane (Figure 3). They form a cylinder with a long axis of 11 nm and a diameter of 8 nm.63 Activation of the receptor by GABA causes a conformational change in the protein subunits leading to the transient creation of a pore along the axis of the cylinder through which chloride ions can flow from one side of the membrane to another.64 The protein subunits making up the GABAA receptor can be grouped into different families based on their amino acid sequence, each family being made up of a restricted number of variants, differing from one another in a restricted number of amino acids.65 The majority of GABAA receptors in the CNS are composed of two α subunits, two β subunits, and a γ subunit. Other families include the δ, ε, and π subunits, which substitute for the γ subunit on certain cell types, the θ subunit, which can substitute for the β subunit, and the ρ subunit, expressed predominantly in the retina, which forms homo-oligomeric GABAA receptors.65
Figure 3.
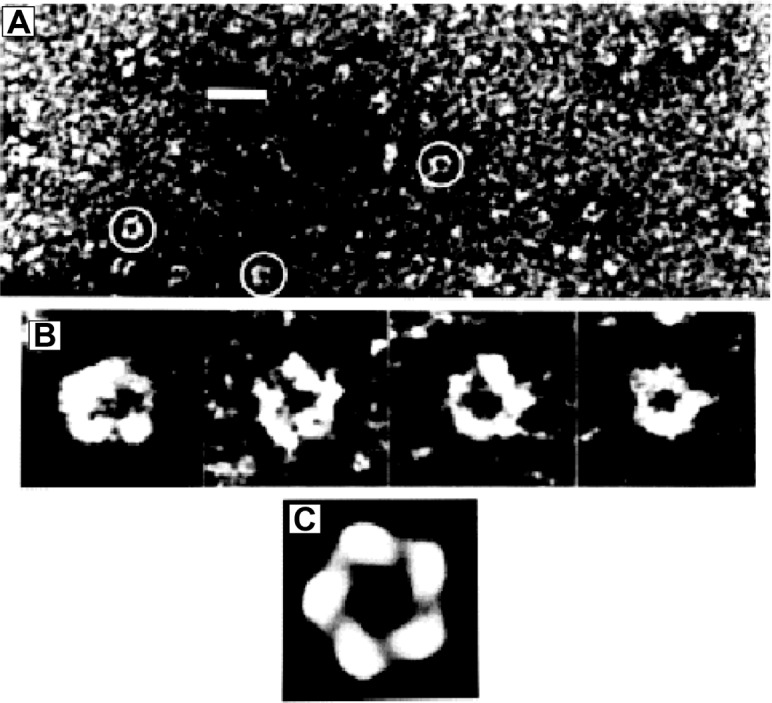
Electron micrograph of the GABAA receptor isolated from a pig’s brain and stained with uranyl acetate.
Note: In (A), the general field of the purified protein stained with uranyl acetate is shown. The receptors are circled in this image taken at an actual magnification of 33,000×; bar =20 nm. In (B), four receptor particles, typical of those exhibiting five-fold symmetry, are shown. (C) shows a rotationally averaged representation of the first particle in (B), in (C) the image is significantly enhanced. Reproduced from Quaternary structure of the native GABAA receptor determined by electron microscopic image analysis, Nayeem N, Green TP, Martin IL, Barnard EA, Journal of Neurochemistry, 1994;62(2):815–818.63 Copyright © 2002, John Wiley and Sons. Available from: http://onlinelibrary.wiley.com/doi/10.1046/j.1471-4159.1994.62020815.x/abstract.
The α subunits confer sensitivity to GABA on the receptor and also determine its pharmacological specificity towards a number of allosteric modulators, notably the benzodiazepines and those that bind to the same site.65 GABA binds within the interface between the α and β subunits; since there are two pairs of α and β subunit on each receptor, two GABA molecules bind to each receptor, and thus allows positive cooperativity in the activation of the chloride conductance, conveying high sensitivity to the response (Figure 4).66 Benzodiazepines and related drugs bind within the interface between the α and γ subunits, and their binding enhances the probability of channel opening in response to GABA.67 Through such a mechanism, benzodiazepines facilitate GABAergic inhibition. The affinity of benzodiazepines for the GABAA receptor is determined by the nature of the α and γ subunit, and since GABAA receptors in different regions of the brain have different subunit structures, their sensitivity to allosteric modulators varies.67
Figure 4.
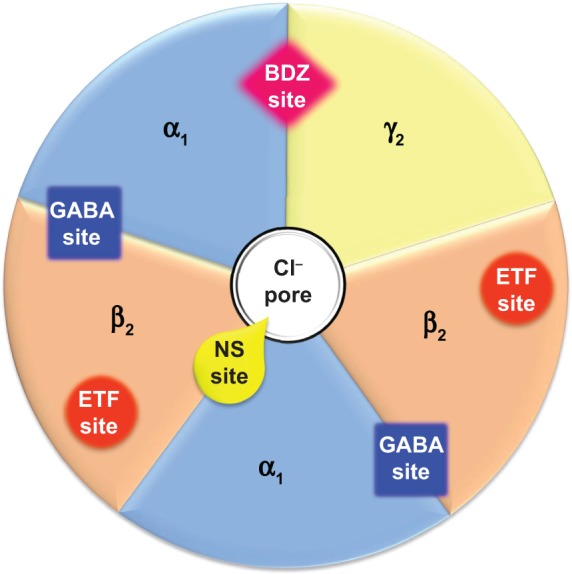
Schematic representation of binding sites on the most common isoform of GABAA receptor.
Note: α1, β2, and γ2 represent the subunits of the most widespread isoform of the GABAA receptor in the central nervous system.
Abbreviations: BDZ, benzodiazepine; ETF, etifoxine; GABA, γ-aminobutyric acid; NS, neurosteroid.
Indeed, the different α subunits also show anatomically specific patterns of expression in the CNS, with functionally different neuronal circuits expressing GABAA receptors of specific α subunit compositions. This differentiation is observed between brain regions, between different subcellular compartments (eg, presynaptic versus postsynaptic, somatic versus dendritic), and even between individual synapses of the same neuron.65 This structural heterogeneity suggests that the different α subunits may confer different behavioral functions and thus may constitute targets for specific therapeutic actions.68 Much effort has been devoted to tailoring the selectivity of molecules targeting these receptors in order to develop more effective medications or to avoid unwanted side-effects such as sedation or dependence. In this respect, it has been suggested, for example, that the relative specificity for α1-containing GABAA receptors of the hypnotic drug zolpidem may explain its relative lack of myorelaxant effects.69 Animal experiments using targeted gene deletion and introduction of point mutations in GABAA receptor genes have indicated that GABAA receptors containing the α2 subunit are particularly relevant for the manifestations of anxiety.70,71 In addition, α2-containing GABAA receptors are the predominant GABAA receptor subtype in the CeA.70
GABAA receptor modulation by neurosteroids
There is growing evidence that neuroactive steroids play an important role as endogenous modulators of neuronal function and behavioral processes, and that alterations of endogenous neuroactive steroid concentrations may contribute to the pathophysiology of anxiety disorders.72 Neurosteroids can modify neuronal excitability through interaction with GABAA receptors, as well as the N-methyl-D-aspartate subtype of excitatory amino acid (glutamate) receptors.73 Certain steroids can act as positive allosteric modulators of the GABAA receptor, as well as directly activating the receptor.74 By binding in the interface between the α and β subunits close to the chloride ion channel (Figure 4), they activate the receptor, allowing chloride ions to traverse the pore. By binding to a separate site on the α subunit, they facilitate channel opening in response to GABA.74 The structure–activity relationships for the two sites are not the same, so different steroids have different relative direct activating and indirect facilitatory effects.75 Paradoxically, some of these agents may also inhibit GABA-mediated responses at higher doses, although the mechanism responsible for this effect remains unclear.75
These steroid sites can be recognized by steroids secreted by the adrenal gland (eg, pregnenolone), by steroids locally synthesized within the CNS, and by synthetic derivatives.76–79 Alfaxolone, which is used as an anesthetic in veterinary medicine, is an example of the latter class. In addition, certain adrenal steroids may be converted to neuroactive derivatives within the CNS. This is the case for progesterone, itself inactive at the GABAA receptor, which is converted to the neuroactive 3α,5α-tetrahydroprogesterone.80,81
From the point of view of understanding the role of GABA receptors in anxiety, an important modulatory system consists of locally produced neurosteroids since their synthesis within glial cells is upregulated by stress.82 They thus represent a potential intrinsic modulatory system to fine-tune inhibition within the CNS in response to anxiogenic stimuli.83 Examples of neurosteroids include allopregnanolone and 3α,5α-tetrahydrodeoxycorticosterone, although adrenal steroids such as pregnenolone and dehydroepiandrosterone are also produced within the CNS.77 These are synthesized by cytochrome P450 enzymes in oligodendrocytes, astrocytes, and possibly in neurons.77 Allopregnanolone is a potent positive allosteric modulator of GABAA receptors, which allows it to facilitate and fine-tune the activity of GABA at these receptors.77 Through its action on GABAA receptors, allopregnanolone exerts anxiolytic, antidepressant, and anticonvulsant activities when administered to experimental animals.84,85 These pharmacological actions are reminiscent of those elicited by barbiturates and benzodiazepines. Interestingly, the synthesis of neurosteroids in the CNS can be upregulated by SSRIs,86 providing a link between these treatments for anxiety disorders and the GABAergic system.
The production of neurosteroids varies between brain regions and, together with the heterogeneity in subunit composition of the GABAA receptor, endows the neurosteroid system with substantial regional and even cellular specificity with regard to allosteric regulation of the GABAA receptor.77 The synthesis of neurosteroids is modulated by stress,87 by changes in the estrous cycle,88 by pregnancy,89 and by alcohol.90,91 Changes in neurosteroid levels and subsequent changes in the sensitivity of GABAA receptors have been implicated in the development of premenstrual dysphoric disorder.92 There have been many studies investigating concentrations of neurosteroids in patients with anxiety disorders, although their results have not always been consistent. For example, serum concentrations of pregnenolone have been shown to be lowered in patients with generalized anxiety disorder93 and with generalized social phobia,94 whereas progesterone concentrations are increased in patients with panic disorder and are correlated with state anxiety.95 In healthy volunteers receiving cholecystokinin tetrapeptide for induction of an experimental panic attack, an increase in adrenocorticotropic hormone and cortisol levels as well as in 3α,5α-tetrahydrodeoxycorticosterone concentrations was observed.96 In experimental animals, direct administration of low concentrations of several neurosteroids, including pregnenolone, dehydroepiandrosterone, and progesterone, has anxiolytic effects.97–99 It has been demonstrated that 3α,5α-tetrahydrodeoxycorticosterone attenuates the stress-induced elevation of plasma adrenocorticotropic hormone and corticosterone in rats.100,101 Moreover, in humans, administration of progesterone also produces sedative–anxiolytic effects.102 Neurosteroids thus represent an attractive target for the development of new anxiolytic or antidepressant drugs.83 Interestingly, functional imaging studies in humans have demonstrated that progesterone and its metabolites may influence states of human anxiety by modulating neuronal activity in the amygdala.103,104 In addition, neuroactive steroids show less abuse potential than benzodiazepines.105 Decreased abuse liability together with the lack of interaction with ethanol would present a potential advantage over currently available benzodiazepine anxiolytics, and rapidity of action would present an advantage over SSRIs. The goal would be to find agents that exploit this molecular pathway, that act with robust anxiolytic-like activity but have a large separation between anxiolytic-like effects and sedation/ataxia, and that show good oral bioavailability. Neurosteroids themselves are perhaps not the ideal candidate – firstly since they are extensively metabolized both in the periphery and within the CNS, and secondly due to the risk of unwanted endocrine side-effects in the periphery. Indeed, in a double-blind, crossover study, treatment with pregnenolone for 4 weeks had no significant effects on mood, memory, self-rated sleep quality, or subjective wellbeing in healthy volunteers.106
On the other hand, small molecules that selectively promote neurosteroid synthesis within the CNS or mimic the action of neurosteroids at GABAA receptors may be more promising.107 For example, etifoxine – a small molecule structurally unrelated to benzodiazepines that demonstrates anxiolytic effects in experimental animals108 and man109 – may be a modulator of neurosteroid action. Randomized clinical trials comparing etifoxine to buspirone, sulpiride, or lorazepam have demonstrated the efficacy of this agent in patients with adjustment disorder.110,111 Moreover, compared to lorazepam, etifoxine has relatively low sedative effects and is associated with less memory impairment and interference with neuropsychological function.112
Etifoxine on the GABAA receptor and neurosteroids
Two distinct but complementary molecular mechanisms have been described for etifoxine, namely a direct effect on the GABAA receptor and a facilitatory effect on the synthesis of 3α,5α-neurosteroids.113–116 These actions would be expected to act in synergy to augment inhibitory GABA neurotransmission.
On the GABAA receptor itself, etifoxine is a positive allosteric modulator which enhances chloride conductance in response to GABA.114 Etifoxine appears to bind to a site on the GABAA receptor distinct from binding sites for benzodiazepines and neurosteroids.113,114 More recent electrophysiological studies using recombinant GABAA receptors of known subunit composition have identified the β subunit of the receptor as being critical for the action of etifoxine.115 Using homomeric GABAA receptors containing β subunits only, etifoxine reduces the spontaneous background chloride flux through these receptor ion channels. Using heteromeric receptors containing either α and β subunits only or β and γ subunits only, etifoxine consistently enhanced the chloride currents evoked by GABA, suggesting that the β subunit of the receptor is necessary and sufficient for the positive allosteric effect of etifoxine.115 The activity of etifoxine was greatest in receptors containing the β2 or β3 isoforms compared to the β1 isoform. On the other hand, the nature of the α subunit did not influence the activity of etifoxine. Interestingly, activity was not abolished by an arginine to serine point mutation in position 289 of the β subunit, as is the case for loreclezole – an anticonvulsant drug that also targets the β subunit.117
The second molecular target of etifoxine is the synthetic machinery for neurosteroids in mitochondria and, in particular, the 18 kDa mitochondrial translocator protein (TSPO) to which it binds.116 TSPO is a transporter protein responsible for presenting steroids in the inner leaflet of mitochondria.118 This process is thought to be the rate-limiting step in the synthesis of neurosteroids.119 In rats, administration of etifoxine leads to a dose-dependent rise in concentrations of pregnenolone, progesterone, 5α-dihydroprogesterone, and allopregnanolone in plasma and cerebrospinal fluid.116 This effect is preserved in adrenalectomized and castrated animals, suggesting that the increase is due to stimulation of neurosteroid synthesis in the CNS.116 The relevance of this effect to the anxiolytic effect of etifoxine is suggested by the observation that these effects are attenuated by the administration of finasteride, an inhibitor of 5α-reductase – a key enzyme in the synthesis of neurosteroids.76,116
The involvement of neurosteroids in the mechanism of action of etifoxine is further supported by studies in neuropathic pain, a condition in which neurosteroids are particularly effective.120 It has been observed that etifoxine attenuates neuropathic pain and that this analgesic effect is blocked by the 5α-reductase inhibitor finasteride.121 It should be noted that etifoxine may have other mechanisms of action independent of GABA, for example, mechanisms related to the neurotrophic factor GDNF.122 However, these GABA-independent mechanisms are currently poorly characterized.
Conclusion
Lines of evidence coming from different branches of neuroscience indicate that anxiety disorders arise not from a hard-wired defect in the brain but rather from a dysfunction in the modulation of brain circuits that regulate the emotional response to potentially threatening stimuli. An integrated modulatory model of anxiety may be represented schematically by an imbalance between overactive bottom-up activity from the amygdala, indicating the presence of potentially threatening stimuli, and dysfunctional top-down control mechanisms originating in the PFC, related to an inappropriate representation of the emotional salience of stimuli. From a neurophysiological perspective, it is now recognized that the neural circuits involved in anxiety comprise inhibitory networks of principally GABAergic interneurons. The presence of allosteric sites on the GABAA receptor allows the level of inhibition of the neuron to be regulated with exquisite precision. These allosteric sites are the molecular targets for the principal classes of anticonvulsant, anxiolytic, and sedative–hypnotic drugs, including benzodiazepines, barbiturates, neurosteroids, certain classes of general anesthetics, and alcohol. In addition, changes in the levels of endogenous modulators of these allosteric sites, in particular the neurosteroids, as well as changes in the subunit composition of the GABAA receptor may represent mechanisms whereby the level of neuronal inhibition is downregulated in pathological anxiety states. Targeting the neurosteroid–GABAA receptor axis may allow specific modulation of anxiety as a function of the anxious stimuli to which the subject is exposed. In this respect, molecules such as etifoxine, which regulate neurosteroid synthesis as well as interact with the GABAA receptor itself, are of particular interest as modulatory agents that could reset the sensitivity of interneuronal regulatory circuits in the amygdala.
Acknowledgments
The author would like to express his gratitude to all the staff at Foxymed (Paris, France) for their help in preparing this review.
Footnotes
Disclosure
Dr Nuss received a research grant and consultancy fees from Biocodex. The author reports no other conflicts of interest in this work.
References
- 1.Uhlenhuth EH, Starcevic V, Warner TD, et al. A general anxiety-prone cognitive style in anxiety disorders. J Affect Disord. 2002;70(3):241–249. doi: 10.1016/s0165-0327(01)00457-8. [DOI] [PubMed] [Google Scholar]
- 2.Ganellen RJ, Matuzas W, Uhlenhuth EH, Glass R, Easton CR. Panic disorder, agoraphobia, and anxiety-relevant cognitive style. J Affect Disord. 1986;11(3):219–225. doi: 10.1016/0165-0327(86)90073-x. [DOI] [PubMed] [Google Scholar]
- 3.Starcevic V, Sammut P, Berle D, et al. Can levels of a general anxiety-prone cognitive style distinguish between various anxiety disorders? Compr Psychiatry. 2012;53(5):427–433. doi: 10.1016/j.comppsych.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 4.American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition – Text Revision: DSM-IV-TR®. Arlington, VA: American Psychiatric Association; 2000. [Google Scholar]
- 5.American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition: DSM-V®. Arlington, VA: American Psychiatric Association; 2013. [Google Scholar]
- 6.Lader M. Benzodiazepines revisited – will we ever learn? Addiction. 2011;106(12):2086–2109. doi: 10.1111/j.1360-0443.2011.03563.x. [DOI] [PubMed] [Google Scholar]
- 7.Canadian Psychiatric Association Clinical practice guidelines. Management of anxiety disorders. Can J Psychiatry. 2006;51(8 Suppl 2):9S–91S. [PubMed] [Google Scholar]
- 8.Bandelow B, Sher L, Bunevicius R, et al. Guidelines for the pharmacological treatment of anxiety disorders, obsessive–compulsive disorder, and posttraumatic stress disorder in primary care. Int J Psychiatry Clin Pract. 2012;16(2):77–84. doi: 10.3109/13651501.2012.667114. [DOI] [PubMed] [Google Scholar]
- 9.Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder, and obsessive–compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(5):403–439. doi: 10.1177/0269881114525674. [DOI] [PubMed] [Google Scholar]
- 10.Generalised anxiety disorder and panic disorder (with or without agoraphobia) in adults: management in primary, secondary, and community care [webpage on the Internet] London: National Institute for Health and Clinical Excellence; 2011. [Accessed September 22, 2014]. Available from: http://www.nice.org.uk/guidance/cg113/chapter/guidance. [Google Scholar]
- 11.Cottraux J. Nonpharmacological treatments for anxiety disorders. Dialogues Clin Neurosci. 2002;4(3):305–319. doi: 10.31887/DCNS.2002.4.3/jcottraux. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Butler AC, Chapman JE, Forman EM, Beck AT. The empirical status of cognitive–behavioral therapy: a review of meta-analyses. Clin Psychol Rev. 2006;26(1):17–31. doi: 10.1016/j.cpr.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 13.Adolphs R, Tranel D, Hamann S, et al. Recognition of facial emotion in nine individuals with bilateral amygdala damage. Neuropsychologia. 1999;37(10):1111–1117. doi: 10.1016/s0028-3932(99)00039-1. [DOI] [PubMed] [Google Scholar]
- 14.Lanteaume L, Khalfa S, Regis J, Marquis P, Chauvel P, Bartolomei F. Emotion induction after direct intracerebral stimulations of human amygdala. Cereb Cortex. 2007;17(6):1307–1313. doi: 10.1093/cercor/bhl041. [DOI] [PubMed] [Google Scholar]
- 15.Etkin A. Functional neuroanatomy of anxiety: a neural circuit perspective. In: Stein MB, Steckler T, editors. Behavioral Neurobiology of Anxiety and Its Treatment. Berlin: Springer Verlag; 2009. pp. 251–277. [DOI] [PubMed] [Google Scholar]
- 16.Forster GL, Novick AM, Scholl JL, Watt MJ. The role of the amygdala in anxiety disorders. In: Ferry B, editor. The Amygdala: A Discrete Multitasking Manager. Rijeka: InTech; 2012. pp. 61–102. [Google Scholar]
- 17.Wager TD, Phan KL, Liberzon I, Taylor SF. Valence, gender, and lateralization of functional brain anatomy in emotion: a meta-analysis of findings from neuroimaging. Neuroimage. 2003;19(3):513–531. doi: 10.1016/s1053-8119(03)00078-8. [DOI] [PubMed] [Google Scholar]
- 18.Phan KL, Wager T, Taylor SF, Liberzon I. Functional neuroanatomy of emotion: a meta-analysis of emotion activation studies in PET and fMRI. Neuroimage. 2002;16(2):331–348. doi: 10.1006/nimg.2002.1087. [DOI] [PubMed] [Google Scholar]
- 19.Carlson JM, Greenberg T, Rubin D, Mujica-Parodi LR. Feeling anxious: anticipatory amygdalo-insular response predicts the feeling of anxious anticipation. Soc Cogn Affect Neurosci. 2011;6(1):74–81. doi: 10.1093/scan/nsq017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Etkin A, Wager TD. Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. Am J Psychiatry. 2007;164(10):1476–1488. doi: 10.1176/appi.ajp.2007.07030504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Straube T, Glauer M, Dilger S, Mentzel HJ, Miltner WH. Effects of cognitive–behavioral therapy on brain activation in specific phobia. Neuroimage. 2006;29(1):125–135. doi: 10.1016/j.neuroimage.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Davis M. Neural circuitry of anxiety and stress disorders. In: Davis KL, Charney D, Coyle JT, Nemeroff C, editors. Neuropsychopharmacology: The Fifth Generation of Progress. Philadelphia: Lippincott, Williams, & Wilkins; 2002. pp. 729–743. [Google Scholar]
- 23.Pitkanen A, Savander V, LeDoux JE. Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci. 1997;20(11):517–523. doi: 10.1016/s0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- 24.Royer S, Martina M, Pare D. An inhibitory interface gates impulse traffic between the input and output stations of the amygdala. J Neurosci. 1999;19(23):10575–10583. doi: 10.1523/JNEUROSCI.19-23-10575.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jongen-Relo AL, Amaral DG. Evidence for a GABAergic projection from the central nucleus of the amygdala to the brainstem of the macaque monkey: a combined retrograde tracing and in situ hybridization study. Eur J Neurosci. 1998;10(9):2924–2933. doi: 10.1111/j.1460-9568.1998.00299.x. [DOI] [PubMed] [Google Scholar]
- 26.Kober H, Barrett LF, Joseph J, Bliss-Moreau E, Lindquist K, Wager TD. Functional grouping and cortical–subcortical interactions in emotion: a meta-analysis of neuroimaging studies. Neuroimage. 2008;42(2):998–1031. doi: 10.1016/j.neuroimage.2008.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim MJ, Loucks RA, Palmer AL, et al. The structural and functional connectivity of the amygdala: from normal emotion to pathological anxiety. Behav Brain Res. 2011;223(2):403–410. doi: 10.1016/j.bbr.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ochsner KN, Gross JJ. The cognitive control of emotion. Trends Cogn Sci. 2005;9(5):242–249. doi: 10.1016/j.tics.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 29.Ochsner KN, Ray RR, Hughes B, et al. Bottom-up and top-down processes in emotion generation: common and distinct neural mechanisms. Psychol Sci. 2009;20(11):1322–1331. doi: 10.1111/j.1467-9280.2009.02459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research – past, present, and future. Biol Psychiatry. 2006;60(4):376–382. doi: 10.1016/j.biopsych.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 31.Campbell-Sills L, Simmons AN, Lovero KL, Rochlin AA, Paulus MP, Stein MB. Functioning of neural systems supporting emotion regulation in anxiety-prone individuals. Neuroimage. 2011;54(1):689–696. doi: 10.1016/j.neuroimage.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalisch R, Wiech K, Critchley HD, et al. Anxiety reduction through detachment: subjective, physiological, and neural effects. J Cogn Neurosci. 2005;17(6):874–883. doi: 10.1162/0898929054021184. [DOI] [PubMed] [Google Scholar]
- 33.Petrovic P, Dietrich T, Fransson P, Andersson J, Carlsson K, Ingvar M. Placebo in emotional processing – induced expectations of anxiety relief activate a generalized modulatory network. Neuron. 2005;46(6):957–969. doi: 10.1016/j.neuron.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 34.Sanders SK, Shekhar A. Regulation of anxiety by GABAA receptors in the rat amygdala. Pharmacol Biochem Behav. 1995;52(4):701–706. doi: 10.1016/0091-3057(95)00153-n. [DOI] [PubMed] [Google Scholar]
- 35.Barbalho CA, Nunes-de-Souza RL, Canto-de-Souza A. Similar anxiolytic-like effects following intra-amygdala infusions of benzodiazepine receptor agonist and antagonist: evidence for the release of an endogenous benzodiazepine inverse agonist in mice exposed to elevated plus-maze test. Brain Res. 2009;1267:65–76. doi: 10.1016/j.brainres.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 36.Heldt SA, Mou L, Ressler KJ. In vivo knockdown of GAD67 in the amygdala disrupts fear extinction and the anxiolytic-like effect of diazepam in mice. Transl Psychiatry. 2012;2:e181. doi: 10.1038/tp.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Del-Ben CM, Ferreira CA, Sanchez TA, et al. Effects of diazepam on BOLD activation during the processing of aversive faces. J Psychopharmacol. 2012;26(4):443–451. doi: 10.1177/0269881110389092. [DOI] [PubMed] [Google Scholar]
- 38.Paulus MP, Feinstein JS, Castillo G, Simmons AN, Stein MB. Dose-dependent decrease of activation in bilateral amygdala and insula by lorazepam during emotion processing. Arch Gen Psychiatry. 2005;62(3):282–288. doi: 10.1001/archpsyc.62.3.282. [DOI] [PubMed] [Google Scholar]
- 39.McDonald AJ, Mascagni F, Guo L. Projections of the medial and lateral prefrontal cortices to the amygdala: a Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience. 1996;71(1):55–75. doi: 10.1016/0306-4522(95)00417-3. [DOI] [PubMed] [Google Scholar]
- 40.Pinto A, Sesack SR. Ultrastructural analysis of prefrontal cortical inputs to the rat amygdala: spatial relationships to presumed dopamine axons and D1 and D2 receptors. Brain Struct Funct. 2008;213(1–2):159–175. doi: 10.1007/s00429-008-0180-6. [DOI] [PubMed] [Google Scholar]
- 41.Busti D, Geracitano R, Whittle N, et al. Different fear states engage distinct networks within the intercalated cell clusters of the amygdala. J Neurosci. 2011;31(13):5131–5144. doi: 10.1523/JNEUROSCI.6100-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manko M, Geracitano R, Capogna M. Functional connectivity of the main intercalated nucleus of the mouse amygdala. J Physiol. 2011;589(Pt 8):1911–1925. doi: 10.1113/jphysiol.2010.201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Royer S, Martina M, Pare D. Bistable behavior of inhibitory neurons controlling impulse traffic through the amygdala: role of a slowly deinactivating K+ current. J Neurosci. 2000;20(24):9034–9039. doi: 10.1523/JNEUROSCI.20-24-09034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marowsky A, Yanagawa Y, Obata K, Vogt KE. A specialized subclass of interneurons mediates dopaminergic facilitation of amygdala function. Neuron. 2005;48(6):1025–1037. doi: 10.1016/j.neuron.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 45.Palomares-Castillo E, Hernandez-Perez OR, Perez-Carrera D, Crespo-Ramirez M, Fuxe K, Perez de la Mora M. The intercalated paracapsular islands as a module for integration of signals regulating anxiety in the amygdala. Brain Res. 2012;1476:211–234. doi: 10.1016/j.brainres.2012.03.047. [DOI] [PubMed] [Google Scholar]
- 46.Lydiard RB. The role of GABA in anxiety disorders. J Clin Psychiatry. 2003;64(Suppl 3):21–27. [PubMed] [Google Scholar]
- 47.Gordon JA, Hen R. The serotonergic system and anxiety. Neuromolecular Med. 2004;5(1):27–40. doi: 10.1385/NMM:5:1:027. [DOI] [PubMed] [Google Scholar]
- 48.Durant C, Christmas D, Nutt D. The pharmacology of anxiety. Curr Top Behav Neurosci. 2010;2:303–330. doi: 10.1007/7854_2009_8. [DOI] [PubMed] [Google Scholar]
- 49.Van’t Veer A, Carlezon WA., Jr Role of κ-opioid receptors in stress and anxiety-related behavior. Psychopharmacology (Berl) 2013;229(3):435–452. doi: 10.1007/s00213-013-3195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hill MN, Gorzalka BB. The endocannabinoid system and the treatment of mood and anxiety disorders. CNS Neurol Disord Drug Targets. 2009;8(6):451–458. doi: 10.2174/187152709789824624. [DOI] [PubMed] [Google Scholar]
- 51.Ruehle S, Rey AA, Remmers F, Lutz B. The endocannabinoid system in anxiety, fear memory, and habituation. J Psychopharmacol. 2012;26(1):23–39. doi: 10.1177/0269881111408958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu G, Feder A, Wegener G, et al. Central functions of neuropeptide Y in mood and anxiety disorders. Expert Opin Ther Targets. 2011;15(11):1317–1331. doi: 10.1517/14728222.2011.628314. [DOI] [PubMed] [Google Scholar]
- 53.Thorsell A. Brain neuropeptide Y and corticotropin-releasing hormone in mediating stress and anxiety. Exp Biol Med (Maywood) 2010;235(10):1163–1167. doi: 10.1258/ebm.2010.009331. [DOI] [PubMed] [Google Scholar]
- 54.Neumann ID, Landgraf R. Balance of brain oxytocin and vasopressin: implications for anxiety, depression, and social behaviors. Trends Neurosci. 2012;35(11):649–659. doi: 10.1016/j.tins.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 55.Bloom FE, Iversen LL. Localizing 3H-GABA in nerve terminals of rat cerebral cortex by electron microscopic autoradiography. Nature. 1971;229(5287):628–630. doi: 10.1038/229628a0. [DOI] [PubMed] [Google Scholar]
- 56.Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321(5885):53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martin DL. Brain glutamate decarboxylase. In: Boulton AA, Baker GB, Yu PH, editors. Neuromethods, Volume 5: Neurotransmitter Enzymes. Clifton, NJ: Humana Press; 1986. pp. 361–388. [Google Scholar]
- 58.Sieghart W. Structure, pharmacology, and function of GABAA receptor subtypes. Adv Pharmacol. 2006;54:231–263. doi: 10.1016/s1054-3589(06)54010-4. [DOI] [PubMed] [Google Scholar]
- 59.Bowery NG. Historical perspective and emergence of the GABAB receptor. Adv Pharmacol. 2010;58:1–18. doi: 10.1016/S1054-3589(10)58001-3. [DOI] [PubMed] [Google Scholar]
- 60.Gorsane MA, Kebir O, Hache G, et al. Is baclofen a revolutionary medication in alcohol addiction management? Review and recent updates. Subst Abus. 2012;33(4):336–349. doi: 10.1080/08897077.2012.663326. [DOI] [PubMed] [Google Scholar]
- 61.Mody I, Pearce RA. Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends Neurosci. 2004;27(9):569–575. doi: 10.1016/j.tins.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 62.Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6(3):215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- 63.Nayeem N, Green TP, Martin IL, Barnard EA. Quaternary structure of the native GABAA receptor determined by electron microscopic image analysis. J Neurochem. 1994;62(2):815–818. doi: 10.1046/j.1471-4159.1994.62020815.x. [DOI] [PubMed] [Google Scholar]
- 64.Horenstein J, Wagner DA, Czajkowski C, Akabas MH. Protein mobility and GABA-induced conformational changes in GABA(A) receptor pore-lining M2 segment. Nat Neurosci. 2001;4(5):477–485. doi: 10.1038/87425. [DOI] [PubMed] [Google Scholar]
- 65.Sieghart W, Sperk G. Subunit composition, distribution, and function of GABA(A) receptor subtypes. Curr Top Med Chem. 2002;2(8):795–816. doi: 10.2174/1568026023393507. [DOI] [PubMed] [Google Scholar]
- 66.Baumann SW, Baur R, Sigel E. Individual properties of the two functional agonist sites in GABA(A) receptors. J Neurosci. 2003;23(35):11158–11166. doi: 10.1523/JNEUROSCI.23-35-11158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sigel E, Luscher BP. A closer look at the high affinity benzodiazepine binding site on GABAA receptors. Curr Top Med Chem. 2011;11(2):241–246. doi: 10.2174/156802611794863562. [DOI] [PubMed] [Google Scholar]
- 68.Grillon C, Morgan CA, 3rd, Davis M, Southwick SM. Effects of experimental context and explicit threat cues on acoustic startle in Vietnam veterans with posttraumatic stress disorder. Biol Psychiatry. 1998;44(10):1027–1036. doi: 10.1016/s0006-3223(98)00034-1. [DOI] [PubMed] [Google Scholar]
- 69.Korpi ER, Mattila MJ, Wisden W, Luddens H. GABA(A)-receptor subtypes: clinical efficacy and selectivity of benzodiazepine site ligands. Ann Med. 1997;29(4):275–282. doi: 10.3109/07853899708999348. [DOI] [PubMed] [Google Scholar]
- 70.Low K, Crestani F, Keist R, et al. Molecular and neuronal substrate for the selective attenuation of anxiety. Science. 2000;290(5489):131–134. doi: 10.1126/science.290.5489.131. [DOI] [PubMed] [Google Scholar]
- 71.Mohler H. Pathophysiological aspects of diversity in neuronal inhibition: a new benzodiazepine pharmacology. Dialogues Clin Neurosci. 2002;4(3):261–269. doi: 10.31887/DCNS.2002.4.3/hmoehler. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eser D, Baghai TC, Schule C, Nothdurfter C, Rupprecht R. Neuroactive steroids as endogenous modulators of anxiety. Curr Pharm Des. 2008;14(33):3525–3533. doi: 10.2174/138161208786848838. [DOI] [PubMed] [Google Scholar]
- 73.Mathews A, Klug F. Emotionality and interference with color-naming in anxiety. Behav Res Ther. 1993;31(1):57–62. doi: 10.1016/0005-7967(93)90043-t. [DOI] [PubMed] [Google Scholar]
- 74.Hosie AM, Wilkins ME, da Silva HM, Smart TG. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature. 2006;444(7118):486–489. doi: 10.1038/nature05324. [DOI] [PubMed] [Google Scholar]
- 75.Wang M. Neurosteroids and GABA-A receptor function. Front Endocrinol (Lausanne) 2011;2:44. doi: 10.3389/fendo.2011.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ugale RR, Sharma AN, Kokare DM, Hirani K, Subhedar NK, Chopde CT. Neurosteroid allopregnanolone mediates anxiolytic effect of etifoxine in rats. Brain Res. 2007;1184:193–201. doi: 10.1016/j.brainres.2007.09.041. [DOI] [PubMed] [Google Scholar]
- 77.Lambert JJ, Belelli D, Peden DR, Vardy AW, Peters JA. Neurosteroid modulation of GABAA receptors. Prog Neurobiol. 2003;71(1):67–80. doi: 10.1016/j.pneurobio.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 78.Mellon SH, Vaudry H. Biosynthesis of neurosteroids and regulation of their synthesis. Int Rev Neurobiol. 2001;46:33–78. doi: 10.1016/s0074-7742(01)46058-2. [DOI] [PubMed] [Google Scholar]
- 79.Paul SM, Purdy RH. Neuroactive steroids. FASEB J. 1992;6(6):2311–2322. [PubMed] [Google Scholar]
- 80.Mellon SH, Griffin LD. Neurosteroids: biochemistry and clinical significance. Trends Endocrinol Metab. 2002;13(1):35–43. doi: 10.1016/s1043-2760(01)00503-3. [DOI] [PubMed] [Google Scholar]
- 81.Rupprecht R. Neuroactive steroids: mechanisms of action and neuropsychopharmacological properties. Psychoneuroendocrinology. 2003;28(2):139–168. doi: 10.1016/s0306-4530(02)00064-1. [DOI] [PubMed] [Google Scholar]
- 82.Barbaccia ML, Serra M, Purdy RH, Biggio G. Stress and neuroactive steroids. Int Rev Neurobiol. 2001;46:243–272. doi: 10.1016/s0074-7742(01)46065-x. [DOI] [PubMed] [Google Scholar]
- 83.Schule C, Eser D, Baghai TC, Nothdurfter C, Kessler JS, Rupprecht R. Neuroactive steroids in affective disorders: target for novel antidepressant or anxiolytic drugs? Neuroscience. 2011;191:55–77. doi: 10.1016/j.neuroscience.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 84.Brot MD, Akwa Y, Purdy RH, Koob GF, Britton KT. The anxiolytic-like effects of the neurosteroid allopregnanolone: interactions with GABA(A) receptors. Eur J Pharmacol. 1997;325(1):1–7. doi: 10.1016/s0014-2999(97)00096-4. [DOI] [PubMed] [Google Scholar]
- 85.Reddy DS. Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog Brain Res. 2010;186:113–137. doi: 10.1016/B978-0-444-53630-3.00008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pinna G, Costa E, Guidotti A. SSRIs act as selective brain steroidogenic stimulants (SBSSs) at low doses that are inactive on 5-HT reuptake. Curr Opin Pharmacol. 2009;9(1):24–30. doi: 10.1016/j.coph.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Purdy RH, Morrow AL, Moore PH, Jr, Paul SM. Stress-induced elevations of γ-aminobutyric acid type A receptor-active steroids in the rat brain. Proc Natl Acad Sci U S A. 1991;88(10):4553–4557. doi: 10.1073/pnas.88.10.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Corpechot C, Collins BE, Carey MP, Tsouros A, Robel P, Fry JP. Brain neurosteroids during the mouse oestrous cycle. Brain Res. 1997;766(1–2):276–280. doi: 10.1016/s0006-8993(97)00749-x. [DOI] [PubMed] [Google Scholar]
- 89.Gilbert Evans SE, Ross LE, Sellers EM, Purdy RH, Romach MK. 3α-reduced neuroactive steroids and their precursors during pregnancy and the postpartum period. Gynecol Endocrinol. 2005;21(5):268–279. doi: 10.1080/09513590500361747. [DOI] [PubMed] [Google Scholar]
- 90.Morrow AL, VanDoren MJ, Penland SN, Matthews DB. The role of GABAergic neuroactive steroids in ethanol action, tolerance, and dependence. Brain Res Brain Res Rev. 2001;37(1–3):98–109. doi: 10.1016/s0165-0173(01)00127-8. [DOI] [PubMed] [Google Scholar]
- 91.Cagetti E, Pinna G, Guidotti A, Baicy K, Olsen RW. Chronic intermittent ethanol (CIE) administration in rats decreases levels of neurosteroids in hippocampus, accompanied by altered behavioral responses to neurosteroids and memory function. Neuropharmacology. 2004;46(4):570–579. doi: 10.1016/j.neuropharm.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 92.Klatzkin RR, Morrow AL, Light KC, Pedersen CA, Girdler SS. Histories of depression, allopregnanolone responses to stress, and premenstrual symptoms in women. Biol Psychol. 2006;71(1):2–11. doi: 10.1016/j.biopsycho.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 93.Semeniuk T, Jhangri GS, Le Melledo JM. Neuroactive steroid levels in patients with generalized anxiety disorder. J Neuropsychiatry Clin Neurosci. 2001;13(3):396–398. doi: 10.1176/jnp.13.3.396. [DOI] [PubMed] [Google Scholar]
- 94.Heydari B, Le Melledo JM. Low pregnenolone sulphate plasma concentrations in patients with generalized social phobia. Psychol Med. 2002;32(5):929–933. doi: 10.1017/s0033291702005238. [DOI] [PubMed] [Google Scholar]
- 95.Brambilla F, Mellado C, Alciati A, et al. Plasma concentrations of anxiolytic neuroactive steroids in men with panic disorder. Psychiatry Res. 2005;135(3):185–190. doi: 10.1016/j.psychres.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 96.Eser D, di Michele F, Zwanzger P, et al. Panic induction with cholecystokinin-tetrapeptide (CCK-4) increases plasma concentrations of the neuroactive steroid 3α, 5α tetrahydrodeoxycorticosterone (3α, 5α-THDOC) in healthy volunteers. Neuropsychopharmacology. 2005;30(1):192–195. doi: 10.1038/sj.npp.1300572. [DOI] [PubMed] [Google Scholar]
- 97.Toufexis DJ, Davis C, Hammond A, Davis M. Progesterone attenuates corticotropin-releasing factor-enhanced but not fear-potentiated startle via the activity of its neuroactive metabolite, allopregnanolone. J Neurosci. 2004;24(45):10280–10287. doi: 10.1523/JNEUROSCI.1386-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Frye CA, Scalise TJ. Anti-seizure effects of progesterone and 3α,5α-THP in kainic acid and perforant pathway models of epilepsy. Psychoneuroendocrinology. 2000;25(4):407–420. doi: 10.1016/s0306-4530(99)00068-2. [DOI] [PubMed] [Google Scholar]
- 99.Maayan R, Lotan S, Doron R, et al. Dehydroepiandrosterone (DHEA) attenuates cocaine-seeking behavior in the self-administration model in rats. Eur Neuropsychopharmacol. 2006;16(5):329–339. doi: 10.1016/j.euroneuro.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 100.Patchev VK, Montkowski A, Rouskova D, Koranyi L, Holsboer F, Almeida OF. Neonatal treatment of rats with the neuroactive steroid tetrahydrodeoxycorticosterone (THDOC) abolishes the behavioral and neuroendocrine consequences of adverse early life events. J Clin Invest. 1997;99(5):962–966. doi: 10.1172/JCI119261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Patchev VK, Shoaib M, Holsboer F, Almeida OF. The neurosteroid tetrahydroprogesterone counteracts corticotropin-releasing hormone-induced anxiety and alters the release and gene expression of corticotropin-releasing hormone in the rat hypothalamus. Neuroscience. 1994;62(1):265–271. doi: 10.1016/0306-4522(94)90330-1. [DOI] [PubMed] [Google Scholar]
- 102.Soderpalm AH, Lindsey S, Purdy RH, Hauger R, Wit de H. Administration of progesterone produces mild sedative-like effects in men and women. Psychoneuroendocrinology. 2004;29(3):339–354. doi: 10.1016/s0306-4530(03)00033-7. [DOI] [PubMed] [Google Scholar]
- 103.van Wingen GA, van Broekhoven F, Verkes RJ, et al. Progesterone selectively increases amygdala reactivity in women. Mol Psychiatry. 2008;13(3):325–333. doi: 10.1038/sj.mp.4002030. [DOI] [PubMed] [Google Scholar]
- 104.van Wingen GA, Zylicz SA, Pieters S, et al. Testosterone increases amygdala reactivity in middle-aged women to a young adulthood level. Neuropsychopharmacology. 2009;34(3):539–547. doi: 10.1038/npp.2008.2. [DOI] [PubMed] [Google Scholar]
- 105.Rowlett JK, Winger G, Carter RB, Wood PL, Woods JH, Woolverton WL. Reinforcing and discriminative stimulus effects of the neuroactive steroids pregnanolone and Co 8-7071 in rhesus monkeys. Psychopharmacology (Berl) 1999;145(2):205–212. doi: 10.1007/s002130051050. [DOI] [PubMed] [Google Scholar]
- 106.Meieran SE, Reus VI, Webster R, Shafton R, Wolkowitz OM. Chronic pregnenolone effects in normal humans: attenuation of benzodiazepine-induced sedation. Psychoneuroendocrinology. 2004;29(4):486–500. doi: 10.1016/s0306-4530(03)00056-8. [DOI] [PubMed] [Google Scholar]
- 107.Nothdurfter C, Baghai TC, Schule C, Rupprecht R. Translocator protein (18 kDa) (TSPO) as a therapeutic target for anxiety and neurologic disorders. Eur Arch Psychiatry Clin Neurosci. 2012;262(Suppl 2):S107–S112. doi: 10.1007/s00406-012-0352-5. [DOI] [PubMed] [Google Scholar]
- 108.Verleye M, Heulard I, Gillardin JM. Investigation of the anticonvulsive effect of acute immobilization stress in anxious Balb/cByJ mice using GABA A-related mechanistic probes. Psychopharmacology (Berl) 2008;197(4):523–534. doi: 10.1007/s00213-007-1066-7. [DOI] [PubMed] [Google Scholar]
- 109.Besnier N, Blin O. Étifoxine: études cliniques récentes [Etifoxine: recent clinical studies] Encephale. 2008;34(Suppl 1):S9–S14. French. [Google Scholar]
- 110.Servant D, Graziani PL, Moyse D, Parquet PJ. Treatment of adjustment disorder with anxiety: efficacy and tolerance of etifoxine in a double-blind controlled study. Encephale. 1998;24(6):569–574. French. [PubMed] [Google Scholar]
- 111.Nguyen N, Fakra E, Pradel V, et al. Efficacy of etifoxine compared to lorazepam monotherapy in the treatment of patients with adjustment disorders with anxiety: a double-blind controlled study in general practice. Hum Psychopharmacol. 2006;21(3):139–149. doi: 10.1002/hup.757. [DOI] [PubMed] [Google Scholar]
- 112.Micallef J, Soubrouillard C, Guet F, et al. A double blind parallel group placebo controlled comparison of sedative and mnesic effects of etifoxine and lorazepam in healthy subjects [corrected] Fundam Clin Pharmacol. 2001;15(3):209–216. doi: 10.1046/j.1472-8206.2001.00025.x. [DOI] [PubMed] [Google Scholar]
- 113.Verleye M, Pansart Y, Gillardin J. Effects of etifoxine on ligand binding to GABA(A) receptors in rodents. Neurosci Res. 2002;44(2):167–172. doi: 10.1016/s0168-0102(02)00121-9. [DOI] [PubMed] [Google Scholar]
- 114.Schlichter R, Rybalchenko V, Poisbeau P, Verleye M, Gillardin J. Modulation of GABAergic synaptic transmission by the non-benzodiazepine anxiolytic etifoxine. Neuropharmacology. 2000;39(9):1523–1535. doi: 10.1016/s0028-3908(99)00253-1. [DOI] [PubMed] [Google Scholar]
- 115.Hamon A, Morel A, Hue B, Verleye M, Gillardin JM. The modulatory effects of the anxiolytic etifoxine on GABA(A) receptors are mediated by the β subunit. Neuropharmacology. 2003;45(3):293–303. doi: 10.1016/s0028-3908(03)00187-4. [DOI] [PubMed] [Google Scholar]
- 116.Verleye M, Akwa Y, Liere P, et al. The anxiolytic etifoxine activates the peripheral benzodiazepine receptor and increases the neurosteroid levels in rat brain. Pharmacol Biochem Behav. 2005;82(4):712–720. doi: 10.1016/j.pbb.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 117.Wafford KA, Bain CJ, Quirk K, et al. A novel allosteric modulatory site on the GABAA receptor β subunit. Neuron. 1994;12(4):775–782. doi: 10.1016/0896-6273(94)90330-1. [DOI] [PubMed] [Google Scholar]
- 118.Nothdurfter C, Rammes G, Baghai TC, et al. Translocator protein (18 kDa) as a target for novel anxiolytics with a favourable side-effect profile. J Neuroendocrinol. 2012;24(1):82–92. doi: 10.1111/j.1365-2826.2011.02166.x. [DOI] [PubMed] [Google Scholar]
- 119.Rupprecht R, Papadopoulos V, Rammes G, et al. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 2010;9(12):971–988. doi: 10.1038/nrd3295. [DOI] [PubMed] [Google Scholar]
- 120.Pathirathna S, Todorovic SM, Covey DF, Jevtovic-Todorovic V. 5α-reduced neuroactive steroids alleviate thermal and mechanical hyperalgesia in rats with neuropathic pain. Pain. 2005;117(3):326–339. doi: 10.1016/j.pain.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 121.Aouad M, Charlet A, Rodeau JL, Poisbeau P. Reduction and prevention of vincristine-induced neuropathic pain symptoms by the non-benzodiazepine anxiolytic etifoxine are mediated by 3α-reduced neurosteroids. Pain. 2009;147(1–3):54–59. doi: 10.1016/j.pain.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 122.Zhou X, He X, He B, et al. Etifoxine promotes glial-derived neurotrophic factor-induced neurite outgrowth in PC12 cells. Mol Med Rep. 2013;8(1):75–80. doi: 10.3892/mmr.2013.1474. [DOI] [PubMed] [Google Scholar]