

Abstract
Failure to maintain mitochondrial integrity is linked to age-related conditions, such as neurodegeneration. Two genes linked to Parkinson's disease, PINK1 and Parkin, play a key role in targeting the degradation of dysfunctional mitochondria (mitophagy). However, the mechanisms regulating the PINK1/Parkin pathway and other processes that impinge on mitochondrial turnover are poorly understood. Two articles in EMBO reports, by the Przedborski and Ganley groups 1,2, shed light on a new role for processed, cytoplasmic PINK1, and show that depletion of cellular iron levels stimulates PINK1/Parkin-independent mitophagy.
Mitochondrial homeostasis is a tightly controlled process that involves mitochondrial dynamics, trafficking and degradation. The discovery of a link between autosomal-recessive forms of Parkinson's disease (PD) and mitochondrial quality control has sparked intense interest in understanding these pathways. The serine-threonine kinase PINK1 and the E3-ubiquitin ligase Parkin act in a common pathway to promote the degradation of failing mitochondria through selective autophagy—a process known as mitophagy. The prevailing model posits that under basal conditions, ‘healthy’ mitochondria import PINK1, which undergoes rapid proteolysis, export and degradation. This process constitutively represses a key degradation signal. Upon mitochondrial damage—modeled by the dissipation of mitochondrial membrane potential (ΔΨm) with CCCP or valinomycin—PINK1 import is blocked, precluding its proteolytic processing and resulting in the stabilization of full-length PINK1 on the outer mitochondrial membrane (OMM). This stimulates the recruitment of cytosolic Parkin to the mitochondrial surface, where it ubiquitinates multiple OMM targets. The mechanism by which this occurs is currently unclear, but ubiquitinated mitochondria are segregated from the network and targeted for safe removal by mitophagy.
… iron depletion specifically triggers mitophagy in a PINK1/Parkin-independent manner
The details of Parkin recruitment remain to be elucidated, but one particular aspect of the pathway has been the subject of much debate; the localization and functional relevance of PINK1 isoforms. Full-length PINK1 is approximately 63 kDa (PINK163); its import into mitochondria leads to processing by several proteases, including the inner mitochondrial membrane (IMM) protease PARL 3, which generates a short PINK1 isoform of approximately 52 kDa (PINK152). PINK163 is localized to mitochondria, consistent with the clear mitochondrial targeting sequence, however, PINK152 localization is more dynamic. PARL-mediated cleavage severs PINK1's transmembrane domain anchor, enabling its re-distribution to other cellular compartments and the possibility for extra-mitochondrial functions.
Early studies considered PINK152 as ‘mature’ PINK1, thought to be the major mediator of PINK1 functionality. Supporting this view was evidence that cytosolic PINK152 was protective against mitochondrial stressors 4, and may perform a distinct role from mitochondrial PINK1 5. However, subsequent data argued that OMM stabilization of PINK163, and not PINK152, is required for Parkin recruitment, E3-ligase activation and mitophagy 6 7. In addition, PINK152 is very short-lived in mammalian cells, being rapidly degraded by the proteasome. In fact, cleavage by PARL exposes an N-terminal phenylalanine residue, promoting N-end rule proteasomal degradation 8. Thus, PINK152 has recently been considered a non-functional intermediate.
Challenging this view, a study in this issue of EMBO reports by Przedborski and colleagues suggests that PINK152 has a direct role in regulating Parkin activity 1. The authors re-assess the subcellular distribution of PINK1 isoforms using multiple approaches. Under basal conditions, both PINK1 isoforms seem to reside on the OMM, with PINK152 more loosely associated than PINK163. However, PINK152 spontaneously exits mitochondria and an N-truncated form (PINK1Δ1–103, representing cytoplasmic PINK152) was found to physically interact with the Parkin RING1 domain. Cytoplasmic PINK152 was also shown to inhibit Parkin translocation. Specifically, the authors show that promoting cytosolic PINK152 accumulation through proteasome inhibition prior to valinomycin treatment significantly decreases Parkin translocation and mitophagy (Fig1). A similar effect is observed upon PINK1Δ1–103 overexpression, suggesting that PINK152 confers a dominant-negative effect (Fig1).
Figure 1. Effects of PINK1 cleavage and low iron on mitophagy.
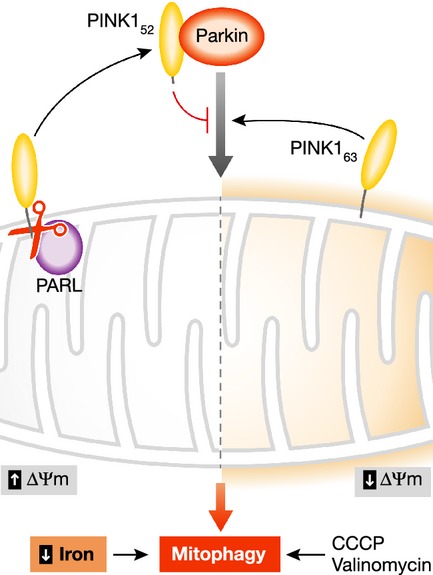
In ‘healthy’ mitochondria with high membrane potential (ΔΨm), PINK1 is imported cleaved by PARL and other proteases, and processed PINK152 released into the cytosol. Upon mitochondrial damage and loss of ΔΨm, induced in vitro by CCCP or valinomycin, full-length PINK163 is stabilized on the outer surface and stimulates the recruitment of Parkin, leading to mitophagy. Przedborski and colleagues present evidence that cytoplasmic PINK152 can inhibit Parkin recruitment. A chemical screen by Ganley and colleagues found that depletion of cellular iron can trigger mitophagy in a new mechanism that does not require PINK1 or Parkin, or the loss of ΔΨm.
Hence, Przedborski and colleagues propose a novel function for cytosolic PINK152 in negatively regulating the PINK1/Parkin-mitophagy pathway. Although the main claim certainly warrants independent verification, the approach used does not perfectly recapitulate the physiological situation. For instance, PINK1Δ1–103 would not be subject to N-end rule degradation, increasing its stability, and potentially overestimating the influence of PINK52 under physiological conditions. Similarly, stabilizing endogenous PINK152 with proteasome inhibitors will of course have non-specific effects, so such experiments should be interpreted with caution. Curiously, the authors find that under basal conditions PINK1Δ1–103 actually promotes Parkin translocation and mitophagy, which is in conflict with the proposed negative regulation of Parkin by PINK152, so further work is required to resolve these details. Nevertheless, the current data suggest that PINK152 may have a distinct cellular function to PINK163 in the cytoplasm.
Not only are additional mechanisms that regulate PINK1/Parkin-mediated mitophagy being identified, but also alternative pathways that influence mitophagy that seem not to rely on these molecules. In the December issue of EMBO reports, Ganley and colleagues reported that iron depletion specifically triggers mitophagy in a PINK1/Parkin independent manner [2; Fig1]. Not only does this study indicate that mitochondrial turnover responds to intracellular iron levels, it presents a novel mechanism through which mitophagy can be initiated. If defective mitophagy is the key underlying defect in PINK1/Parkin-related PD, stimulating mitophagy via iron depletion may offer a therapeutic approach.
Allen et al 2 performed a screen for chemical inducers of mitophagy using an elegant mitophagy sensor system consisting of an OMM-bound, tandem GFP-mCherry. Upon lysosomal localization of mitochondria, GFP is quenched whilst the mCherry signal persists, providing a simple but effective read-out of mitophagy. In this screen, in addition to known inducers of mitophagy—such as CCCP and valinomycin—the iron chelator Deferiprone (DFP) robustly induced mitophagy. An increase in transferrin receptor levels and a rescue by the addition of exogenous iron support the notion that iron depletion stimulates mitophagy.
One intriguing feature of DFP-induced mitophagy is that affected organelles maintain their ΔΨm, in contrast to the PINK1/Parkin-mediated mitophagy discussed above, which requires the dissipation of ΔΨm. Membrane potential is generally regarded as a read-out of mitochondrial health, therefore, DFP-treated cells would appear to have a healthy mitochondrial network and, indeed, ATP-levels remain stable. However, the cells had switched from oxidative phosphorylation to glycolysis, previously suggested to be a permissive event in the induction of mitophagy 9.
The maintenance of ΔΨm in DFP-induced mitophagy is immediately at odds with the model for the PINK1/Parkin pathway, suggesting this is triggered by a separate mechanism. Ganley and colleagues show that DFP-induced mitophagy does not lead to PINK163 stabilization on the OMM, and occurs in cells lacking PINK1 or Parkin. These results firmly uncouple this mechanism from the PINK1/Parkin pathway. However, notably DFP still potently induces mitophagy in these cells, supporting the idea that this may provide a therapeutic angle.
These findings have broader implications in the context of PD, as dysregulated iron metabolism is associated with PD and other neurodegenerative diseases 10, through its involvement in generating oxidative radicals and in the biosynthesis of iron-sulfur clusters and heme in mitochondria. Here Ganley and colleagues suggest another way in which iron dysregulation may impact on neuroprotective mechanisms.
Taken together, these two studies provide new insights in our understanding of mitophagy and its induction. PINK1/Parkin-mediated mitophagy is clearly a complex pathway and much still needs to be resolved, particularly in physiological settings. It is encouraging that mitophagy can be pharmacologically stimulated, although the physiological impact of this will require careful consideration.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Fedorowicz MA, et al. EMBO Rep. 2013;15:86–93. doi: 10.1002/embr.201337294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen GF, et al. EMBO Rep. 2013;14:1127–1135. doi: 10.1038/embor.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas E, et al. Hum Mol Genet. 2011;20:867–879. doi: 10.1093/hmg/ddq526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque ME, et al. Proc Natl Acad Sci USA. 2008;105:1716–1721. doi: 10.1073/pnas.0705363105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, et al. J Neurochem. 2013 doi: 10.1111/jnc.12494. [Google Scholar]
- Matsuda N, et al. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, et al. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K. Youle RJ. Autophagy. 2013;9:1758–1769. doi: 10.4161/auto.24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laar VS, et al. Hum Mol Genet. 2011;20:927–940. doi: 10.1093/hmg/ddq531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadzhieva M, Kirches E, Mawrin C. Neuropathol Appl Neurobiol. 2013 doi: 10.1111/nan.12096. doi: 10.1111/nan.12096. [DOI] [PubMed] [Google Scholar]