

Abstract
PINK1 is a mitochondrial kinase proposed to have a role in the pathogenesis of Parkinson's disease through the regulation of mitophagy. Here, we show that the PINK1 main cleavage product, PINK152, after being generated inside mitochondria, can exit these organelles and localize to the cytosol, where it is not only destined for degradation by the proteasome but binds to Parkin. The interaction of cytosolic PINK1 with Parkin represses Parkin translocation to the mitochondria and subsequent mitophagy. Our work therefore highlights the existence of two cellular pools of PINK1 that have different effects on Parkin translocation and mitophagy.
Keywords: mitochondria, mitophagy, Parkin, Parkinson's disease, PINK1
Introduction
Parkinson's disease (PD) is the second most prevalent neurodegenerative disorder 1. The discovery of mutations in the genes encoding the PTEN-induced putative kinase-1 (PINK1) and Parkin, which are linked to rare familial forms of PD, has led to the hypothesis that a defect in mitochondrial quality control may contribute to PD 2. Upon reduction of the mitochondrial membrane potential (ΔΨm) by chemicals such as Carbonyl cyanide m-chlorophenyl hydrazone (CCCP) or valinomycin, cytosolic Parkin translocates to the mitochondria 3 in a PINK1-dependent manner 4 5 6 7 8 9. Once the mitochondria are decorated with Parkin, they cluster and migrate toward the perinuclear area of the cell where they co-localize with autophagy/lysosomal markers 4 5. Eventually, these mitochondria disappear, leaving cells, such as HeLa, alive but devoid of mitochondria 3. Yet, if instead of expressing wild-type Parkin and PINK1, cells express PD pathogenic forms of Parkin and/or PINK1, Parkin translocation to the mitochondria and ensuing mitophagy are no longer observed, even if mitochondria have a low ΔΨm 4 5 6 7 8 9. These findings support the notions that both Parkin and PINK1 contribute to the normal turnover of mitochondria 10 11 and that PD mutations, by affecting this quality control mechanism, ultimately cause neurodegeneration 2.
Although PINK1 appears necessary for the recruitment of Parkin, its subcellular distribution and turnover remain debated. Together with our previous findings 12 and those of others 13 14 15 16, we show here that, following the processing of full length PINK1 (PINK163) inside the mitochondria, cleaved PINK1 (PINK152) accumulates at the mitochondrial outer membrane (MOM) to ultimately end up in the cytosol. We also show that once in the cytosol PINK152 represses the translocation of Parkin to the mitochondria and the ensuing mitophagy by physically binding to cytosolic Parkin. Ultimately, cleaved PINK1 is degraded by the proteasome. We believe that our data provide further details about the life cycle of PINK1, which should be taken into consideration if PINK1 becomes a therapeutic target 17. In addition, this study further supports the notion that PINK1 may be acting as a non-canonical mitochondrial protein, like fumarase and aconitase 18, and thus has a dual subcellular localization and a dual function.
Results and Discussion
Protease resistance of cleaved PINK1 is not due to its sheltering inside the mitochondria
We sought to revisit the question of the sub-mitochondrial localization of the PINK1 main cleavage product, PINK152, since debates about where PINK152 resides within the mitochondria have re-emerged 13 14 19 20 21 22. Adding to the debate surrounding PINK152 topology is the uncertainty about whether this cleaved fragment is the mature form of PINK1 endowed with functional roles or is merely a byproduct destined to be degraded 16 21 22 23.
To address these questions, we exposed crude mitochondrial preparations from HeLa cells transiently transfected with HA-tagged human PINK1 to increasing concentrations of Proteinase K (PK) with and without detergent as described previously 12 20. In intact mitochondria (no detergent), Western blot analysis revealed that PINK152 was more resistant to proteolysis than PINK163 (supplementary Fig S1A), a finding that is consistent with that of Jin et al 21. After permeabilization of membranes, evidenced by the PK digestion of SMAC/Diablo, the differential susceptibility of PINK152 versus PINK163 persisted (supplementary Fig S1A). Greater resistance of PINK152 over PINK163 was also noted previously in permeabilized mitochondria isolated from HeLa cells overexpressing PINK1 when exposed to trypsin 12, hence excluding the possibility that the greater resistance of PINK152 was restricted to PK only. Yet, the PK assay performed on purified mitochondria from untransfected HeLa cells that were subjected to an in vitro import assay 12, showed that PINK163 and PINK152 were equally susceptible to proteolysis (supplementary Fig S1B), irrespective of the membrane permeabilization status. These results indicate that the reported differential sensitivity to PK between PINK163 and PINK152 21, which is most detectable at high PINK1 expression levels, does not reflect differential submitochondrial localization, but rather an intrinsic lower susceptibility of PINK152 to proteolysis.
Mitochondrial PINK52 is loosely attached to the MOM and translocates to the cytosol
Several studies, including our own 4 13 16 20, have shown that proteasome inhibitors, such as MG132 or epoxomicin, lead to the accumulation of PINK152, as evidenced on Western blots of whole cell lysates. Here, we transiently transfected HeLa cells with HA-tagged human PINK1 and treated them with either MG132, valinomycin or both for 8 h. Incubation with MG132 was associated with a marked increase in PINK152, not only in the crude mitochondrial fraction but also in the cytosolic fraction (Fig1A), confirming that PINK152 is present in both cellular compartments 12 13 14 15 16. Conversely, incubation with valinomycin was associated with a decrease in PINK152 in both subcellular fractions while there was an increase in PINK163 in the mitochondrial fractions (Fig1A). Finally, the combination of valinomycin and MG132 decreased the content of PINK152 in both subcellular fractions compared to MG132 alone (Fig1A). PINK1 signal quantification is provided in supplementary Fig S2A. These results are consistent with the notion that PINK152 is generated within mitochondria and that collapsing ΔΨm hinders PINK1 cleavage.
Figure 1. Subcellular localization of full-length and cleaved PINK1.
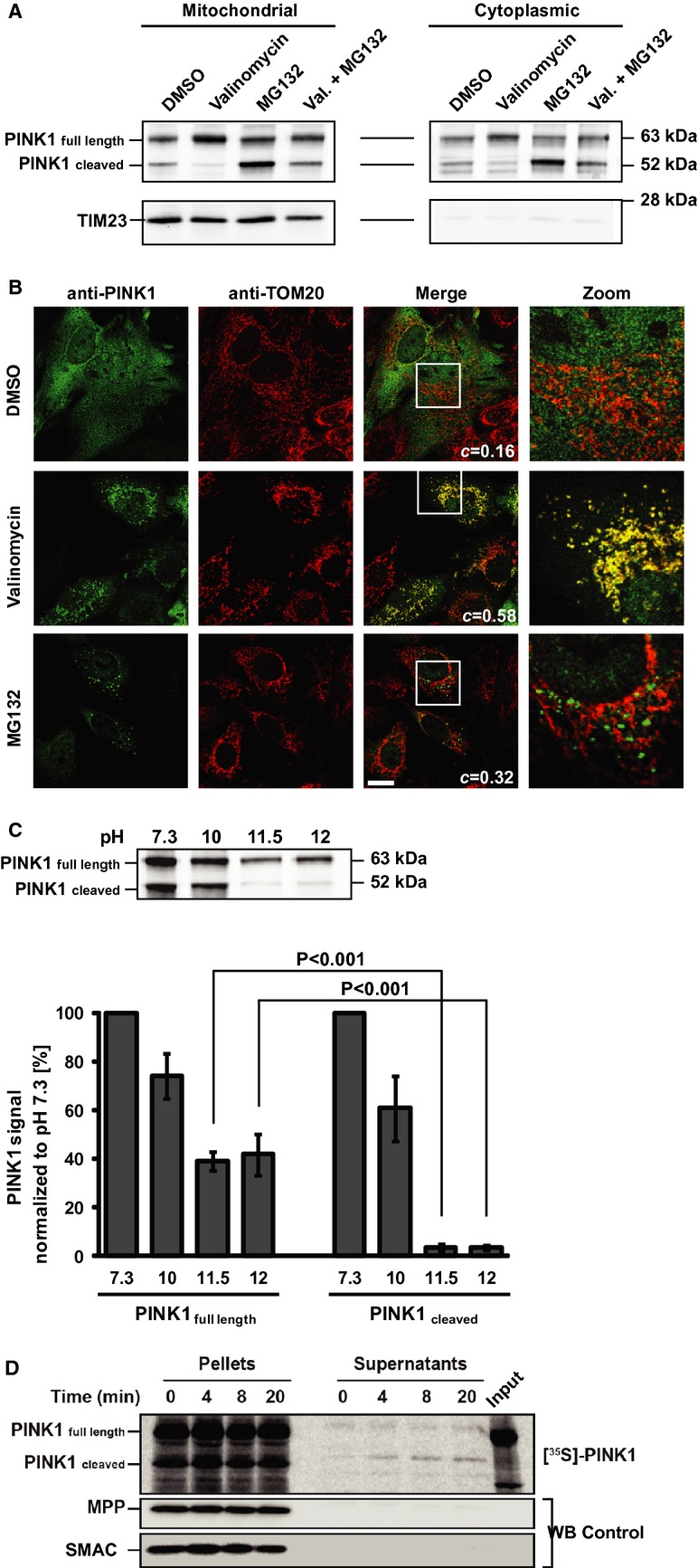
- PINK1 immunoblot of cytoplasmic and mitochondrial fractions from HeLa cells transfected with PINK1 after 8 h exposure to either DMSO, valinomycin, MG132 or both. TIM23 = mitochondrial marker.
- Representative images of PINK1-transfected HeLa cells after 2 h valinomycin or MG132. TOM20 = mitochondrial marker. c = co-localization coefficient. Scale bar = 20 μm.
- Alkaline extraction of PINK1 from isolated mitochondria. Upper panel: Representative immunoblot. Lower panel: Quantification of PINK1 in the particulate fraction (not extractable portion) at varying pH. There is a significant interaction between PINK1 fragments and pH (2W-ANOVA, F3,16 = 253.11, P < 0.001). Newman–Keuls test indicates that PINK1 full length is significantly less extractable than PINK1 cleaved at both pH 11.5 and 12. Values are means ± s.e.m. of three independent experiments.
- Autoradiogram of in vitro export assay of [35S]-labeled PINK1 in mitochondria and supernatants (see Materials and Methods). Immunoblots for mitochondrial markers MPP and SMAC.
As illustrated by immunocytochemistry (Fig1B), under basal conditions (DMSO control), there was marginal co-localization between PINK1 and the mitochondrial marker TOM20 (co-localization coefficient c = 0.16). In contrast, after 2 h exposure to valinomycin, there was a greater co-localization between PINK1 and TOM20 (c = 0.58), with PINK1 immunoreactivity displaying a prominent punctate distribution. Following the incubation of cells with MG132, we saw cytosolic PINK1+ aggregates, many of which were TOM20−. Consequently, the mitochondrial co-localization was limited (c = 0.32), again suggesting that a significant portion of PINK152 is localized in the cytosol.
Once PINK163 or at least its N-terminal part is imported into mitochondria, it undergoes two sequential proteolytic processing steps which, according to Greene et al 24, are mediated first by matrix processing peptidase (MPP) and then by presenilin-associated rhomboid-like protease (PARL) and the m-AAA protease. Previously, we have estimated that the second cleavage should be between amino acids 91 and 104 20, a prediction confirmed by Deas et al 25 who showed that PINK1 is indeed cleaved within the mitochondria by PARL between A103 and F104.
Given that PINK152 is generated by cleavage within the transmembrane (TM) domain, we hypothesized that it may be less firmly integrated into the MOM. We thus subjected mitochondria isolated from HeLa cells which transiently express HA-tagged PINK1 to alkaline extraction and assessed the relative amounts of PINK163 and PINK152 (Fig1C). We found that approximately 40% of PINK163 remained in the particulate fraction, even at pH 12, whereas < 4% of PINK152 did so. Comparable results were obtained for the alkaline extraction assay on isolated mitochondria after in vitro import of radiolabeled [35S]-PINK1 (supplementary Fig S2B). This suggests that PINK163 is more strongly integrated into the MOM than PINK152.
To determine if PINK152 can exit mitochondria, we performed an in vitro export experiment with radiolabeled PINK1 and intact mitochondria from HeLa cells. This experiment revealed the accumulation of a signal for PINK152 in the supernatants (Fig1D) indicating that at least some PINK152 does exit the mitochondria spontaneously.
PINK152 binds Parkin
Although cleaved PINK1 is degraded by the proteasome, we doubt that the sole reason for mitochondrial PINK152 to enter the cytosol is to be disposed of. Instead, we predict that cytosolic PINK152 plays a role in the cytosol. Germane to this possibility, we wondered whether cytosolic PINK152 might bind to cytosolic Parkin, and in doing so, whether it might prevent Parkin translocation to mitochondria and ensuing mitophagy. To test this question, we first performed co-immunoprecipitation experiments of whole cell extracts and confirmed that PINK1 physically interacts with Parkin 5 26 27 (Fig2A). Since specific domains of Parkin are necessary to allow cytosolic Parkin to translocate to mitochondria 27 28, we next sought to determine the PINK1 and Parkin binding domains. Accordingly, immunoprecipitation experiments with several truncated forms of PINK1 carrying a C-terminal Flag-tag were performed (Fig2A). In transfected HEK293T cells, endogenous Parkin was predominantly pulled-down by two PINK1 fragments (Fig2A, lower panel), both containing the middle section of the kinase domain (aa 156–507 and aa 310–428) and, to a lesser extent, by the truncation encompassing the first part of the kinase domain (aa 156–309). The N-terminal part of the protein that encompasses the mitochondrial targeting sequence and the TM (aa 1–155) and the C-terminal part of PINK1 (aa 429–581) did not bind to endogenous Parkin. The kinase domain of PINK1 is thus essential for the physical interaction between PINK1 and Parkin. We then used the PINK1 kinase domain fragment (aa 156–507) to determine the PINK1 binding domain of Parkin. Co-immunoprecipiation was performed in HEK293T cells co-transfected with PINK1156-507-Flag and various truncated forms of Parkin carrying an N-terminal Myc-tag (Fig2B). Although not all our Parkin fragments were expressed and/or successfully pulled-down, the results revealed that only the products comprising the R1 domain of Parkin did bind to the PINK1 kinase domain (Fig2B). To confirm that cleaved PINK1 is indeed able to bind Parkin, we transfected HeLa cells stably expressing YFP-Parkin with PINKΔ1-103, mimicking PINK152. This co-immunorecipitation experiment demonstrates (Fig2C) that PINKΔ1–103 effectively binds YFP-Parkin.
Figure 2. Identification of the PINK1-Parkin binding sites.
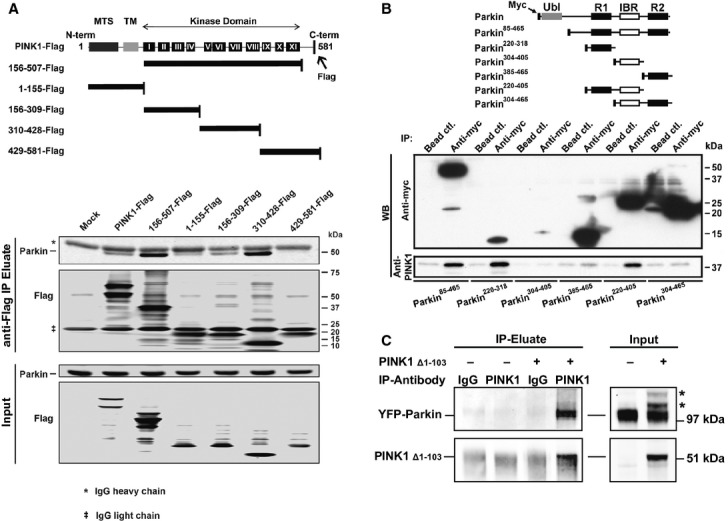
- Co-immunoprecipitation assay on lysates of HEK293T cells transfected with C-terminal Flag-tagged human PINK1 or PINK1 fragment constructs (schematic) with an anti-Flag antibody, followed by immunoblotting for Flag and Parkin.
- Co-immunoprecipitation assay on lysates of HEK293T cells co-transfected with PINK1 kinase domain (PINK1156–507-HA) and selected myc-tagged Parkin domain constructs (schematic).
- PINK1Δ1–103 (used to mimic cleaved PINK1) binds to YFP-Parkin. Co-immunoprecipitation assay on YFP-Parkin HeLa cells transiently transfected or not with PINK1Δ1–103. Rabbit IgG = nonspecific binding control; *Parkin high-molecular weight species.
Cytosolic cleaved PINK1 places a break on Parkin translocation and mitophagy
Next, we wondered whether such protein-protein interactions that were to take place in the cytosol, could interfere with Parkin translocation. To test this idea, we first pretreated YFP-Parkin HeLa cells with MG132 for 5 h, to increase endogenous levels of PINK152 in the cytosol. We then induced Parkin translocation by dissipating ΔΨm with valinomycin for 2 h in the presence of MG132. After fixation, cells were immunostained for PINK1 and for the mitochondrial marker TOM20. As shown in Fig3A, MG132 treatment significantly impaired Parkin translocation to mitochondria. Using the same pretreatment conditions but extending the time of the exposure to valinomycin/MG132 to 19 h, we also found that the proportions of cells with either reduced or abolished TOM20+ mitochondrial network, used as a surrogate of mitophagy, were significantly less than control (Fig3B).
Figure 3. Cleaved PINK1 attenuates Parkin translocation and mitophagy.
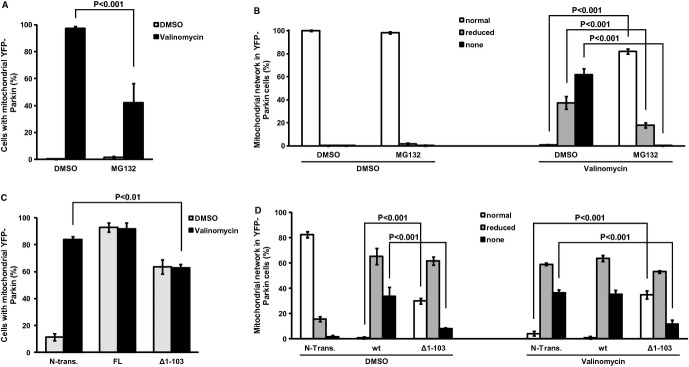
- A, B YFP-Parkin HeLa cells were pretreated with MG132 (5 h) to increase endogenous cleaved PINK1 and then exposed to valinomycin/MG132 for: (A) Parkin translocation (2 h); (B) mitophagy (19 h). Mitophagy was inferred by the disappearance of the mitochondrial network (see representative images in supplementary Fig S3). In (A), there is a significant interaction between membrane potential and proteasome inhibition (2W-ANOVA,F[1,8] = 16.12, P < 0.001). Newman–Keuls test indicates that the number of cells with mitochondrial YFP-Parkin is significantly higher in the DMSO/valinomycin than in the MG132/valinomycin group. In (B), there is a significant interaction among membrane potential, proteasome inhibition, and mitochondrial network (3W-ANOVA,F[2,24] = 239.01, P < 0.001). Newman–Keuls test indicates that the number of cells with normal mitochondrial network is significantly lower in the DMSO/valinomycin than in the MG132/valinomycin group. It also shows that the number of cells with reduced mitochondrial network or no mitochondrial network is significantly higher in the DMSO/valinomycin than in the MG132/valinomycin group.
- C, D YFP-Parkin HeLa cells were either transfected with full-length (FL), cleaved mimicking (Δ1–103) PINK1 or not transfected (N-Trans.) and then exposed to valinomycin (C: Parkin translocation [1.5 h]; D: mitophagy [16 h]). In (C), there is a significant interaction between membrane potential and PINK1 fragment (2W-ANOVA,F[2,12] = 68.4, P < 0.001). Newman–Keuls test indicates that the number of cells with mitochondrial YFP-Parkin is significantly higher in N-trans cells than cells expressing PINK1 Δ1–103 after valinomycin treatment. In (D), there is a significant interaction among membrane potential, PINK1 fragment, and mitochondrial network (3W-ANOVA,F[4,36] = 96.01, P < 0.001). Newman–Keuls test indicates that, after valinomycin treatment, the number of cells without mitochondrial network is significantly higher in N-Trans. cells than in cells expressing PINK1 Δ1–103.
Data information: All values are means ± s.e.m. of 3 independent experiments.
Since MG132 alters the cellular content of a variety of proteins, we next sought to confirm the relationship between cytosolic PINK152 and Parkin translocation and the ensuing mitophagy by overexpressing PINKΔ1–103. Consistent with the valinomycin/MG132 data (Fig3A and 3B), we found that the proportions of YFP-Parkin HeLa cells overexpressing PINKΔ1–103 that displayed Parkin translocation or an abolished TOM20+ mitochondrial network after valinomycin exposure were significantly less than controls (Fig3C and 3D). Since all cells express endogenous levels of PINK1, the attenuation of Parkin translocation and of mitochondrial disappearance in PINK1Δ1–103-expressing cells (Fig3C and 3D) suggests that cytosolic PINK152 exerts a dominant negative effect on Parkin recruitment and subsequent mitophagy. However, overexpression of PINK1Δ1–103, even in absence of valinomycin exposure, promotes some Parkin translocation and mitophagy (Fig3C and 3D). This apparent paradoxical finding perhaps stems from the fact that a fraction of cleaved PINK1 still associates with the mitochondria, as we have previously shown 20. We believe that in doing so, mitochondrial PINK1Δ1–103, which retains its kinase function, might be able to stimulate Parkin translocation and mitophagy. However, the rest of overexpressed PINK1Δ1–103 is cytosolic and thus could interfere with most, but not all, Parkin molecules. Consequently, we hypothesize that, even under the stimulating effect of either high mitochondrial PINK1 levels or valinomycin, only some Parkin molecules can translocate and induce mitophagy. Although more work is required to fully examine this idea, it is consistent with the type of response we observed (Fig3C and 3D).
Conclusions
The findings presented here, together with those available in the current literature, suggest the following model of PINK1 function, topology and turnover (Fig4). According to this model, PINK163 or at least its N-terminal part is imported into mitochondria by a TOM20-dependent mechanism. PINK1 is then processed sequentially by MPP and PARL/m-AAA. PINK152 then gains access to the cytosol and once there, it attenuates cytosolic Parkin translocation and ensuing mitophagy by binding to Parkin. Yet, overexpressed Parkin, does not overcome the inhibition by endogenous PINK152 through a mass effect, and does not cause mitophagy (Fig3). This is thus consistent with the finding that in addition to mitochondrial translocation, Parkin has to be activated 29 to induce mitophagy. Eventually, PINK152 is rapidly degraded by the proteasome to ensure a fast turnover and a fast response of the system.
Figure 4. Model of the PINK1 cycle.
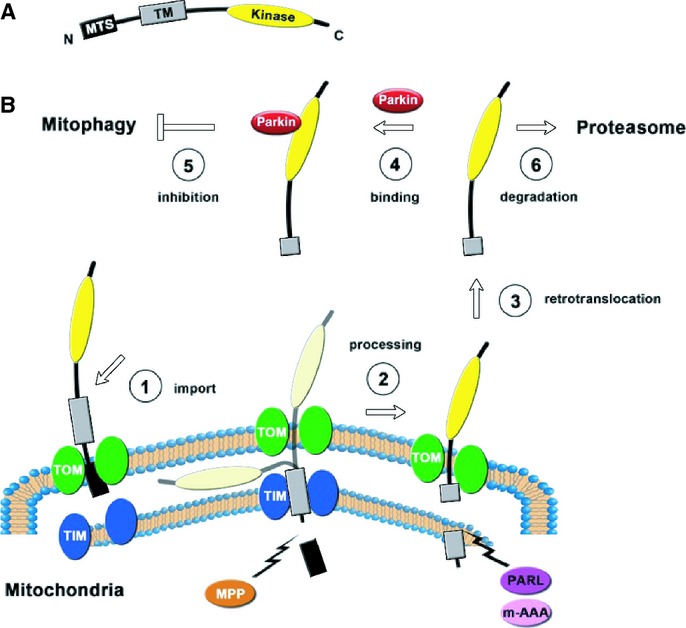
- Schematic representation of PINK1. MTS: mitochondrial targeting sequence; TM: transmembrane domain; Kinase: kinase domain.
- Proposed life cycle of PINK1.
If mitochondria are damaged and exhibit a loss of ΔΨm, no import of PINK1 occurs and thus all of the PINK163 remains at the surface of the mitochondria. As PINK152 is degraded and no-longer replaced, its cytosolic content decreases, hence reducing the repressive action of PINK1 on cytosolic Parkin, and eventually allowing Parkin to translocate to the mitochondria and to contribute to mitophagy. Since the loss of PINK1 repression on Parkin translocation will rely on PINK1 proteasomal degradation, a time-lag between the loss of ΔΨm and the translocation of Parkin is expected, which is what is experimentally observed. Upon exposure to CCCP the loss of ΔΨm, evidenced with fluorescent probes such as tetramethyl rhodamine methyl ester, is almost instantaneous while overt Parkin translocation is only noticeable after approximately 30 min.
Finally, it has been reported that there is an increased expression of PINK152 protein in PD brains 29. If our model is correct, this suggests that in PD there may be a deficit in mitophagy by virtue of the fact that cleaved PINK1 may prevent Parkin translocation, hence hampering the elimination of damaged mitochondria.
Materials and Methods
Export assay
Isolation of mitochondria and in vitro import of radiolabeled PINK1 was described before 12. Following 25 min of import at 37°C mitochondria were re-isolated by centrifugation (12 min, 12 000 × g, 4°C), re-suspended in fresh mitochondria isolation buffer and incubated at 37°C. After the indicated times, an aliquot was taken and separated into mitochondrial pellet and supernatant by centrifugation (12 min, 12 000 × g, 4°C). Samples were analyzed by SDS-PAGE and digital autoradiography.
Alkaline extraction
Crude mitochondria isolated from HeLa cells transfected with HA-tagged wild-type PINK1 were re-suspended in 1 mL of 0.1 mM Na2CO3 at indicated pH. After 30 min incubation on ice, samples were centrifuged (1 h, 100 000 × g, 4°C). The not extractable portion of PINK1 in the pellet was analysed by SDS-PAGE and Western blotting.
Co-Immunoprecipitation
Flag-IP: HEK293T cells were transfected with PINK1-Flag or Flag-tagged PINK1 fragments. After 24 h, cells were harvested and resuspended in lysis buffer (50 mM Tris pH 7.4, 50 mM NaCl, 1 mM EDTA, 0.1% Triton X-100) supplemented with 2 × protease inhibitor (Roche Diagnostics Corporation, Indianapolis, IN, USA) and agitated (4°C, 1 h). Homogenates were centrifuged (10 min, 11 000 × g) and lysate supernatants were collected. Lysates were then incubated with prewashed anti-FLAG M2 antibody affinity gel (A2220; Sigma, St. Louis, MO, USA) overnight (4°C) with constant agitation, followed by washes with lysis buffer. The resins that captured PINK1 were eluted in 2 × SDS sample buffer. Myc-IP: Cells were lysed in buffer containing 1% Triton X-100, 150 mM NaCl, 20 mM Tris (pH 7.5), 0.5 mM PMSF, 0.5 mM EDTA, and 0.5% protease inhibitor cocktail (Sigma). After centrifugation at 14 000 × g (10 min), the supernatants were incubated with anti-myc (9E10) antibody-conjugated CNBr-activated Sepharose 4B (Pharmacia Biotech, Piscataway, NJ, USA) or control beads. PINK1-IP: HeLa cells stably expressing YFP-Parkin transiently transfected with PINK1Δ1-103 or not transfected as a control were lysed in 20 mM HEPES, 100 mM NaCl, 1 mM EDTA pH 7.4 and 1% Triton X-100 in the presence of protease inhibitors (Roche). After sonication and incubation (4°C, 30 min) cell lysates were obtained by centrifugation (20 min, 15 000 × g, 4°C). 800 μg lysate protein was used for IP with the Dynabeads ® Protein G Immunoprecipitation kit from Invitrogen (Carlsbad, CA, USA) according to manufacturer's protocol. The anti-PINK1 antibody was used at 1:25 dilution. Normal rabbit IgG (Santa Cruz Biotechnology, Inc., Dallas, TX, USA, sc-2027) was used as a negative control.
Statistical analysis
Difference among means was analyzed by 2-or 3-way ANOVA followed by a Newman–Keuls post-hoc test. Cell counts were generated for ≥ 100 cells per condition/construct.
Acknowledgments
We would like to thank Dr. Sudarshan Phani for his assistance in creating Fig4 and Eric A. Schon for his comments on the manuscript. The YFP-Parkin expressing HeLa cells were kindly provided by Richard J. Youle (NINDS, Bethesda, MD 20892, USA). MAF is supported by a grant from the Deutsche Forschungsgemeinschaft (Grant 805231). SP is supported by grants from the Parkinson's Disease Foundation, the National Institutes of Health (NS38370-12; ES017384; NS070276), US Department of Defense (W81WXWH-08-1-0465 and W81XWH-10-1-053) and the Thomas Hartman Foundation. SP is the Page and William Black Professor of Neurology. DMAW was supported by an award from the NIH (2 TL1 RR 24158-6).
Author contributions
MAF, RdVS, CR, DB, YH, CZ, DMAW, WV and SP designed research; MAF, RdVS, CR, YH, CZ, and YL performed research; MAF, RdVS, CR, DB, YH, CZ, WV, YL and SP analyzed data; MAF, RdVS, CR, YH, CZ, and SP wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://embor.embopress.org
Supplementary Figure S1-S3
Review Process File
References
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RLA, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrané J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Kawajiri S, Saiki S, Sato S, Sato F, Hatano T, Eguchi H, Hattori N. PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett. 2010;584:1073–1079. doi: 10.1016/j.febslet.2010.02.016. [DOI] [PubMed] [Google Scholar]
- Rakovic A, Grunewald A, Seibler P, Ramirez A, Kock N, Orolicki S, Lohmann K, Klein C. Effect of endogenous mutant and wild-type PINK1 on Parkin in fibroblasts from Parkinson disease patients. Hum Mol Genet. 2010;19:3124–3137. doi: 10.1093/hmg/ddq215. [DOI] [PubMed] [Google Scholar]
- Ziviani E, Tao RN, Whitworth AJ. Drosophila Parkin requires PINK1 for mitochondrial translocation and ubiquitinates Mitofusin. Proc Natl Acad Sci USA. 2010;107:5018–5023. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci USA. 2010;107:11835–11840. doi: 10.1073/pnas.0914569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkerson RW, De Vries RL, Lebot P, Wikstrom JD, Torgyekes E, Shirihai OS, Przedborski S, Schon EA. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum Mol Genet. 2012;21:978–990. doi: 10.1093/hmg/ddr529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker D, Richter J, Tocilescu MA, Przedborski S, Voos W. Pink1 kinase and its membrane potential (Deltaψ)-dependent cleavage product both localize to outer mitochondrial membrane by unique targeting mode. J Biol Chem. 2012;287:22969–22987. doi: 10.1074/jbc.M112.365700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatori S, Ito G, Iwatsubo T. Cytoplasmic localization and proteasomal degradation of N-terminally cleaved form of PINK1. Neurosci Lett. 2008;430:13–17. doi: 10.1016/j.neulet.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Beilina A, van der Burg M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, Cookson MR. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci USA. 2005;102:5703–5708. doi: 10.1073/pnas.0500617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihofen A, Ostaszewski B, Minami Y, Selkoe DJ. Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum Mol Genet. 2008;17:602–616. doi: 10.1093/hmg/ddm334. [DOI] [PubMed] [Google Scholar]
- Lin W, Kang UJ. Characterization of PINK1 processing, stability, and subcellular localization. J Neurochem. 2008;106:464–474. doi: 10.1111/j.1471-4159.2008.05398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz NT, Berthet A, Sos ML, Thorn KS, Burlingame AL, Nakamura K, Shokat KM. A neo-substrate that amplifies catalytic activity of Parkinson's-disease-related kinase PINK1. Cell. 2013;154:737–747. doi: 10.1016/j.cell.2013.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev-Rudzki N, Yogev O, Pines O. The mitochondrial targeting sequence tilts the balance between mitochondrial and cytosolic dual localization. J Cell Sci. 2008;121(14):2423–2431. doi: 10.1242/jcs.029207. (Pt. [DOI] [PubMed] [Google Scholar]
- Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet. 2005;14:3477–3492. doi: 10.1093/hmg/ddi377. [DOI] [PubMed] [Google Scholar]
- Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci USA. 2008;105:12022–12027. doi: 10.1073/pnas.0802814105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque ME, Thomas KJ, D'Souza C, Callaghan S, Kitada T, Slack RS, Fraser P, Cookson MR, Tandon A, Park DS. Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc Natl Acad Sci USA. 2008;105:1716–1721. doi: 10.1073/pnas.0705363105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata H, Sakaguchi M, Jin Y, Sakaguchi Y, Futami J, Yamada H, Kataoka K, Huh NH. A new cytosolic pathway from a Parkinson disease-associated kinase, BRPK/PINK1: activation of AKT via mTORC2. J Biol Chem. 2011;286:7182–7189. doi: 10.1074/jbc.M110.179390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, Abramov AY, Wood NW. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet. 2011;20:867–879. doi: 10.1093/hmg/ddq526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha D, Chin LS, Li L. Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Hum Mol Genet. 2010;19:352–363. doi: 10.1093/hmg/ddp501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba K, Arai T, Sato S, Kubo S, Ohba Y, Mizuno Y, Hattori N. Parkin stabilizes PINK1 through direct interaction. Biochem Biophys Res Commun. 2009;383:331–335. doi: 10.1016/j.bbrc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, Kitada T, Kim JM, Chung J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377:975–980. doi: 10.1016/j.bbrc.2008.10.104. [DOI] [PubMed] [Google Scholar]
- Muqit MM, Abou-Sleiman PM, Saurin AT, Harvey K, Gandhi S, Deas E, Eaton S, Payne Smith MD, Venner K, Matilla A, Healy DG, Gilks WP, Lees AJ, Holton J, Revesz T, Parker PJ, Harvey RJ, Wood NW, Latchman DS. Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J Neurochem. 2006;98:156–169. doi: 10.1111/j.1471-4159.2006.03845.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1-S3
Review Process File