

Abstract
Defective autophagy is implicated in the pathogenesis of nonalcoholic fatty liver diseases (NAFLD) through poorly defined mechanisms. Cardiolipin is a mitochondrial phospholipid required for bioenergetics and mitophagy from yeast to mammals. Here, we investigated a role for ALCAT1 in the development of NAFLD. ALCAT1 is a lysocardiolipin acyltransferase that catalyzes pathological cardiolipin remodeling in several aging-related diseases. We show that the onset of diet-induced NAFLD caused autophagic arrest in hepatocytes, leading to oxidative stress, mitochondrial dysfunction, and insulin resistance. In contrast, targeted deletion of ALCAT1 in mice prevented the onset of NAFLD. ALCAT1 deficiency also restored mitophagy, mitochondrial architecture, mtDNA fidelity, and oxidative phosphorylation. In support for a causative role of the enzyme in mitochondrial etiology of the disease, hepatic ALCAT1 expression was significantly up-regulated in mouse models of NAFLD. Accordingly, forced expression of ALCAT1 in primary hepatocytes led to multiple defects that are highly reminiscent of NAFLD, including hepatosteatosis, defective autophagy, and mitochondrial dysfunction, linking pathological cardiolipin remodeling by ALCAT1 to the pathogenesis of NAFLD.
Oxidative stress causes mitochondrial dysfunction, which is implicated in the etiology of NAFLD and its progression to nonalcoholic steatohepatitis (NASH). Although the precise molecular mechanisms remain elusive, defective autophagy contributes to the pathogenesis of NAFLD, which is supported by the phenotypes of mouse models of impaired autophagy (1). Autophagy is required for the mitochondrial quality control process by engaging in a crosstalk with reactive oxygen species (ROS) to eliminate damaged mitochondria through mitophagy (2). Hence, defective mitophagy leads to accumulation of damaged mitochondria, mitochondrial fragmentation, and oxidative stress, which are highly reminiscent of metabolic defects in patients with NASH (3). Furthermore, recent development in the field demonstrates that autophagic degradation of lipid droplets plays a key role in lipid homeostasis by releasing free fatty acids for oxidation (1). Thus, the onset of obesity, the major cause of NAFLD, is associated with severe down-regulation of hepatic autophagy, whereas restoration of hepatic autophagy enhances insulin sensitivity and glucose tolerance in the liver of obese mice (4).
Cardiolipin (CL) is a mitochondrial phospholipid required for mitochondrial bioenergetics, membrane structure, and dynamics. Recent studies suggest that CL also plays a key role in autophagy from yeast to mammals (5, 6). CL is required for autophagosome biogenesis and cargo recognition by supporting the membrane structure of autophagosomes and the activity of autophagy proteins (6, 7). Additionally, CL mediates a cross-talk between mitochondria and lysosomes, a key step in autophagic process (5). Furthermore, externalization of CL from mitochondrial inner membrane to mitochondrial surface was recently shown to act as a recognition signal that directs damaged mitochondria to mitophagy (6). Among all the phospholipids, CL is highly sensitive to oxidative damage of its double bonds by ROS due to its exclusive location in mitochondria. Hence, CL is the only phospholipid in mitochondria that undergoes early oxidation during apoptosis, and detachment of CL from cytochrome c triggers apoptosis (8). Oxidized CL itself also functions as a major source of ROS, triggering a self-destruction process known as “CL peroxidation”. Thus, CL peroxidation is a major defect associated with aging and age-related diseases including obesity, NAFLD, diabetes, cardiovascular diseases, cancer, and neurodegenerative diseases (9–11).
We recently identified a novel pathway by which oxidative stress causes mitochondrial dysfunction in metabolic diseases (9, 12, 13). This pathway is mediated by ALCAT1, an acyl-CoA dependent lysocardiolipin acyltransferase (9). ALCAT1 catalyzes the remodeling of CL with aberrant acyl composition commonly found in obesity, diabetes, and cardiovascular disease, leading to TLCL depletion and enrichment of DHA in CL (12). Additionally, ALCAT1 is localized at mitochondria-associated membrane (MAM) where autophagosome biogenesis takes place (9, 14). Overexpression of ALCAT1 causes dilation of MAM (9), suggesting a potential role of the enzyme in regulating autophagy. Using gain and loss function studies, this report demonstrated a key role of the enzyme in the etiology of NAFLD by regulating mitochondrial architecture and autophagy.
Experimental Procedures
The generation of the ALCAT1 knockout mice and measurement of oxygen consumption rate (OCR) were as previously described (9). All experiments used littermate control of matched age and sex and in accordance with approval of institutional animal care and use protocols according to NIH guidelines (NIH publication No. 86–23, 1985). Details on reagents, animal care, and experimental procedures, including RT-PCR, measurement of ROS, lipid peroxidation, EM analysis, hepatocyte isolation and culture, mtDNA mutation assay, mitochondrial dynamics, and statistical analysis were described in supplemental information.
Results
Ablation of ALCAT1 prevents the onset of diet-induced NAFLD
Using mice with targeted deletion of ALCAT1 (12), we investigated the role of ALCAT1 in the development of NAFLD associated with diet-induced obesity. Wild type (WT) control mice developed severe NAFLD after 18-weeks on high-fat diet (HFD), as evidenced by the presence of an enlarged and pale liver (Fig. 1A), hepatocyte vacuolation (Fig. 1C), steatosis (Fig. 1E), and fibrosis (Fig. 1G, highlighted by an arrow). Strikingly, these defects were completely absent in the liver of ALCAT1 knockout (ALCAT1−/−) mice (Fig. 1B, 1D, 1F, and 1H), which is further supported by decreased liver weight and hepatic triglyceride levels (Fig. 1J & 1K). In contrast, ALCAT1−/− mice was indistinguishable from WT controls in hepatic morphology when fed a regular chow (Fig. S1A–1H).
Fig. 1.
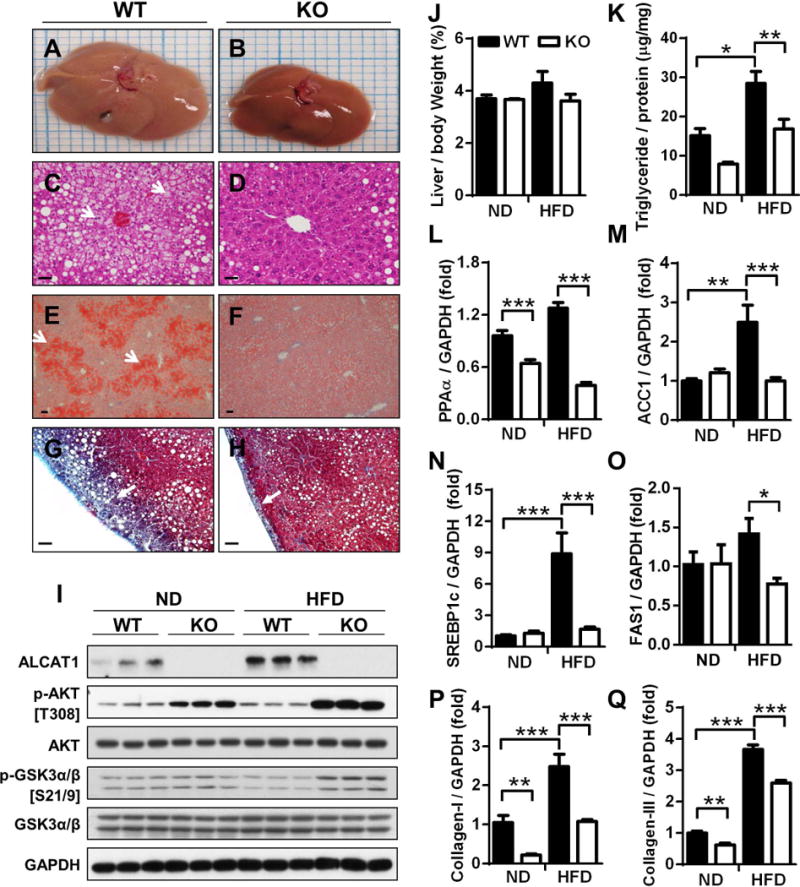
Ablation of ALCAT1 prevents NAFLD and its related metabolic complications. ALCAT1−/− mice and WT controls were fed a high-fat diet (HFD) for 18 consecutive weeks, and were analyzed for: (A–B) gross morphology; (C–D) liver sections by H&E staining; (E–F) hepatosteatosis by Oil Red staining; and (G–H) hepatic fibrosis by trichrome staining. (I) Western blot analysis of ALCAT1 expression and insulin-stimulated phosphorylation of AKT and GSK3α/β in the liver. (J–K) Analysis of liver weight and triglyceride level. (L–O), RT-PCR analysis of genes that promote lipogenesis in the liver, including PPARα, ACC1, SREBP1c and FAS1. (P–Q) RT-PCR analysis of hepatic fibrosis biomarkers, including collagen-I (P) and collagen-III (Q). Data are expressed as mean ± SEM; n = 6–8, *p < 0.05, **p < 0.01 and ***p < 0.001. Scale Bar: 50μm.
Up-regulated ALCAT1 expression is implicated in the pathogenesis of NAFLD
To assess a role for ALCAT1 in the etiology of NAFLD, we next examined whether hepatic ALCAT1 protein expression is regulated by the onset of NAFLD in mice with diet-induced obesity and in db/db mice, a mouse model of obesity, type 2 diabetes, and NAFLD. As shown in Fig 1I (quantified in Fig. S2A), ALCAT1 protein expression in the liver was dramatically up-regulated by NAFLD in WT control mice. Likewise, ALCAT1 protein expression is also stimulated by NAFLD in db/db mice (Fig. S3A, quantified in Fig. S3C), suggesting a potential role of ALCAT1 in the etiology of NAFLD. In support for a role of up-regulated ALCAT1 expression in the pathogenesis of NAFLD, ALCAT1 deficiency significantly improved insulin sensitivity, as evidenced by increased phosphorylation of AKT and GSK3α/β in response to insulin treatment (Fig. 1I, quantified in Fig. S2B and Fig. S2C). Accordingly, ALCAT1 deficiency significantly improved glucose tolerance and decreased serum insulin level (Fig. S1J and Fig. S1K).
ALCAT1 deficiency attenuates hepatic lipogenesis and fibrosis
Increased de novo lipogenesis plays an important role in the accumulation of triglyceride in NAFLD. To uncover molecular mechanisms by which ALCAT1 causes hepatosteatosis, we next analyzed the expression of genes that promote lipogenesis by RT-PCR analysis. As shown in Fig. 1L–1O, NAFLD significantly increased expression of lipogenic genes, including PPARα, ACC1, SREBP1c and FAS1 in the liver of the WT control mice. Consistent with a lack of hepatosteatosis in ALCAT1−/− mice, ALCAT1 deficiency also normalized the expression of these genes. Consistent with severe hepatofibrosis in WT control mice (Fig 1G, highlighted by arrow), NAFLD also significantly up-regulated expression of genes encoding matrix proteins, including collagen-I and collagen-III (Fig. 1P–Q), in the liver of WT control mice. These findings were further underscored by the increased presence of collagen fiber bundles in the liver of WT mice (Fig. S2D, highlighted by arrows in Fig. S2E). In contrast, these defects were prevented by ALCAT1 deficiency (Fig. 1P and 1Q).
ALCAT1 deficiency prevents mitochondrial dysfunction in the liver
Mitochondrial architecture is regulated by a family of mitochondrial GTPases, including MFN2, OPA1, and DRP1. MFN2 and OPA1 are required for the fusion of outer and inner mitochondrial membranes, respectively, whereas DRP1 is required for mitochondrial fission. MFN2 depletion is recently implicated in mitochondrial fragmentation in obesity and type 2 diabetes (15). We next questioned whether the onset of NAFLD caused mitochondrial fragmentation, and if so whether up-regulated ALCAT1 expression was implicated in the process. The results showed that NAFLD caused mitochondrial fragmentation in isolated hepatocytes (Fig. 2A, highlighted in Fig. 2C), which is likely caused by depletion of MFN2 expression (Fig. 2E, quantified in Fig. S2F) in WT control mice. In support for a causative role of ALCAT1 in mitochondrial fragmentation, ALCAT1 deficiency completely prevented mitochondrial fragmentation (Fig. 2B, highlighted in Fig. 2D) by increasing MFN2 expression in the liver (Fig. 2E, quantified in Fig. S2F). In contrast, ALCAT1 deficiency did not affect the expression of other regulators of mitochondrial architecture, including OPA1, DRP1, and VDAC1 (Fig. 2E).
Fig. 2.
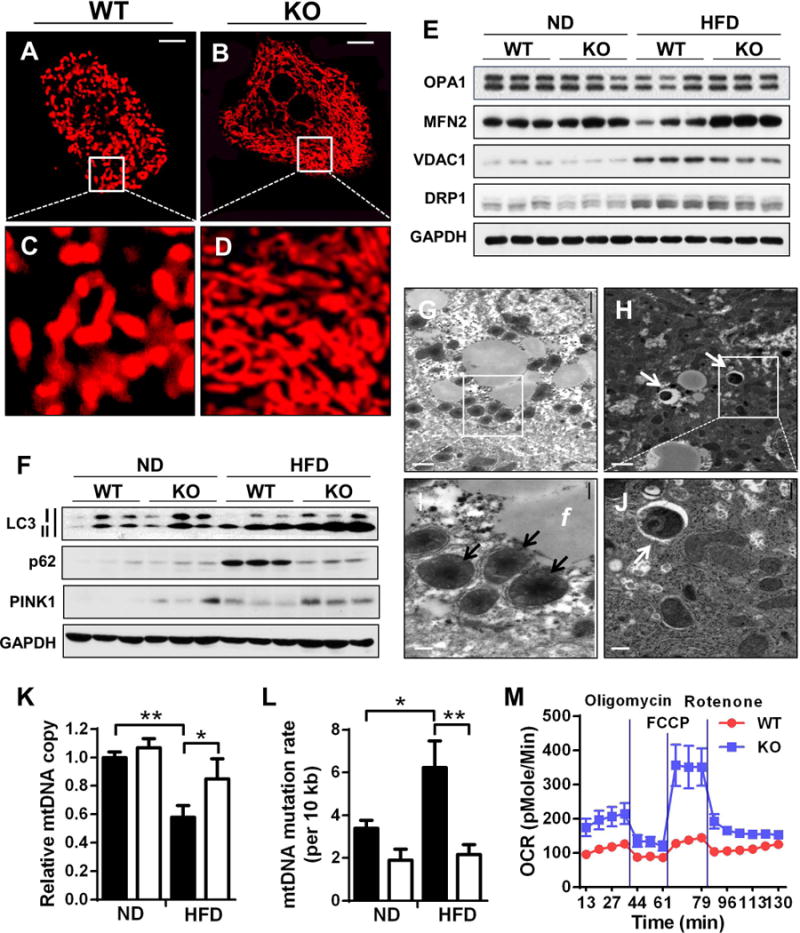
ALCAT1 deficiency prevents mitochondrial dysfunction in hepatocytes. (A–D) Analysis of mitochondrial architecture in isolated hepatocytes from WT controls (A, highlighted in C) and ALCAT1−/− mice (B, highlighted in D) stained by MitoTracker Red using liver samples from Fig. 1. Scale Bar: 10 μm. (E) Western blot analysis of mitochondrial fusion and fission proteins, including OPA1, MFN2, VDAC1 and DRP1, using GAPDH as loading control and liver samples from Fig. 1. (F) Western blot analysis of autophagic biomarkers, including LC3, p62, and PINK1. (G–J) Electron micrographic (EM) analysis of liver sections from ALCAT1−/− mice (H, highlighted in J) and WT mice (G, highlighted in I). Arrows highlight deformed mitochondria (I) or mitophagosomes (H and J); f, fat droplets. Scale Bars: 2 μm (G & H) and 0.5 μm (I & J). (K–L) RT-PCR analysis of mtDNA copy number and mtDNA mutation rate. (M) Primary hepatocytes from ALCAT1−/− and WT mice on HFD were analyzed for mitochondrial respiration by Seahorse XF-24 to determine changes in oxygen consumption rate (OCR) in response to treatment with indicated mitochondrial inhibitors, including oligomycin, FCCP, and rotenone. Mean ± SEM; n=5, *P<0.05, **P<0.01, and ***P<0.001.
Defective autophagy is a common defect in NALFD, but the underlying causes remain elusive. We next investigated a role for ALCAT1 in defective autophagy in the NAFLD, since CL is required for autophagosome biogenesis and cargo recognition (6, 7, 16). We first determined the effect of ALCAT1 on the expression of autophagic biomarkers in the liver by western blot analysis. The results show that NAFLD significantly increased the protein level of p62 in the liver of WT mice (Fig. 2F, quantified in Fig. S2H). The increased p62 level could either be caused by increased transcription of the p62 gene or by impaired autophagic consumption of the p62 protein. To address this issue, we next analyzed p65 mRNA expression level in the liver. The results from RT-PCR analysis of the p62 mRNA level support the later (Fig. S2J), since WT control mice exhibited significantly lower p62 mRNA level than ALCAT1−/− mice. In contrast, ALCAT1−/− mice exhibited an increased autophagic flux, which is supported by increased level of active form of LC3 protein (LC3-II) and decreased p62 protein level, while p62 mRNA level is much higher than WT control mice. In further support for the role of impaired autophagy in the pathogenesis of NAFLD, the db/db mice also exhibited defective autophagy in the liver, as evidenced by more than 95% depletion of LC3-II and increased p62 expression (Fig. S3B, quantified in Fig. S3D, and S3E).
PINK1 is a protein kinase required for autophagic degradation of damaged mitochondria and is depleted in Parkinson’s diseases (17). We next investigated a role for ALCAT1 in regulating PINK1 expression and mitophagy in NAFLD. The results show that PINK1 expression was significantly up-regulated in the liver of ALCAT1−/− mice on HFD (Fig. 2F, quantified in Fig. S2I). Likewise, PINK1 expression was depleted more than 70% in the liver of db/db mice (Fig. S3B, quantified in S3F), suggesting a role of the kinase in defective mitophagy in NAFLD. Consistent with these findings, the number of mitophagosomes was significantly higher in hepatocytes of ALCAT1−/− mice on HFD (Fig. 2H, highlighted in Fig. 2J by arrows) relative to WT controls (Fig. 2G, highlighted in Fig. 2I, quantified in Fig. S2K). In contrast, liver sections from WT mice exhibited a large number of deformed and swollen mitochondria (highlighted in Fig. 2I by arrows). In support of a protective role of mitophagy in safeguarding mtDNA fidelity, NAFLD caused mtDNA depletion and mutations in WT control mice, which were also mitigated in ALCAT1−/− mice (Fig. 2K and Fig. 2L).
Targeted deletion of ALCAT1 restores mitochondrial respiration
Using the Seahorse XF-24 Extracellular Flux analyzer, we next determined a role for ALCAT1 in mitochondrial oxidative phosphorylation in primary hepatocytes. The results show that the onset of NAFLD drastically impaired mitochondrial respiration capacity, as evidenced by decreased mitochondrial oxygen consumption rate (OCR) and blunted responses to the treatments with different mitochondrial inhibitors, including oligomycin (an ATPase inhibitor), FCCP (a mitochondrial uncoupler), and rotenone (a complex | inhibitor) (Fig. 2M, quantified in Fig. S2L). In contrast, ALCAT1 depletion significantly improved mitochondrial functions, which is supported by significantly higher basal level of OCR and dramatic changes in OCRs in response to treatment with mitochondrial inhibitors. Furthermore, ALCAT1 depletion also remarkably enhanced mitochondrial respiratory capacity, which is supported by a dramatic increase in OCR in response to treatment with FCCP.
ALCAT1 depletion promotes autophagosome biogenesis in hepatocytes
Using MitoTraker red and recombinant adenoviruses expressing LC3-II fused with GFP, we examined the effects of NAFLD on mitochondrial architecture and biogenesis of autophagosome. The analysis is based on the principles that GFP-tagged LC3 precursors are proteolytically processed to form LC3-I, which is diffusely distributed in the cytosol. Upon initiation of autophagy, the C-terminal glycine of LC3-I is modified by addition of a phosphatidylethanolamine to form LC3-II, which translocates rapidly to nascent autophagosomes in a punctate distribution. The results show that NAFLD caused severe mitochondrial fragmentation in isolated hepatocytes from WT control mice (Fig. 3A–3D). In contrast, ALCAT1 deficiency not only preserved mitochondrial architecture, but also significantly promoted autophagosome biogenesis, as evidenced by increased number of puncta (Fig. 3E–3H). Additionally, ALCAT1 deficiency stimulated the biogenesis of mitophagosomes (highlighted by arrows in Fig. 3H). In contrast, there were no significant differences between ALCAT1−/− mice and WT controls in mitochondrial architecture and autophagy when fed a regular chow diet (Fig. S4A–4F). In direct support for a causative role of oxidative stress in autophagic arrest in NAFLD, treatment of primary hepatocytes with H2O2 led to mitochondrial fragmentation and defective mitophagy (Fig. 3I–3L). These defects were also mitigated by ALCAT1 depletion, as evidenced by normal mitochondrial architecture and increased number of autophagosomes (Fig. 3M and 3N). ALCAT1 deletion also stimulated mitophagy which is known to eliminate damaged mitochondria (Fig. 3O and 3P, highlighted with arrows, quantified in Fig. 3Q). Conversely, overexpression of ALCAT1 in primary hepatocytes led to defective autophagy (Fig. S4G–L), further corroborating a key role of ALCAT1 in defective autophagy in NAFLD. In support of a protective role for mitophagy in attenuating oxidative stress, the onset of NAFLD dramatically increased oxidative stress and lipid peroxidation in WT control mice, as evidenced by increased levels of H2O2 and malondialdehyde (MDA), a byproduct of lipid peroxidation (Fig. 3R and 3S). Again, these defects were absent in the liver of ALCAT1−/− mice.
Fig. 3.
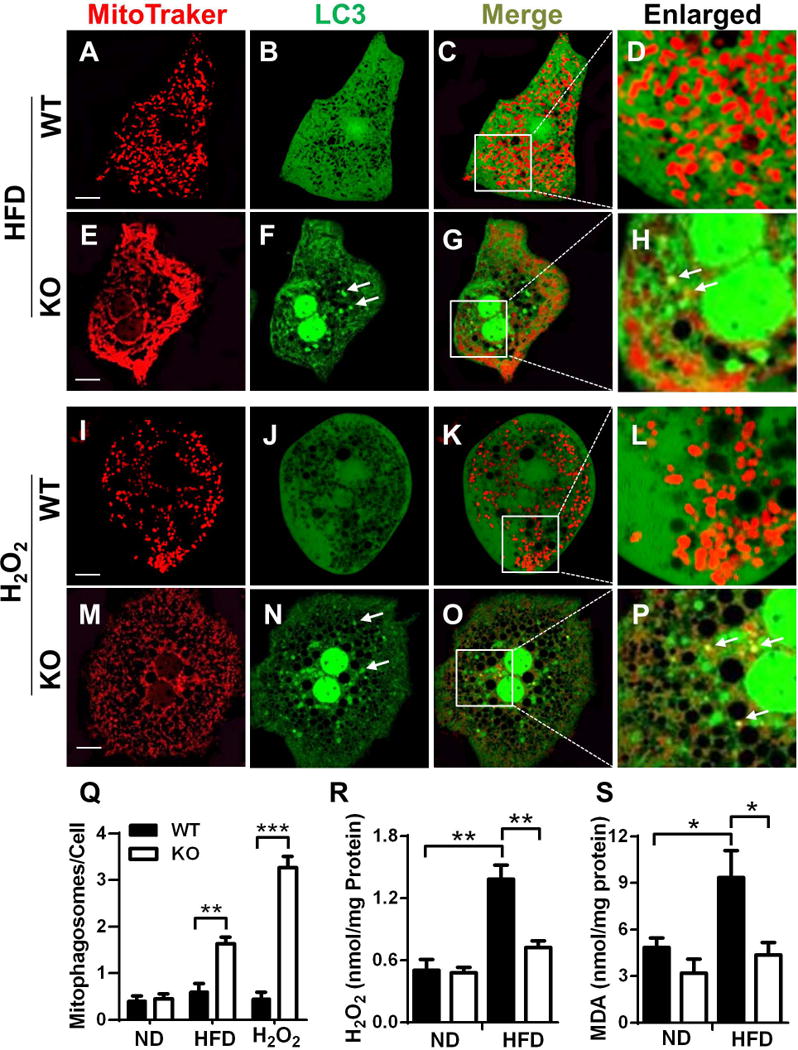
ALCAT1 deficiency promotes autophagosome biogenesis through attenuation of oxidative stress. Primary hepatocytes from ALCAT1−/− mice (KO) and WT controls were infected with recombinant adenoviruses expressing LC3-GFP fusion protein, stained by MitoTracker Red, and followed by confocal imaging analysis of mitochondrial network and autophagosome biogenesis. (A–H) Primary hepatocytes from WT and ALCAT1−/− mice on HFD were analyzed for mitochondrial architecture (A & E), autophagosomes (B & F), and mitophagosomes (C & G, enlarged in D & H). (I–P) Primary hepatocytes from WT and ALCAT1−/− mice on chow diet were treated with 0.5 mM H2O2 for 2 hours, followed analysis of mitochondrial architecture (I & M), autophagosome biogenesis (J & N), and mitophagosomes (K & O, enlarged in L & P). (Q) Quantitative analysis of autophagosomes per hepatocyte (n=100). (R–S) Analysis of oxidative stress and lipid peroxidation in the forms of H2O2 and malondialdehyde (MDA), a byproduct of lipid peroxidation. Mean ± SEM; n=6–8, *P<0.05, **P<0.01, and ***P<0.001. Scale Bar: 10 μm.
Ectopic ALCAT1 overexpression leads to hepatosteatosis in response to oxidative stress
As previously reported (12), ALCAT1−/− mice are resistant to the onset of diet-induced obesity (Fig. S1I), raising a key question whether ALCAT1−/− mice are protected from HFD-induced NAFLD secondary to mitigation of obesity. We addressed this critical issue by determining the direct effect of ALCAT1 overexpression on hepatosteatosis in isolated primary hepatocytes and in HepG2 human hepatic cells. In direct support for a causative role of up-regulated ALCAT1 expression in NAFLD, adenoviral overexpression of ALCAT1 in primary hepatocytes led to steatosis (Fig. 4B, highlighted in 4F) relative to vector control (Fig. 4A, highlighted in 4E), as evidenced by the increased number and size of lipid droplets (highlighted by arrows in Fig. 4F). These defects were remarkably exacerbated in response to treatment with H2O2, leading to severe steatosis (Fig. 4D, highlighted in Fig. 4H) relative to vector control (Fig. 4C, highlighted in Fig. 4G). Likewise, similar findings were also discovered in HepG2 cells (Fig. 4I–4P). The results suggest that up-regulated ALCAT1 expression causes heptosteatosis in response to oxidative stress.
FIG. 4.
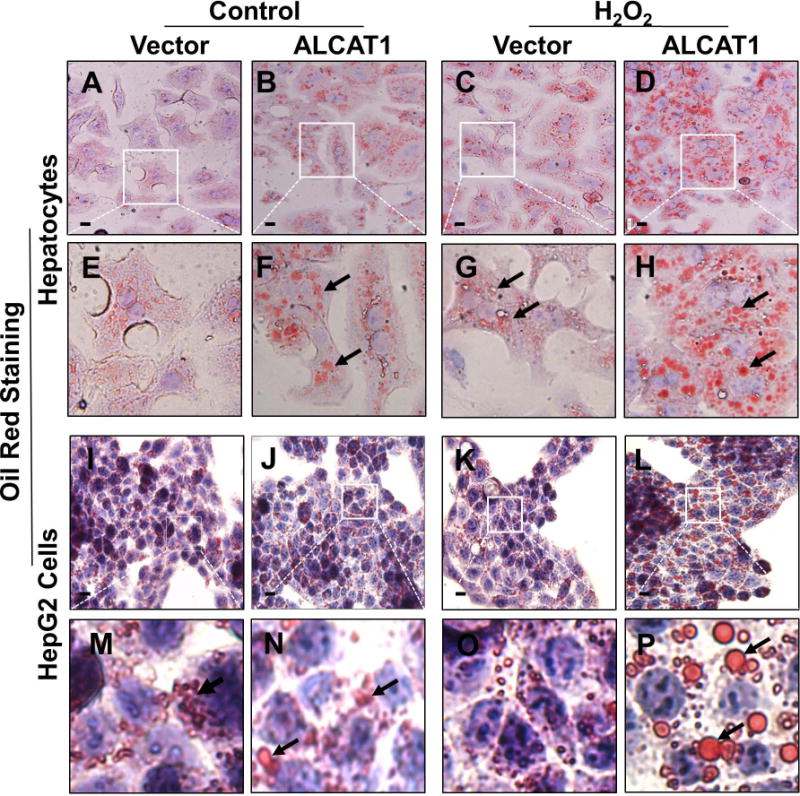
Overexpression of ALCAT1 leads to hepatosteatosis in response to oxidative stress. Primary hepatocytes and HepG2 cells were infected with recombinant adenoviruses overexpressing ALCAT1 or vector control, and analyzed for lipid droplets by Oil Red staining. (A–H) Overexpression of ALCAT1 (B, highlighted in F) in primary hepatocytes led to hepatosteatosis relative to vector control (A, highlighted in E), as evidenced by increased number and size of oil droplets (highlighted by arrows). The defects were exacerbated in response to treatment with 0.05 mM H2O2 for 2 hours (C and D, highlighted in G and H). (I–P) Overexpression of ALCAT1 (J, highlighted in N) in HepG2 cells significantly increased the number and size of lipid droplets relative to vector control (I, highlighted in M). The defects were exacerbated in response to treatment with 0.05 mM H2O2 for 12h (K and L, highlighted in O and P). Scale Bar: 20 μm.
Hepatic ALCAT1 overexpression causes mitochondrial dysfunction
To identify molecular mechanism by which up-regulated ALCAT1 causes hepatosteatosis, we next examined the effect of forced expression of ALCAT1 on mitochondrial architecture and function in primary hepatocytes and HepG2 cells. Adenoviral overexpression of ALCAT1 disrupted mitochondrial network, leading to mitochondrial fragmentation in primary hepatocytes (Fig. 5B, highlighted in 5F) relative to the vector control (Fig. 5A, highlighted in 5E). In support for a causative role of oxidative stress in mitochondrial fragmentation, treatment of hepatocytes with H2O2 also caused mitochondrial fragmentation (Fig. 5C, highlighted in 5G), which was further exacerbated by ALCAT1 overexpression (Fig. 5D, highlighted in 5H, quantified in Fig. 5I). We next examined the effect of ALCAT1 overexpression on oxidative stress, lipid peroxidation, mtDNA copy number, and mtDNA fidelity in HepG2 cells. The results show that adenoviral overexpression of ALCAT1 in HepG2 cells dramatically increased oxidative stress and lipid peroxidation, as evidenced by increased levels of H2O2 production and lipid peroxidation (Fig. 5J and 5K). The defects were again exacerbated in response to oxidative stress induced by H2O2. Consistent with the findings, ALCAT1 overexpression also depleted mtDNA copy number (Fig. 5L) and increased mtDNA mutation rate (Fig. 5M) in response to treatment with H2O2, linking oxidative stress by ALCAT1 to poor mtDNA fidelity.
FIG. 5.
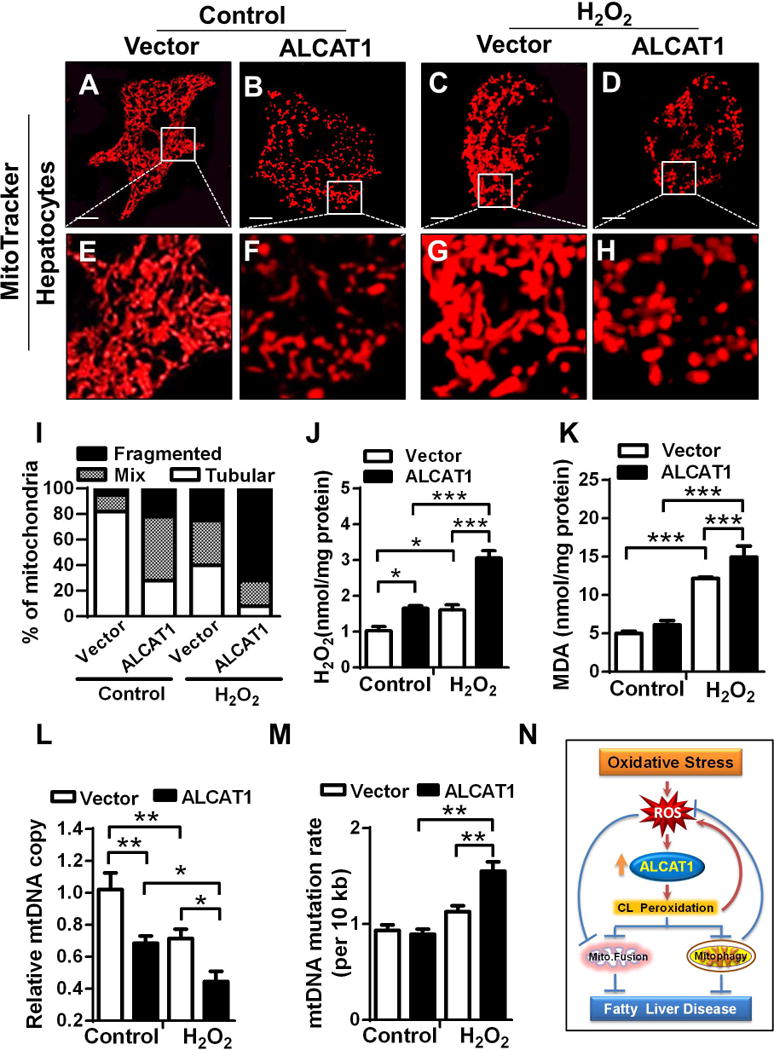
Overexpression of ALCAT1 leads to mitochondrial fragmentation, oxidative stress, lipid peroxidation, mtDNA depletion, and mtDNA mutation. (A–H) Primary hepatocytes from WT mice on a chow diet were infected with recombinant adenoviruses overexpressing ALCAT1 or vector control, stained with MitoTracker Red or Oil Red, and followed by confocal imaging analysis. Overexpression of ALCAT1 in primary hepatocytes caused mitochondrial fragmentation (B, highlighted in F) relative to vector control (A, highlighted in E), which was exacerbated in response to treatment with H2O2 (C and D, highlighted in G and H). Scale Bar: 10 μm (A–D). (I) Quantification of mitochondrial architecture in panel 5A–5D, n=100. (J–M) HepG2 cells were infected with recombinant adenoviruses overexpressing ALCAT1 or vector control and treated with vehicle (control) or 0.5 mM H2O2, followed by analysis for changes in levels of oxidative stress (J), lipid peroxidation (K), mtDNA depletion (L), and mtDNA mutation rate (M). Mean ± SEM; n= 5, *P<0.05, **P<0.01, and ***P<0.001. (N) A hypothetical model depicting a causative role of ALCAT1 in NAFLD in response to oxidative stress associated with diet-induced obesity.
Discussion
Defective autophagy is implicated in the pathogenesis of NAFLD, but the underlying causes remain elusive. One of the striking findings from this study is the identification of a key role for ALCAT1 in regulating autophagosome biogenesis and mitophagy. Accordingly, we showed that NAFLD caused autophagic arrest and accumulation of deformed mitochondria in the liver. These defects were mitigated in ALCAT1−/− mice. Additionally, ALCAT1 deficiency significantly promoted autophagic flux, as evidenced by increased LC3-II level and decreased p62 level. Furthermore, ALCAT1 deficiency also increased expression of PINK1, a mitochondrial kinase that directs damaged mitochondria to autophagosomes (18). PINK1 deficiency impairs mitophagy, leading to oxidative stress and accumulation of damaged mitochondria (17). Consequently, ablation of ALCAT1 significantly enhanced mitophagy, whereas ALCAT1 overexpression dramatically impaired autophagosome biogenesis in hepatocytes. These changes are likely regulated by changes in CL levels and acyl composition, since ALCAT1 deficiency significantly increases the level of CL and the content of TLCL (9). Our results are consistent with recent reports that CL is required for multiple steps of the autophagic process, from membrane structure of autophagosomes, the activities of autophagic proteins, recognition of damaged mitochondria, to a cross talk between mitochondria and lysosomes (5–7, 16). A key role of ALCAT1 in autophagy is further underscored by our previous findings that ALCAT1 is localized at ER–mitochondria contact sites where autophagosomes biogenesis takes place (9, 14). Overexpression of ALCAT1 leads to disruption of mitochondria/ER connections (9), which is known to dramatically impair starvation-induced autophagy (7).
Excessive accumulation of triglyceride and decreased fatty acid oxidation are the major defects in NAFLD. CL is required for fatty acid oxidation by supporting the activity of the mitochondrial trifunctional protein (MTP) which catalyzes β-oxidation of fatty acids (19). Recently, a splice variant of MTP was reported to encode a monolysocardiolipin acyltransferase (MLCL AT) that catalyzes physiological CL remodeling in mitochondria (20). In contrast to ALCAT1, CL remodeling by MLCL AT significantly increases the content of TLCL, a unique CL species required for mitochondrial function in the liver. Thus, the two enzymes have opposing effect on fatty acid oxidation. Overexpression of ALCAT1 diminishes fatty acid oxidation by depleting MTP expression, whereas ablation of ALCAT1 significantly enhances mitochondrial fatty acid oxidation by up-regulating MTP expression in the liver (9). Consistent with a protective role of MTP in NAFLD, MTP mutations in humans and experimental animals lead to early onset of hepatosteatosis and mitochondrial fragmentation (21), which resemble those defects caused by ALCAT1 overexpression in primary hepatocytes. In support for a role of CL remodeling by ALCAT1 in NAFLD, we show in this report that ALCAT1 deficiency significantly attenuated the expression of genes involved in de novo lipogenesis, including PPARα, SREBP1c, FAS1, and ACC1 which were up-regulated by the onset of NAFLD.
Oxidative stress disrupts mitochondrial architecture, which is implicated in mitochondrial ideology of age-related metabolic diseases (22). Mitochondria go through frequent cycles of fusion and fission, a process required for mitochondrial quality control by eliminating ROS-damaged mitochondria through mitophagy (23). We show in this study that the onset of NALFD caused mitochondrial fragmentation and the depletion of MFN2 which is required for mitochondrial fusion. MFN2 depletion in liver leads to glucose intolerance and hepatosteatosis (24). MFN2 deficiency is also a common defect in obesity and type 2 diabetes (25). Hence, targeted deletion of MFN2 causes mitochondrial fragmentation, oxidative stress, mtDNA instability, and severe mtDNA depletion (26), which are highly reminiscent of the defects caused by ALCAT1 overexpression. In final support for a causative role for ALCAT1 in the mitochondrial etiology of NAFLD, ALCAT1 deficiency prevents mitochondrial fragmentation associated with NAFLD by increasing MFN2 expression, leading to restoration of mitochondrial oxidative capacity. Collectively, our data support a key role of ALCAT1 in the etiology of NAFLD, as depicted in Fig. 5N. Accordingly, up-regulated ALCAT1 expression by ROS leads to CL peroxidation, which triggers a vicious cycle of oxidative stress and mitochondrial dysfunction, culminating in the onset of NAFLD.
Supplementary Material
Acknowledgments
We would like to thank Sharon Rannels for technical assistance in hepatocyte isolation. This study was supported in part by grants from NIH (DK076685, Y.S. and DK13499, L.S.J.) and National Natural Science Foundation of China (31371192, Y.S).
Footnotes
The authors disclose no financial conflict of interest.
References
- 1.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 3.Sanyal AJ. Mechanisms of Disease: pathogenesis of nonalcoholic fatty liver disease. Nat Clin Pract Gastroenterol Hepatol. 2005;2:46–53. doi: 10.1038/ncpgasthep0084. [DOI] [PubMed] [Google Scholar]
- 4.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen S, Tarsio M, Kane PM, Greenberg ML. Cardiolipin mediates cross-talk between mitochondria and the vacuole. Mol Biol Cell. 2008;19:5047–5058. doi: 10.1091/mbc.E08-05-0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–1205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 9.Li J, Romestaing C, Han X, Li Y, Hao X, Wu Y, Sun C, et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010;12:154–165. doi: 10.1016/j.cmet.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi Y. Emerging roles of cardiolipin remodeling in mitochondrial dysfunction associated with diabetes, obesity, and cardiovascular diseases. J Biomed Res. 2010;24:6–15. doi: 10.1016/S1674-8301(10)60003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petrosillo G, Portincasa P, Grattagliano I, Casanova G, Matera M, Ruggiero FM, Ferri D, et al. Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I, reactive oxygen species and cardiolipin. Biochim Biophys Acta. 2007;1767:1260–1267. doi: 10.1016/j.bbabio.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Liu X, Wang H, Zhang W, Chan DC, Shi Y. Lysocardiolipin acyltransferase 1 (ALCAT1) controls mitochondrial DNA fidelity and biogenesis through modulation of MFN2 expression. Proc Natl Acad Sci U S A. 2012;109:6975–6980. doi: 10.1073/pnas.1120043109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X, Ye B, Miller S, Yuan H, Zhang H, Tian L, Nie J, et al. Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol Cell Biol. 2012;32:4493–4504. doi: 10.1128/MCB.01092-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013 doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 15.Bach D, Naon D, Pich S, Soriano FX, Vega N, Rieusset J, Laville M, et al. Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes. 2005;54:2685–2693. doi: 10.2337/diabetes.54.9.2685. [DOI] [PubMed] [Google Scholar]
- 16.Huang W, Choi W, Hu W, Mi N, Guo Q, Ma M, Liu M, et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012;22:473–489. doi: 10.1038/cr.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chu CT. A pivotal role for PINK1 and autophagy in mitochondrial quality control: implications for Parkinson disease. Hum Mol Genet. 2010;19:R28–37. doi: 10.1093/hmg/ddq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor WA, Mejia EM, Mitchell RW, Choy PC, Sparagna GC, Hatch GM. Human trifunctional protein alpha links cardiolipin remodeling to beta-oxidation. PLoS One. 2012;7:e48628. doi: 10.1371/journal.pone.0048628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor WA, Hatch GM. Identification of the human mitochondrial linoleoyl-coenzyme A monolysocardiolipin acyltransferase (MLCL AT-1) J Biol Chem. 2009;284:30360–30371. doi: 10.1074/jbc.M109.048322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ibdah JA, Perlegas P, Zhao Y, Angdisen J, Borgerink H, Shadoan MK, Wagner JD, et al. Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology. 2005;128:1381–1390. doi: 10.1053/j.gastro.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sebastian D, Hernandez-Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, Waget A, et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci U S A. 2012;109:5523–5528. doi: 10.1073/pnas.1108220109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molina AJ, Wikstrom JD, Stiles L, Las G, Mohamed H, Elorza A, Walzer G, et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes. 2009;58:2303–2315. doi: 10.2337/db07-1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.