

Abstract
Background
Nuclear factor kappa B (NF-κB) is often implicated in contributing to the detrimental effects of cardiac injury. This ostensibly negative view of NF-κB competes with its important role in the normal host inflammatory and immune response. We have previously demonstrated that pharmacologic inhibition of NF-κB at the time of acute pressure-overload accelerates the progression of left ventricular hypertrophy (LVH) to heart failure in mice. NF-κB regulates angiogenesis and other factors responsible for compensatory reaction to intracellular hypoxia. We hypothesized that impaired angiogenesis may be the trigger, not the result, of pathologic LVH through NF-κB related pathways.
Methods and Results
Transgenic mice (KO) were generated with cardiomyocyte (CMC)-specific deletion of the p65 subunit of NF-κB. Mice underwent transverse aortic constriction and serially followed with echocardiography for 6 weeks. CMC p65 NF-κB deletion promoted maladaptive LVH and accelerated progression towards heart failure, as measured by ejection fraction, LV mass, and lung congestion. Transgenic mice had higher levels of fibrosis and periostin expression. Whole-field digital microscopy revealed increased capillary domain areas in KO mice while concurrently demonstrating decreased microvessel density. This observation was associated with decreased expression of hypoxia-inducible factor 1α (HIF-1α).
Conclusions
Rather than developing compensatory LVH, pressure overload in CMC NF-κB deficient mice resulted in functional deterioration that was associated with increased fibrosis, decreased HIF expression, and decreased microvessel density. These observations mechanistically implicate NF-κB, and its regulation of hypoxic stress, as an important factor determining the path between adaptive hypertrophy and maladaptive heart failure.
Keywords: nuclear factor-kappa B, pressure overload, remodeling heart failure, hypoxia-inducible factor-1, microcirculation
NF-κB is a family of transcription factors intimately involved with the regulation of genes involved in the innate immune response, inflammation, cell survival, and proliferation.1, 2 Within the heart, NF-κB activation can promote both survival and death pathways. This toggling is related, in part, to the type of stimulus, local cellular environment, canonical versus non-canonical signaling, and activation of cofactors.3, 4 Multiple studies have demonstrated that strategies aimed at inhibiting NF-κB activity in the face of ischemia-reperfusion can protect the injured heart.5-8 Conversely, other evidence supports a cardioprotective role of NF-κB in models of ischemic preconditioning and coronary ligation.9, 10 As such, understanding the adaptive and maladaptive balance of NF-κB activation remains a largely unanswered question.11
Transverse aortic constriction (TAC) is a well-known model with which to study the influence of pressure overload on left ventricular hypertrophy (LVH) and cardiac remodeling. Our group has extended this model by removing the constricting band in order to examine processes related to pressure-overload induced HF and its subsequent regression with pressure relief.12-14 Within this system, we identified several transcriptional pathways that appear to be significantly involved with both forward and reverse remodeling.15 In particular, we observed the presence of multiple genes driven by NF-κB, with particular pathways corresponding to different functional phases of the heart. Early after TAC, more genes were associated with cellular proliferation and differentiation; with longer periods of pressure overload and the development of heart failure, there were more genes involved with death pathways. Cumulatively, these studies demonstrate the phenotypic diversity of NF-κB related signaling.
Conceptually, since NF-κB is such an important component of our innate immunity, the presence of this transcription factor should be essential for normal physiologic responses to stress.16 We tested this hypothesis by administering a potent upstream canonical antagonist of NF-κB activation at the time of TAC.17 Compared to control animals that developed compensatory LVH, mice that received IKKβ-inhibition more quickly developed decompensated heart failure. While others have demonstrated a necessary role of NF-κB in the development of compensatory hypertrophy,18-21 many studies point to its importance in promoting cardiac remodeling and heart failure following pressure overload.22-24
Multiple factors have been implicated in the transition from compensatory LVH to decompensated heart failure.25, 26 Metabolically, with increased myocardial mass, the cardiomyocyte can outgrow its nutrient vessels, thus subjecting the cell to increased oxidant stress, contractile dysfunction, and death. We have previously demonstrated that the failing human heart has decreased microvascular density.27 Impaired angiogenesis and dysregulation of hypoxia-inducible factor (HIF) signaling have been implicated in the maladaptive progression of pressure-induced heart failure.28, 29 Currently, the relationship between NF-κB and HIF signaling with regards to cardiac remodeling remains largely unknown.
Although a number of strategies have been employed to study the influence of NF-κB on cardiovascular biology, few have provided the specificity to directly interrogate its role. Within its canonical signaling cascade, the sentinel event is generally considered to be the translocation and subsequent binding of the p65 subunit and its activation of its target gene. We recently developed a mouse that has cardiomyocyte-specific absence of p65-NF-κB and demonstrated that when subjected to ischemia-reperfusion, these hearts were protected.30 In the present study, we sought to determine the effect of pressure overload on mice deficient in p65 NF-κB. We hypothesized that NF-κB is necessary for the development of compensatory adaptation and its absence will accelerate the remodeling process and functional development of heart failure.
Methods
Animal model
Mice were generated with cardiomyocyte-specific ablation of p65 NF-κB as previously described.30 The Cre-loxP system was used in C57Bl/6 mice (Jackson Laboratories). Mice homozygous for the floxed p65 alleles (p65flox/flox) were crossed with mice in which Cre recombinase is expressed under the control of alpha myosin heavy chain promoter (MHCCre/+). Both mice were backcrossed with C57BL/6J mice eight times. All animal studies were approved by the IACUC, University of Utah.
Surgical model
Minimally-invasive transverse arch banding was performed in 12-16 week-old transgenic and wild-type mice as previously described by our group.14 In brief, animals were weighed and administered Ketamine/Xylazine (100 mg/Kg; 10 mg/Kg). A suprasternal incision was used to expose the transverse arch. TAC was performed using a titanium microclip and applier specifically customized for this procedure (Horizon™; Teleflex) and calibrated to a 30G needle. Following wound closure, mice were administered 0.15 mg/Kg of Buprenorphine SR LAB (Wildlife Pharmaceuticals/Zoopharm) SQ immediately post-operative.
Transthoracic Echocardiography
Transthoracic echocardiography was performed using the Vevo 2100 high-resolution echocardiography machine equipped with a 22-55 MHz transducer (VisualSonics). Please see additional details in the Supplemental Material. The standard images included LV dimensions, wall thickness, volumes, ejections fraction, mass, as well as pressure gradients across the constricted arch were recorded. Coronary blood flow across the left circulation was also calculated (Supplemental Figure 1). All acquisitions were performed by the same experienced operator and analyzed using the VevoStrain software (VisualSonics).
Western immunoanalysis
Whole hearts or cardiomyocytes were isolated from 8-12-week-old wild type or p65 knockout mouse as previously described.30, 31 Western immunoblotting was performed on tissue and cell preparations with the following primary antibodies: p65, phosphorylated p65-NF-κB, GAPDH (Cell Signaling Technology, Inc), β-myosin heavy chain (Sigma), and HIF-1α (Novus Bilologics). Secondary peroxidase-conjugated antibody (Cell Signaling Technology, Inc) was applied.
Quantitative RT-PCR
After RNA isolation, real time PCR was performed using the QuantStudio™12K Flex System with Power SYBR® Green PCR Master Mix (Applied Biosystems). Gene expression levels were calculated in accordance with the 2-ΔΔct method and normalized to 16s.
Histology
Hearts were harvested, fixed for 24 hours at 10% formaldehyde. Seven micrometer longitudinal or cross sections of paraffin-embedded specimens were subjected to H&E, PAS (Periodic Acid-Schiff), Masson’s Trichrome staining. Whole-field digital microscopy was performed using the Aperio ScanScope XT (Aperio Technologies) as previously described.27 A pixel count algorithm with optimized color hue and saturation values (ImageScope 10.0, Aperio Technologies, Inc.) was set to accurately identify blue colored collagen fibers versus red cardiomyocytes, thereby allowing for calculation of percent fibrosis. All histopathologic data was confirmed by a blinded pathologist.
Microvascular density evaluation
Endothelial-specific immunohistochemistry staining was performed with Biotinylated GSL I–isolectin B4 (Vector Laboratories, Inc.) on freshly cut sections. Whole slide images were analyzed for microvascular density using the Image-Scope 10.0 microvessel analysis algorithm. For capillary domain area studies, analyzed images of the tissue for microvessel density were transferred to Image J software (4). Capillary domain areas (CDA) to determine tissue cross-sectional area that is closer to a given capillary than to any other were defined by creating Voronoi polygons around each vessel using Voronoi tessellation. Capillary domain surface area was measured for each capillary centered polygon and exported to Microsoft excel 2010 (Microsoft) for further analysis (Supplemental Figure 2).
Statistical Analysis
If not specified in the figure legends, exact animal numbers per experiment are provided in Supplemental Table 1. Comparisons between experimental groups for serial echocardiographic measurements (Table 1 and Supplemental Table 2) were made using two-way ANOVA with Tukey’s multiple comparison post hoc testing using the statistical package, Prism 6.04 (GraphPad). Two-tailed Student’s t-test accommodating for unequal variances was used for comparisons between the four experimental groups. Data are expressed as mean ± SEM. Statistical significance was declared when we obtained -<0.05 from a two-sided test.
Table.
Echocardiographic details 6 weeks following pressure overload. p<0.05:
| Wild Type | Knock-Out | WT Sham | KO Sham | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Base | Post TAC | Base | Post TAC | Base | Post TAC | Base | Post TAC | |
| N= | 13 | 13 | 13 | 13 | 6 | 6 | 4 | 4 |
| EF (%) | 49.5 ± 2.1 | 33.6 ± 2.9 *‡ | 49.4 ± 1.9 | 21.3 ± 2.6 *†§ | 43.8 ± 2.4 | 46.7 ± 4.2 | 48.0 ± 3.3 | 50.8 ± 1.9 |
| FS (%) | 29.8 ± 1.3 | 21.8 ± 2.6 * | 31.4 ± 1.4 | 12.9 ± 2.3 *†§ | 26.2 ± 1.8 | 25.2 ± 1.9 | 28.9 ± 1.9 | 30.5 ± 1.8 |
| LVIDs | 2.89 ± 0.1 | 3.77 ± 0.3 * | 2.90 ± 0.1 | 4.49 ± 0.2 *†§ | 3.05 ± 0.2 | 3.00 ± 0.2 | 2.79 ± 0.2 | 2.70 ± 0.1 |
| LVIDd | 4.10 ± 0.1 | 4.66 ± 0.2 *‡ | 4.18 ± 0.1 | 5.12 ± 0.2 *† | 4.19 ± 0.1 | 4.03 ± 0.12 | 3.97 ± 0.1 | 3.94 ± 0.1 |
| LV Mass | 83.5 ± 2.9 | 151 ± 12. *‡ | 83.8 ± 1.6 | 173 ± 10 *† | 85.7 ± 4.5 | 90.2 ± 4.3 | 81.8 ± 1.5 | 90.5 ± 9.4 |
vs Baseline,
vs KO sham,
vs WT sham,
vs WT TAC
(LVID, left ventricular internal dimensions during (s) systole or (d) diastole (mm); PW, posterior wall (mm); FS, fractional shortening; EF, ejection fraction).
Results
We had previously generated this transgenic mouse and demonstrated adequacy of cardiomyocyte p65 NF-κB deletion embryologically, in vitro, and in vivo.30 However, these studies were performed in the context of ischemia-reperfusion injury. Therefore, we first sought to determine the influence of pressure overload on global NF-κB expression. Whole hearts were evaluated both before and after 6 weeks of pressure-overload. Baseline myocardial protein expression for both total and phosphorylated p65 NF-κB was less in transgenic animals (Figure 1). Both groups of animals had increased expression of the active form of NF-κB following TAC, but phosphorylated p65 NF-κB remained lower in the transgenic mouse. The basal and inducible expression of p-p65 in transgenic mice is not unexpected as there are multiple sources of NF-κB besides the cardiomyocyte. Nevertheless, the cardiomyocyte contribution to total NF-κB production appears to be significant and its absence can be correlated to our subsequent phenotypic findings.
Figure 1.

Pressure overload and p65 NF-κB expression. Western Blots on whole heart tissue for total and phosphorylated p65 NF-κB six-weeks after TAC. (pp65 samples were performed on the same blot and modified to align with the heart sequence in the upper panels). n=3-5; p<0.05, *vs. WT sham, †vs. KO sham, ‡vs. WT post-TAC.
To investigate the functional influence of p65 NF-κB deficiency in vivo, mice were subjected to pressure-overload and followed for 6 weeks. We have previously demonstrated the progression of LVH and HF within this model system.12-15 Echocardiography demonstrated more profound loss of systolic function in transgenic mice after 6 weeks of pressure overload (Table 1). This was associated with left ventricular dilation and increased LV mass and lung congestion (Figure 2). As opposed to the smooth transition into failure seen typically with wild-type mice, TAC in the p65-deficient mice resulted in a steep loss of function within 1-week (Figure 2B, Supplemental Table 2). While TAC increased mRNA expression of classic hypertrophic markers in both groups, β-MHC, ANP, and BNP levels were markedly increased in transgenic mice compared to wild-types (Figure 2C).
Figure 2.
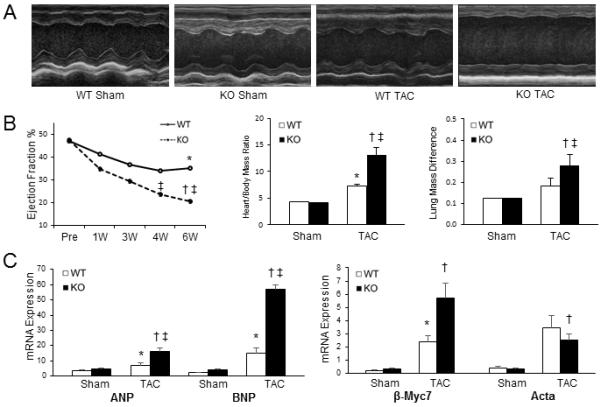
Functional assessment of cardiomyocyte p65 NF-κB deletion with pressure-overload. TAC was performed on sham, WT, and KO mice and followed for 6 weeks. (A) Characteristic M-mode transthoracic echocardiography at mid-papillary level. (B) Graphical representation of ejection fraction, heart weight/body mass ratio (×10−3), and wet/dry lung mass in respective animals. (C) Real-time PCR for hypertrophic markers (β-MHC - β-myosin heavy chain; Acta - smooth muscle α-actinin; ANP - atrial natriuretic peptide; BNP - brain natriuretic peptide). (p<0.05, *vs. WT sham, †vs. KO sham, ‡vs. WT post-TAC).
We next sought to determine the influence of p65-deficiency on myocardial fibrosis and remodeling. Six-weeks after pressure-overload, and correlating with our echocardiographic data, p65-deficient hearts were grossly enlarged (Figure 3A). Whole heart tissue was stained with Masson’s Trichrome in order to examine levels of fibrosis. As previously described in our human heart samples,27 digital microscopy with specialized software was utilized to quantitate extracellular collagen deposits compared to whole tissue volume (Figure 3B – microscopic view; Supplemental Figure 3 – macroscopic view). Compared to wild-type animals, transgenic mice had a trend towards increased baseline fibrosis (p=0.07). Following TAC, p65-deficiency resulted in more fibrosis compared to its baseline and wild-type mice (Figure 3C).
Figure 3.
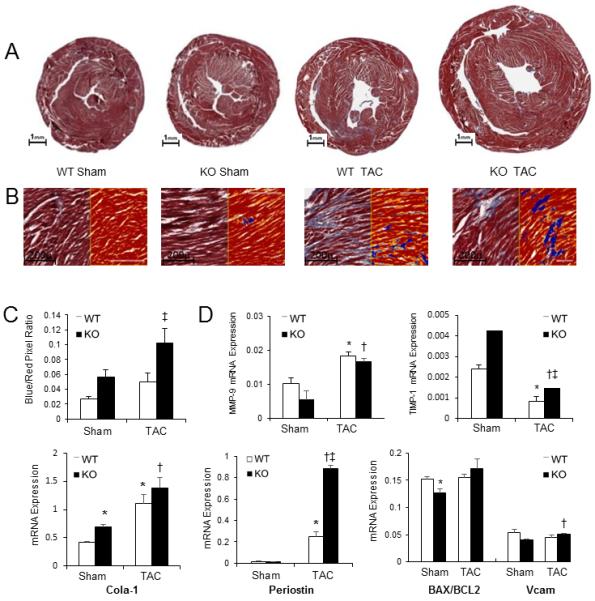
Influence of absence of cardiomyocyte p65 NF-κB on myocardial fibrosis 6-weeks following TAC. (A) Mid-papillary cross-section of mouse hearts stained with Masson’s trichrome. (B) Positive pixel count digital analysis for fibrosis quantification (20x magnification). Blue represents collagen fibers; red represents cardiomyocytes. (C) Quantification of fibrosis as assessed by pixel count ratio. (D) Real-time PCR for fibrotic markers matrix metallopeptidase 9 (MMP9), tissue inhibitor of metallopeptidase 1 (TIMP1), collagen 1α1 (Col1α1), vascular cell adhesion molecule (VCAM), Bax/Bcl2, and periostin. (p<0.05, *vs. WT sham, †vs. KO sham, ‡vs. WT post-TAC).
We next examined the expression of several important fibrosis markers (Figure 3D). Absence of cardiomyocyte p65 NF-κB caused no basal differences in Bax/Bcl2, VCAM, and periostin expression. There was a trend towards less MMP-9 and more TIMP1 expression, but a significant increase in collagen1-α1 (Col1α1). After TAC, transgenic mice had enhanced expression of periostin compared to wildtypes. These results suggest a propensity of mice with cardiomyocytes lacking p65 to have a more aggressive fibrotic response. With regards to apoptosis, we observed no differences after TAC in all groups by TUNEL staining (Supplemental Figure 4) or Bax/Bcl2 ratios.
While fibrosis is a well-known element of cardiac remodeling, we sought to better understand the mechanism of maladaptive hypertrophy by examining the angiogenic response to pressure overload. Indeed, the imbalance between angiogenesis and muscle volume expansion can provoke oxygen delivery/consumption mismatch with subsequent cellular hypoxia and oxidative stress, further promoting functional decline and heart failure.32 Six-weeks following TAC, whole heart sections were stained with endothelial specific markers to quantify microvessel density (Figure 4A). There were no baseline differences observed in the respective sham animals. Following TAC, mice deficient in p65 NF-κB had lower levels of microvessel density compared to wild-types (Figure 4A, C).
Figure 4.
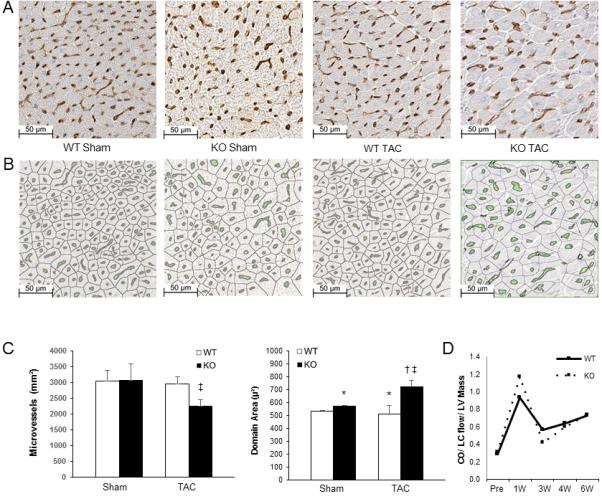
Microvascular study of myocardial tissue 6 weeks after TAC. (A) Immunohistochemical staining with biotinylated GSL I - isolectin B4. (B) Microvessel density digital analysis (green) and sites of capillary domain area measurement. (C) Quantification of microvessel density and capillary domain area. (D) Quantification of echocardiographically determined coronary flow. (*p<0.05 vs. WT)
In addition to vascular density, spatial distribution of microvessels is also an important factor influencing tissue oxygen delivery.33 As such, we examined capillary distribution within myocardial layers. Traversing from the endocardium to the epicardium, there were less endocardial microvessels in transgenic animals after TAC compared to sham, but no differences were noted compared to WT animals after TAC (Supplemental Figure 5). We next sought to determine if there were changes in geometric capillary spacing by quantifying capillary domain areas (Figure 4B,C). Compared to wild type hearts, p65-deficiency resulted in a larger capillary domain area. Thus, we can speculate that the larger distances from feeding capillaries to cells contribute to the maladaptive phenotype.
In order to evaluate the influence of epicardial (inflow) blood flow to the microcirculation, we calculated left main coronary blood flow by echocardiography. These values were normalized to cardiac output and LV mass so as to not confound flow rates by lower ejection fraction and variability in LV mass. As demonstrated in Figure 4D, higher coronary flows were seen immediately after TAC that normalized by 3 weeks and subsequently increased over the ensuing weeks. No differences were observed between groups. These results suggest that changes in coronary inflow blood supply did not effect changes seen at the microvasculature.
Having demonstrating that p65-deficient mice have decreased microvascular density associated with enhanced capillary domain area, we next sought to relate NF-κB to mechanisms of angiogenesis (Figure 5). Compared to wild type, transgenic mice had decreased baseline expression of several angiogenic markers including vascular endothelial growth factor (VEGF), VEGF receptor-2 (VEGFr-2), and VE-cadherin (VE-CAD). After TAC, no differences were observed between groups in the expression levels of VEGF and VEGFr-2, but VE-CAD remained lower in the transgenic animals. Finally, baseline protein expression of HIF1α was markedly lower in mice deficient in cardiomyocyte p65-NF-κB. While increased after TAC in wild type mice, HIF1α levels remained lower after TAC in transgenic mice and, furthermore, did not increase from basal expression.
Figure 5.
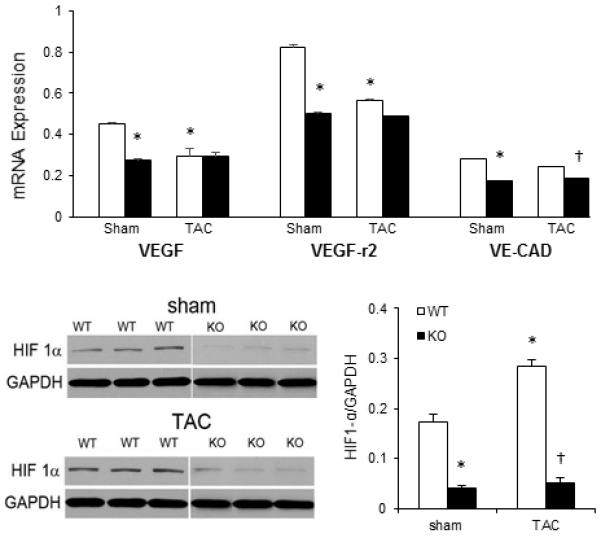
Angiogenesis markers in whole heart tissue. (A) qPCR gene expression of VEGF, VEGF-r2, and VE-CAD 6 weeks after TAC. (B) Western Blot for HIF1α protein following TAC (HIF1α samples were performed on the same blot and modified to align with the appropriate headings). p<0.05, *vs. WT Sham, †vs. WT TAC.
In order to investigate the earlier events in remodeling, TAC was performed on wild-type and knockout mice. Hearts were then analyzed by qPCR after 5 days of pressure overload. At this early stage both groups had similar expression of the previously described hypertrophic, fibrosis, and angiogenic markers (data not shown, n=5-animals/group). Quite possibly, this time point was too early to detect significant difference to correlate functional decline with microvascular changes, especially within whole heart tissues. As such, we next sought to determine if p65 deletion created a microenvironment that predisposed the heart to maladaption. TAC was performed and 48 hours later, cardiomyocytes were isolated and examined for p65 NF-κB and HIF1α expression (Figure 6). Compared to wild type animals at baseline, transgenic mice had less p65 NF-κB and HIF1α expression. Following TAC, HIF1α expression increased in wild types but the expression of HIF1α in knockout mice remained low.
Figure 6.
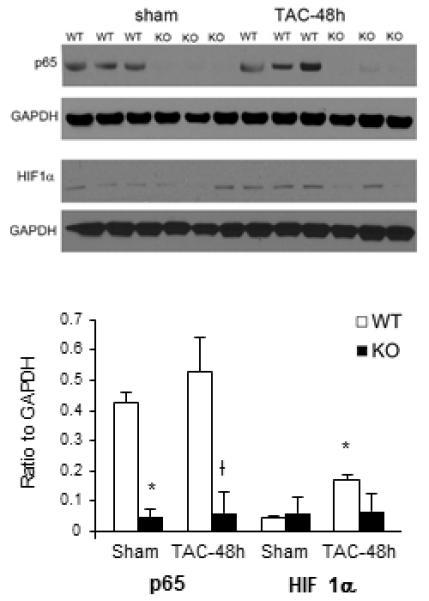
Influence of cardiomyocyte p65 NF-κB deletion early after pressure overload. Cardiomyocytes were isolated 48 hours after TAC. Western Blot for p65 NF-κB and HIF1α was performed. p<0.05, *vs. WT Sham, †vs. WT TAC.
Discussion
We demonstrate that cardiomyocyte p65 NF-κB is necessary for the development of compensatory LVH in mice subjected to acute pressure-overload. Similar to what we observed with mice that received systemic IKK inhibition,17 the transgenic animals functionally sped through the adaptive phase of remodeling and went in to heart failure. This observation was associated with increased fibrosis, decreased microvascular density, increased capillary domain area, and decreased expression of HIF1α.
Deciphering the contribution of NF-κB, and its multiple components, to the development of LVH and pressure-overload induced heart failure remains perplexing and the subject of many reviews over the last decade.1, 11, 34, 35 Using a variety of approaches, some have demonstrated that NF-κB is required for adaptive hypertrophy both at the cellular and whole heart level.18-20 Conversely, others have implicated NF-κB as promoting adverse cardiac remodeling.5, 22, 23 Our group is responsible for some of this confusion, as we have previously shown that isoproterenol or angiotensin-II induced LVH is abrogated with NF-κB blockade.36 A number of factors likely are in play, including the microenvironment as well as the intensity and chronicity of stimulus.11
Others have recently demonstrated the significant role of NF-κB signaling with a similar model system by constructing cardiomyocyte-specific deletions of IKK-β and NEMO/IKKγ.21, 37 Both of these proteins are part of the IKK complex and their conditional absence promoted an aggressive development of dilated cardiomyopathy mediated, in part, by dysregulation of the oxidant stress response. Conversely, a gain-of-function approach to overexpress cardiomyocyte-specific IKK-β demonstrated an exaggerated inflammatory response and a dilated cardiomyopathy.24 These transgenic studies are confounded by the propensity of the mouse hearts to naturally dilate as well as compensatory upregulation of other NF-κB related genes, potentially distorting the effects of induced stress (pressure-overload) that occurs to non-genetically manipulated animals. Furthermore, IKK subunit deletions influence the function of the entire complex thereby removing specificity that we observed with cardiomyocyte p65-ablation.
One other group has studied a cardiomyocyte-specific p65-deficient mouse.38 In that elegant study, the authors observed that the absence of p65-NF-κB conferred protection from deleterious remodeling, including decreased cardiomyocyte hypertrophy, heart mass, and fibrosis with preservation of contractile function. A number of caveats when comparing our findings must be considered. In their study, TAC was performed on 8-week old mice and studied after 4 weeks of pressure overload. The response to pressure overload can vary with a number of factors including age and gender. In our study, we used 12 week-old mice to avoid some of the variability that we have seen with younger animals. As we have previously shown, the duration of pressure-overload is directly associated with the physiologic phenotype.15 As such, one would expect that findings in a 4-week post-TAC mouse would differ from that of a 6-week post-TAC mouse. That said, we saw a precipitous drop in function within a week of TAC (as assessed by 2D echocardiography), as opposed to the preservation seen in the Liu study (as assessed by fractional shortening). Finally, their transgenic mouse used Cre recombinase under the control of Nkx2.5, whereas our mice were under the control of α-MHC. Quite possibly, a Cre associated with an earlier phase of cardiomyocyte developmental could promote pathways and signals that would not be associated with that seen with α-MHC.
We demonstrated that the p65 deficient animals had increased fibrosis. While the observation that decompensated hearts associated with pressure overload have enhanced fibrosis is not surprising, we sought to implicate the angiogenic influence of cardiomyocyte p65 NF-κB on the maladaptive phenotype. In particular, after TAC, the transgenic animals had decreased microvascular density and enhanced capillary domain area, despite similar epicardial coronary flow. Interesting, the capillary domain area was increased at baseline in the knock out hearts, portending a larger distance to feed a given cell. In addition to absolute numbers, the quality, location, and size of the vessels likely play an important role in distribution of nutrients.33, 39 Indeed, increased microvascular density might not be beneficial if its distribution pattern is disorganized, thereby making the distance from the center of a myofibril to the closest vessel beyond the capacity of oxygen diffusion and transport.32, 39
While the vascular changes in the transgenic animals might represent epiphenomenon of any maladaptive heart, we further examined the expression of several classic angiogenic factors. Although no differences were observed between groups in the expression levels of VEGF and VEGFr-2, VE-CAD was lower in the knockout mice. VE-CAD is a vascular endothelial adhesion molecule that is implicated in capillary leak associated with inflammation.40 Decreased VE-CAD expression in transgenic mice associated with pressure overload highlights the complex web of the local capillary environment that includes not only microvascular density and geometry, but vessel integrity as well. Importantly, the p65 deficient mice had decreased baseline expression of VEGF, VEGF-r2, and VE-CAD. Taken together, these hearts were potentially predisposed to have an altered angiogenic response to hypertrophic stress.
As the master regulator of intracellular oxygen homeostasis, cardiomyocyte HIF family proteins activate different intra/extra cellular pathways to regulate oxygen delivery and utilization. HIF-induced angiogenesis will result in mature non-leaky micro vessels and influences nitric oxide production, erythropoiesis, cell growth and differentiation.41 NF-κB subunit promoter sites have been identified on the HIF1α gene and can regulate the HIF1α response to hypoxic stress in multiple cell types.42, 43 Although deductively identified in the present study, the relationship linking NF-κB, maladaptive LVH, and HIF1α has heretofore not been convincingly described. HIF1α is an important player in cardiac remodeling after pressure overload, and is regulated, in part, by p53 accumulation.28 Additional circumstantial evidence supports a role between HIF1α and NF-κB as well as p53 and NF-κB.44, 45 In addition to that indirect linkage, we used a novel approach to provide hypertrophic stress to isolated cardiomyocytes and demonstrate the p65 deletion abolishes the normal HIF1α response to pressure overload (Figure 6).
Although we have targeted the classic, canonical pathway of NF-κB stimulation, we must acknowledge that other non-canonical signals are active. Indeed, the p50-p105 axis has been shown to alternatively protect from and promote cardiac remodeling in ischemic models.46, 47 Conversely, we have previously demonstrated with an infarct model that there was no compensatory upregulation of other NF-κB related proteins in our transgenic mouse.30
In conclusion, this report adds to the evidence that NF-κB plays an important homeostatic role in cardiac physiology. This approach to NF-κB biology is in line with those that believe in the protective influence of NF-κB and its importance in promoting survival pathways.48 Mice with cardiomyocyte-specific deletion of p65 NF-κB were unable to mount an adequate angiogenic response to pressure overload and developed rapid, severe heart failure. As depicted in Figure 7, this report highlights the influence of NF-κB in the normal, compensatory response to hypertrophic stress. It furthermore provides insight into the pathogenesis of maladaptive heart failure, and in particular, the regulation of the angiogenic response necessary to maintain a physiologic adaptive state.
Figure 7.
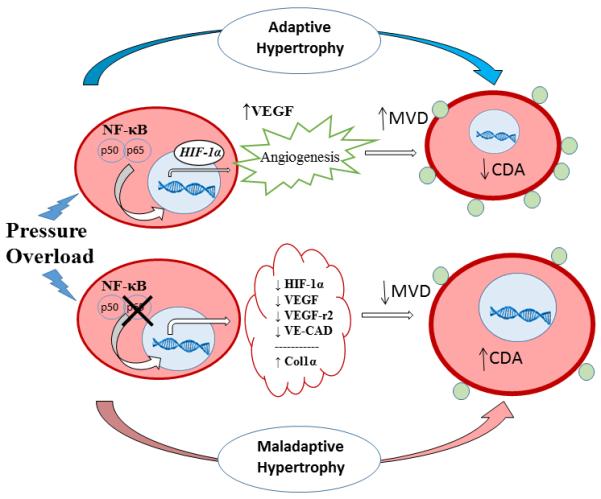
Working model. In a normal cardiomyocyte, hypertrophic stress promotes p65 NF-κB translocation to the nucleus and binding of its target transcription factors, including HIF1α. HIF1α initiates angiogenic pathways including upregulation of VEGF that increases microvascular density (MVD) and decreases capillary domain area (CDA), thereby allowing for adequate nutrient/oxygen delivery and adaptive physiologic response. In the absence of p65 NF-κB, the cardiomyocyte is unable to enhance HIF1α expression and the angiogenic response is blunted (decreased MVD, and increased CDA). In addition to baseline increased collagen 1α synthesis, the p65-deleted cardiomyocyte outgrows its vascular supply, thereby more rapidly progressing to a maladaptive state.
Supplementary Material
Acknowledgments
Sources of Funding
This work was funded in part from the National Institute of Health R01HL089592 (CHS).
Footnotes
Disclosures
None.
References
- 1.Jones WK, Brown M, Wilhide M, He S, Ren X. NF-kappaB in cardiovascular disease: diverse and specific effects of a "general" transcription factor? Cardiovasc Toxicol. 2005;5:183–202. doi: 10.1385/ct:5:2:183. [DOI] [PubMed] [Google Scholar]
- 2.Valen G. Signal transduction through nuclear factor kappa B in ischemia-reperfusion and heart failure. Basic Res Cardiol. 2004;99:1–7. doi: 10.1007/s00395-003-0442-7. [DOI] [PubMed] [Google Scholar]
- 3.Hall G, Hasday JD, Rogers TB. Regulating the regulator: NF-kappaB signaling in heart. J Mol Cell Cardiol. 2006;41:580–591. doi: 10.1016/j.yjmcc.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 4.van Empel VP, De Windt LJ. Myocyte hypertrophy and apoptosis: a balancing act. Cardiovasc Res. 2004;63:487–499. doi: 10.1016/j.cardiores.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn B, Prabhu SD. Cardiomyocyte NF-kappaB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc Res. 2011;89:129–138. doi: 10.1093/cvr/cvq274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ling H, Gray CB, Zambon AC, Grimm M, Gu Y, Dalton N, Purcell NH, Peterson K, Brown JH. Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circ Res. 2013;112:935–944. doi: 10.1161/CIRCRESAHA.112.276915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moss NC, Stansfield WE, Willis MS, Tang RH, Selzman CH. IKKbeta inhibition attenuates myocardial injury and dysfunction following acute ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;293:H2248–2253. doi: 10.1152/ajpheart.00776.2007. [DOI] [PubMed] [Google Scholar]
- 8.Stansfield WE, Moss NC, Willis MS, Tang R, Selzman CH. Proteasome inhibition attenuates infarct size and preserves cardiac function in a murine model of myocardial ischemia-reperfusion injury. Ann Thorac Surg. 2007;84:120–125. doi: 10.1016/j.athoracsur.2007.02.049. [DOI] [PubMed] [Google Scholar]
- 9.Tranter M, Ren X, Forde T, Wilhide ME, Chen J, Sartor MA, Medvedovic M, Jones WK. NF-kappaB driven cardioprotective gene programs; Hsp70.3 and cardioprotection after late ischemic preconditioning. J Mol Cell Cardiol. 2010;49:664–672. doi: 10.1016/j.yjmcc.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilhide ME, Tranter M, Ren X, Chen J, Sartor MA, Medvedovic M, Jones WK. Identification of a NF-kappaB cardioprotective gene program: NF-kappaB regulation of Hsp70.1 contributes to cardioprotection after permanent coronary occlusion. J Mol Cell Cardiol. 2011;51:82–89. doi: 10.1016/j.yjmcc.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res. 2011;108:1122–1132. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- 12.Stansfield WE, Charles PC, Tang RH, Rojas M, Bhati R, Moss NC, Patterson C, Selzman CH. Regression of pressure-induced left ventricular hypertrophy is characterized by a distinct gene expression profile. J Thorac Cardiovasc Surg. 2009;137:232–238. doi: 10.1016/j.jtcvs.2008.08.019. 238e231-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stansfield WE, Rojas M, Corn D, Willis M, Patterson C, Smyth SS, Selzman CH. Characterization of a model to independently study regression of ventricular hypertrophy. J Surg Res. 2007;142:387–393. doi: 10.1016/j.jss.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 14.Zhang X, Javan H, Li L, Szucsik A, Zhang R, Deng Y, Selzman CH. A modified murine model for the study of reverse cardiac remodelling. Exp Clin Cardiol. 2013;18:e115–117. [PMC free article] [PubMed] [Google Scholar]
- 15.Andersen NM, Stansfield WE, Tang RH, Rojas M, Patterson C, Selzman CH. Recovery from decompensated heart failure is associated with a distinct, phase-dependent gene expression profile. J Surg Res. 2012;178:72–80. doi: 10.1016/j.jss.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valen G. Innate immunity and remodelling. Heart Fail Rev. 2011;16:71–78. doi: 10.1007/s10741-010-9187-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersen NM, Tang R, Li L, Javan H, Zhang XQ, Selzman CH. Inhibitory kappa-B kinase-beta inhibition prevents adaptive left ventricular hypertrophy. J Surg Res. 2012;178:105–109. doi: 10.1016/j.jss.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freund C, Schmidt-Ullrich R, Baurand A, Dunger S, Schneider W, Loser P, El-Jamali A, Dietz R, Scheidereit C, Bergmann MW. Requirement of nuclear factor-kappaB in angiotensin II- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation. 2005;111:2319–2325. doi: 10.1161/01.CIR.0000164237.58200.5A. [DOI] [PubMed] [Google Scholar]
- 19.Purcell NH, Tang G, Yu C, Mercurio F, DiDonato JA, Lin A. Activation of NF-kappa B is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc Natl Acad Sci U S A. 2001;98:6668–6673. doi: 10.1073/pnas.111155798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zelarayan L, Renger A, Noack C, Zafiriou MP, Gehrke C, van der Nagel R, Dietz R, de Windt L, Bergmann MW. NF-kappaB activation is required for adaptive cardiac hypertrophy. Cardiovasc Res. 2009;84:416–424. doi: 10.1093/cvr/cvp237. [DOI] [PubMed] [Google Scholar]
- 21.Kratsios P, Huth M, Temmerman L, Salimova E, Al Banchaabouchi M, Sgoifo A, Manghi M, Suzuki K, Rosenthal N, Mourkioti F. Antioxidant amelioration of dilated cardiomyopathy caused by conditional deletion of NEMO/IKKgamma in cardiomyocytes. Circ Res. 2010;106:133–144. doi: 10.1161/CIRCRESAHA.109.202200. [DOI] [PubMed] [Google Scholar]
- 22.Cook SA, Novikov MS, Ahn Y, Matsui T, Rosenzweig A. A20 is dynamically regulated in the heart and inhibits the hypertrophic response. Circulation. 2003;108:664–667. doi: 10.1161/01.CIR.0000086978.95976.41. [DOI] [PubMed] [Google Scholar]
- 23.Gupta S, Young D, Maitra RK, Gupta A, Popovic ZB, Yong SL, Mahajan A, Wang Q, Sen S. Prevention of cardiac hypertrophy and heart failure by silencing of NF-kappaB. J Mol Biol. 2008;375:637–649. doi: 10.1016/j.jmb.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maier HJ, Schips TG, Wietelmann A, Kruger M, Brunner C, Sauter M, Klingel K, Bottger T, Braun T, Wirth T. Cardiomyocyte-specific IkappaB kinase (IKK)/NF-kappaB activation induces reversible inflammatory cardiomyopathy and heart failure. Proc Natl Acad Sci U S A. 2012;109:11794–11799. doi: 10.1073/pnas.1116584109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation. 2013;128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diwan A, Dorn GW., 2nd Decompensation of cardiac hypertrophy: cellular mechanisms and novel therapeutic targets. Physiology (Bethesda) 2007;22:56–64. doi: 10.1152/physiol.00033.2006. [DOI] [PubMed] [Google Scholar]
- 27.Drakos SG, Kfoury AG, Hammond EH, Reid BB, Revelo MP, Rasmusson BY, Whitehead KJ, Salama ME, Selzman CH, Stehlik J, Clayson SE, Bristow MR, Renlund DG, Li DY. Impact of mechanical unloading on microvasculature and associated central remodeling features of the failing human heart. J Am Coll Cardiol. 2010;56:382–391. doi: 10.1016/j.jacc.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 29.Oka T, Akazawa H, Naito AT, Komuro I. Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ Res. 2014;114:565–571. doi: 10.1161/CIRCRESAHA.114.300507. [DOI] [PubMed] [Google Scholar]
- 30.Zhang XQ, Tang R, Li L, Szucsik A, Javan H, Saegusa N, Spitzer KW, Selzman CH. Cardiomyocyte-specific p65 NF-kappaB deletion protects the injured heart by preservation of calcium handling. Am J Physiol Heart Circ Physiol. 2013;305:H1089–1097. doi: 10.1152/ajpheart.00067.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang RH, Zheng XL, Callis TE, Stansfield WE, He J, Baldwin AS, Wang DZ, Selzman CH. Myocardin inhibits cellular proliferation by inhibiting NF-kappaB(p65)-dependent cell cycle progression. Proc Natl Acad Sci U S A. 2008;105:3362–3367. doi: 10.1073/pnas.0705842105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dorn GW., 2nd Myocardial angiogenesis: its absence makes the growing heart founder. Cell Metab. 2007;5:326–327. doi: 10.1016/j.cmet.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 33.Egginton S, Gaffney E. Tissue capillary supply--it's quality not quantity that counts! Exp Physiol. 2010;95:971–979. doi: 10.1113/expphysiol.2010.053421. [DOI] [PubMed] [Google Scholar]
- 34.Gupta S, Sen S. Role of the NF-kappaB signaling cascade and NF-kappaB-targeted genes in failing human hearts. J Mol Med. 2005;83:993–1004. doi: 10.1007/s00109-005-0691-z. [DOI] [PubMed] [Google Scholar]
- 35.Purcell NH, Molkentin JD. Is nuclear factor kappaB an attractive therapeutic target for treating cardiac hypertrophy? Circulation. 2003;108:638–640. doi: 10.1161/01.CIR.0000085362.40608.DD. [DOI] [PubMed] [Google Scholar]
- 36.Stansfield WE, Tang R, Moss NC, Baldwin AS, Willis MS, Selzman CH. Proteasome inhibition promotes regression of left ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2008;294:H645–650. doi: 10.1152/ajpheart.00196.2007. [DOI] [PubMed] [Google Scholar]
- 37.Hikoso S, Yamaguchi O, Nakano Y, Takeda T, Omiya S, Mizote I, Taneike M, Oka T, Tamai T, Oyabu J, Uno Y, Matsumura Y, Nishida K, Suzuki K, Kogo M, Hori M, Otsu K. The I{kappa}B kinase {beta}/nuclear factor {kappa}B signaling pathway protects the heart from hemodynamic stress mediated by the regulation of manganese superoxide dismutase expression. Circ Res. 2009;105:70–79. doi: 10.1161/CIRCRESAHA.108.193318. [DOI] [PubMed] [Google Scholar]
- 38.Liu Q, Chen Y, Auger-Messier M, Molkentin JD. Interaction between NFkappaB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ Res. 2012;110:1077–1086. doi: 10.1161/CIRCRESAHA.111.260729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldman D. Theoretical models of microvascular oxygen transport to tissue. Microcirculation. 2008;15:795–811. doi: 10.1080/10739680801938289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sidibe A, Imhof BA. VE-cadherin phosphorylation decides: vascular permeability or diapedesis. Nat Immunol. 2014;15:215–217. doi: 10.1038/ni.2825. [DOI] [PubMed] [Google Scholar]
- 41.Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol. 2014;76:39–56. doi: 10.1146/annurev-physiol-021113-170322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonello S, Zahringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, Kietzmann T, Gorlach A. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol. 2007;27:755–761. doi: 10.1161/01.ATV.0000258979.92828.bc. [DOI] [PubMed] [Google Scholar]
- 43.Belaiba RS, Bonello S, Zahringer C, Schmidt S, Hess J, Kietzmann T, Gorlach A. Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol Biol Cell. 2007;18:4691–4697. doi: 10.1091/mbc.E07-04-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chatterjee A, Mir SA, Dutta D, Mitra A, Pathak K, Sarkar S. Analysis of p53 and NF-kappaB signaling in modulating the cardiomyocyte fate during hypertrophy. J Cell Physiol. 2011;226:2543–2554. doi: 10.1002/jcp.22599. [DOI] [PubMed] [Google Scholar]
- 45.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frantz S, Hu K, Bayer B, Gerondakis S, Strotmann J, Adamek A, Ertl G, Bauersachs J. Absence of NF-kappaB subunit p50 improves heart failure after myocardial infarction. FASEB J. 2006;20:1918–1920. doi: 10.1096/fj.05-5133fje. [DOI] [PubMed] [Google Scholar]
- 47.Timmers L, van Keulen JK, Hoefer IE, Meijs MF, van Middelaar B, den Ouden K, van Echteld CJ, Pasterkamp G, de Kleijn DP. Targeted deletion of nuclear factor kappaB p50 enhances cardiac remodeling and dysfunction following myocardial infarction. Circ Res. 2009;104:699–706. doi: 10.1161/CIRCRESAHA.108.189746. [DOI] [PubMed] [Google Scholar]
- 48.Dhingra R, Gang H, Wang Y, Biala AK, Aviv Y, Margulets V, Tee A, Kirshenbaum LA. Bidirectional regulation of nuclear factor-kappaB and mammalian target of rapamycin signaling functionally links Bnip3 gene repression and cell survival of ventricular myocytes. Circ Heart Fail. 2013;6:335–343. doi: 10.1161/CIRCHEARTFAILURE.112.000061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.