

Abstract
Intracellular ISG15 is an interferon (IFN)-α/β-inducible ubiquitin-like modifier which can covalently bind other proteins in a process called ISGylation; it is an effector of IFN-α/β-dependent antiviral immunity in mice1–4. We previously published a study describing humans with inherited ISG15 deficiency but without unusually severe viral diseases5. We showed that these patients were prone to mycobacterial disease and that human ISG15 was non-redundant as an extracellular IFN-γ-inducing molecule. We show here that ISG15-deficient patients also display unanticipated cellular, immunological and clinical signs of enhanced IFN-α/β immunity, reminiscent of the Mendelian autoinflammatory interferonopathies Aicardi–Goutières syndrome and spondyloenchondrodysplasia6–9.We further show that an absence of intracellular ISG15 in the patients’ cells prevents the accumulation of USP1810,11, a potent negative regulator of IFN-α/β signalling, resulting in the enhancement and amplification of IFN-α/β responses. Human ISG15, therefore, is not only redundant for antiviral immunity, but is a key negative regulator of IFN-α/β immunity. In humans, intracellular ISG15 is IFN-α/β-inducible not to serve as a substrate for ISGylation-dependent antiviral immunity, but to ensure USP18-dependent regulation of IFN-α/β and prevention of IFN-α/β-dependent autoinflammation.
Calcification of the cerebral basal ganglia during childhood is an important radiological sign associated with a range of genetic and non-genetic states12,13. In Fahr’s disease, also known as idiopathic basal ganglia calcification (IBGC)14–17, the genetic causes identified are germline mutations of the SLC20A2, PDGFB and PDGFRB genes15,16. Intracranial calcification is also a well recognized feature of a group of Mendelian autoinflammatory diseases associated with upregulation of IFN-α/β signalling8,18, including Aicardi–Goutières syndrome (AGS) and spon-dyloenchondromatosis (SPENCD), in particular. We investigated three siblings from China with IBGC. The eldest child (P4) died during an episode of epileptic seizures at the age of 13 years (Supplementary Information, case report). The other two siblings, currently aged 11 (P6) and 13 (P5) years, have suffered only occasional seizures. Despite having been exposed to common childhood viruses (Extended Data Table 1), these children have experienced no severe infectious disease. Whole-exome sequencing (WES) of P5 and P6 and their healthy mother identified only one common nonsense homozygous mutation, which had not been reported in public databases or in in-house WES data for 1,500 other individuals (Extended Data Table 2). This variant was in exon 2 of ISG15: c.163C>T/163C>T (p.Gln 55*/Gln 55*; asterisks denote stop codons). Familial segregation was consistent with an autosomal recessive mode of inheritance (Fig. 1a, b and Extended Data Fig. 1a).
Figure 1. Familial segregation of the ISG15 allele and CT scans for the affected families.

a, Familial segregation in a family from Turkey (Kindred A), a family from Iran (Kindred B) (previously reported) and a family from China (Kindred C). Asterisks denote stop codons; E denotes unknown genotype; fs, frameshift; WT, wild type. b, Graphical representation of the proISG15 protein, with the LRLRGG motif required for substrate ISGylation and the eight-amino-acid sequence (black) cleaved to yield ISG15, and the putative proteins synthesized in the patients. c, Axial view cerebral CT scans of P1, P2, P3, P4, P5, P6 and the healthy mother of P4, P5 and P6 (ISG15−/+).
This observation was surprising, as we recently described three unrelated children from two families from Turkey and Iran who were homozygous for loss-of-function mutations of ISG155. These patients (P1, P2 and P3, now aged 17, 14 and 17 years, respectively) displayed clinical disease caused by the BCG vaccine, which, in otherwise healthy individuals, defines Mendelian susceptibility to mycobacterial disease (MSMD), a rare disorder characterized by severe clinical disease following infection with weakly virulent mycobacteria19,20. In these patients, MSMD resulted from insufficient ISG15-dependent IFN-γ production21. These patients displayed no severe viral disease2. Following the identification of ISG15 mutations in Chinese children with a putative diagnosis of IBGC, we performed computed tomography (CT) scans on the Iranian and Turkish ISG15-deficient MSMD patients. We found that P1 and P2 displayed calcification of the basal ganglia, with CT imaging in P3 also showing calcification along the cerebral falx (Fig. 1c).The ISG15-deficient patients from China were not vaccinated with BCG at birth, consistent with their current lack of an MSMD phenotype. The c.163C>T/163C>T mutant allele showed no ISG15 protein on western blots (Extended Data Fig. 1b). In total, six patients (P4 with inferred genotype) with IBGC from three kindreds from China, Iran and Turkey were found to have only homozygous rare and null ISG15 mutant alleles in common (Extended Data Table 3). These findings strongly suggest that the three ISG15 alleles identified are disease-causing, not only for MSMD, but also for IBGC, an intracranial calcification phenotype unrelated to any obvious cerebral infection.
We next investigated whether enhanced IFN-α/β immunity might account for both the lack of antiviral immunodeficiency5 and the intracranial calcification, as in patients with autoinflammatory AGS and SPENCD18. AGS- and SPENCD-causing mutations can lead to high levels of IFN-α/β in the blood and cerebrospinal fluid, and an upregulation of interferon-stimulated genes9. Such an IFN-α/β signature is also observed in patients with the autoimmune disorder systemic lupus erythematosus (SLE), which is sometimes associated with basal ganglia calcification and may be allelic with AGS in rare cases6,22,23. We determined the expression levels of six interferon-stimulated genes reported to be upregulated in patients with AGS7. IFI27, IFI44L, IFIT1, ISG15, RSAD2 and SIGLEC1 transcripts were measured in peripheral blood or peripheral blood mononuclear cells from ISG15-deficient patients and controls. Notably, all ISG15-deficient patients, like patients with AGS, had significantly higher levels of interferon-stimulated gene messenger RNA than unaffected relatives or healthy controls (Fig. 2a). The two Chinese patients who displayed seizures (P5 and P6) also had detectable IFN-α antiviral activity in their plasma (Extended Data Fig. 1c, d). These findings indicate that the constitutive enhancement of IFN-α/β activity characterizes ISG15 deficiency, in addition to AGS and SPENCD. All three of these conditions have the same prominent radiological sign, intracranial calcification.
Figure 2. High levels of interferon-stimulated gene expression in ISG15-deficient individuals.

a, Relative mRNA levels for IFI27, IFI44L, IFIT1, ISG15, RSAD2 and SIGLEC1 in peripheral blood from patients (n = 3) or controls (C) (n = 24) or in peripheral blood mononuclear cells (PBMCs) from family members (wild type and heterozygous for ISG15 deficiency) (n = 2) and patients (n = 2), as assessed by RT–qPCR, comparison done with unpaired t-tests. ***P < 0.0001; horizontal bars represent means; RQ defined in reference to C1 unstimulated condition. b, hTert-immortalized fibroblasts from C1, C16, C18, a NEMO−/− subject (as a negative control due to known hyporesponsiveness), P1 and P2 were treated with the indicated doses of IFN-α2b for 12 h, washed with PBS and left to rest for 36 h, after which relative mRNA levels were assessed. b shows a representative experiment of three performed. GUS is used as a housekeeping control gene. NS, not stimulated. c, The same experimental procedure as in b was followed, but the mRNA was used for a microarray experiment, for C1, C6, C16, P1, P2 and P3 cells, with green indicating relative upregulation and red indicating downregulation of the probe concerned. d, In the same experimental setup, we used lentiviral particles containing luciferase–RFP (red fluorescent protein) or wild-type ISG15–RFP genes to transduce hTert-immortalized fibroblasts and then assessed mRNA levels for IFIT1 and MX1 by RT–qPCR. Panel d shows one representative experiment of three performed, where (−) denotes not transduced conditions.
ISG15 is an IFN-α/β-inducible ubiquitin-like protein that can be conjugated to many intracellular substrates via ISGylation1,3,4,24–26; we wondered how and why ISG15 deficiency would be associated with enhanced IFN-α/β immunity. We investigated the relationship between ISG15 deficiency and enhanced IFN-α/β immunity by monitoring the IFN-α response of hTert-immortalized fibroblasts from P1, P2, P3 and controls by reverse transcription with quantitative PCR (RT–qPCR) and genome-wide microarray analyses (Fig. 2b, c and Extended Data Table 4). The relative levels of 78 transcripts, including 20 canonical interferon-stimulated genes (including RSAD2, IFI27, OAS3, IFI44, USP18, IFIT1 and MX1; Extended Data Table 4) were significantly higher in patients than in controls. This in vitro phenotype was rescued by transduction with wild-type ISG15 (Fig. 2d) or with ISG15 (ΔGG), a mutant incapable of conjugation (Extended Data Fig. 2a)5.We excluded the involvement of secreted ISG15 (Extended Data Fig. 2b). Consistent with mRNA levels, cells from patients expressedmoreMX1 and IFIT1 proteins than control cells (Fig. 3a, b). Remarkably, however, levels of USP18 were low in patients’ cells (Fig. 3a, b), despite their higher levels of USP18 mRNA (Fig. 2c and Extended Data Table 4). Similar data were obtained for patient-derived Epstein–Barr virus (EBV)-transformed B cells and Simian virus 40 (SV40)-transformed cell lines, as well as for fibrosarcoma cells (HLLR1-1.4) and a human epithelial cell line, WISH, in which ISG15 was silenced (Extended Data Fig. 3a, b, c). These results demonstrate a positive impact of ISG15 deficiency on the IFN-α/β response and suggest the possible involvement of USP18.
Figure 3.

a, hTert-immortalized fibroblasts from a control (C18) and patient P3 were treated with 100pM IFN-α2 for 0.5 to 36 h. Cell lysates were analysed by western blot for levels of phosphorylated STAT (pSTAT) proteins and proteins encoded by interferon-stimulated genes. b, hTert-immortalized fibroblasts from controls (C1, C18) and patients P1 and P2 were treated with 100pM IFN-α2 for 12 h, washed and left to rest for 24 or 36 h. Protein levels were assessed by western blot. c, HLLR1-1.4 cells were transfected with control short interfering RNA (siRNA) or with siRNA targeting USP18, ISG15 or both. One day later, cells were left untreated (naive) or were primed for 8 h with 500 pM IFN-β, washed and left to rest for 16 h, and then restimulated for 30 min with 100 pM IFN-α2or IFN-β. Lysates were analysed with the indicated antibodies. d, HLLR1-1.4 cells were transfected with control siRNA, ISG15 siRNA or UBE1L siRNA. One day later, IFN-β (500 pM) was added for various periods of time. Lysates were analysed as indicated. e, hTert-immortalized fibroblasts from P3 transduced with lentiviral particles expressing RFP and luciferase (LV), wild-type ISG15 (LV ISG15 WT) or the IS15(ΔGG) mutant (LV ISG15(ΔGG)) were stimulated with IFN-β (500 pM) for 8 to 36 h and protein levels assessed by western blot.
USP18 is an interferon-stimulated gene, an isopeptidase and a potent negative regulator of IFN-α signalling10,27,28. We therefore hypothesized that, in the absence of ISG15, IFN-α/β signalling would be inadequately attenuated due to insufficient USP18. Indeed, in patients’ cells, STAT1 and STAT2 were more persistently phosphorylated and IFIT1 andMX1 levels were higher than in controls, whereas USP18 levels were low (Fig. 3a, b and Extended Data Fig. 3a). The same was observed following the knockdown of USP18 and/or ISG15 expression in HLLR1-1.4 cells primed with IFN-β (Fig. 3c). ISG15 appeared to act in its unconjugated free form, since silencing of UBE1L or of other ISGylation enzymes failed to reduce USP18 levels (Fig. 3d and Extended Data Fig. 3d) and patients’ cells transduced with wild-type ISG15 or ISG15 (ΔGG) exhibited attenuated levels of interferon-stimulated-gene transcripts and proteins (Extended Data Fig. 2a and Fig. 3e). These data indicate that intracellular free ISG15 downregulates the IFN-α/β response by maintaining levels of the negative-feedback regulator USP18.
We then investigated the mechanism by which ISG15 regulates USP18. We measured the translation-independent decay of USP18 in hTert fibroblasts. In cells lacking ISG15, unlike control cells, USP18 levels began to decline after as little as one hour of cycloheximide (CHX) treatment (Fig. 4a and Extended Data Fig. 4a, b). This phenotype was reversed by transduction of patients’ cells with wild-type ISG15 or ISG15 (ΔGG) constructs (Fig. 4b and Extended Data Fig. 4c). In HEK293T cells, co-transfection of USP18 with either wild-type ISG15 or ISG15 (ΔGG) resulted in augmented USP18 protein levels (Fig. 4c). Co-transfection of USP18 with ubiquitin led to strong ubiquitination of USP18 (Fig. 4d, lane 5 and Extended Data Fig. 4d), as previously reported11. The coexpression of either wild-type ISG15 or ISG15 (ΔGG) with USP18 and ubiquitin resulted in markedly lower levels of USP18 ubiquitination (Fig. 4d, lanes 9–11 and Extended Data Fig. 4e) and larger total amounts of USP18.Overall, these data indicate that free intracellular ISG15 antagonizes USP18 ubiquitination and degradation, thereby promoting the stability and function of this protein.
Figure 4. Free ISG15 stabilizes USP18 by preventing SKP2-dependent ubiquitination.

a, hTert-immortalized fibroblasts from control (C18) and patient P3 were left untreated (−) or treated for 6 h with IFN-β (500 pM). Cycloheximide (CHX) was added for an additional 0.5 to 5 h. Lysates were analysed as indicated. b, hTert fibroblasts from P3 transduced with lentiviral particles expressing RFP and luciferase (LV) conjugated to wild-type ISG15 (LV ISG15 WT) or the non-conjugatable ISG15(ΔGG) mutant (LV ISG15(ΔGG)) were processed as in a. c, HEK293T cells were transfected with USP18 expression vector alone or in combination with various amounts of ISG15 (Flag–ISG15), either wild-type or ΔGG. Two days later, lysates were analysed as indicated. Ratios of USP18 or ISG15 to AKT are shown below gels. d, HEK293T cells were cotransfected with USP18 and haemagglutinin (HA)–ubiquitin in the presence of wild-type ISG15 (Flag–ISG15). Two days later, lysates were subjected to immunoprecipitation (IP) with anti-USP18 antibodies. Lysates (left panels) were analysed with antibodies against HA and actin. Immunoprecipitates (right panels) were analysed with antibodies against HA and USP18. Asterisk indicates IgG heavy chain. e, HEK293T cells were transfected with USP18, Flag–SKP2 and Flag–ISG15 expression vectors, as indicated. Two days later, USP18 was immunoprecipitated. Lysates (left panels) and immunoprecipitates (right panels) were analysed with antibodies against SKP2, USP18 and ISG15. Arrowheads indicate two endogenous SKP2 isoforms; arrow indicates ectopic Flag–SKP2; asterisk, background band (lanes 6, 7 and 9, 10). f, HLLR1-1.4 cells were transfected with control siRNA or SKP2 siRNA and 24 h later IFN-β (500 pM) was added and cells were incubated for various times. Lysates were analysed as indicated. USP18 and ISG15 levels were quantified relative to actin levels.
USP18 is subject to S-phase kinase-associated protein 2 (SKP2)-mediated proteolysis11,29. We therefore analysed the impact of ISG15 on SKP2-mediated USP18 degradation. The formation of the USP18–SKP2 complex (Fig. 4e, lane 8), was prevented by co-expression of ISG15 (lane 10 versus lane 8) and ISG15 co-immunoprecipitated with USP18 independently of SKP2 expression (lanes 9 and 10). Finally, we assessed IFN-induced USP18 accumulation in cells silenced for SKP2. At 24 h and beyond, USP18 levels were higher in SKP2-silenced than in unsilenced cells (Fig. 4f and Extended Data Fig. 4f). These results suggest that ISG15 can antagonize SKP2-mediated proteolysis of USP18, although the involvement of other E3 ligases cannot be excluded. In the absence of ISG15, USP18 proteolysis is more rapid, driving the dysregulated IFN-α/β response and resulting in both the blood IFN-α/β signature and brain calcifications seen in the patients.
In conclusion, we have shown that all six ISG15-deficient children identified to date display abnormally strong IFN-α/β immunity, as demonstrated by their high levels of circulating IFN-α and/or leukocyte interferon-stimulated genes. As in patients with AGS and SPENCD30, ISG15-deficient individuals display marked intracranial calcification. Three of the six ISG15-deficient individuals experienced epileptic seizures, which were lethal in one case. These patients also had autoantibody levels higher than those in age-matched controls (Extended Data Fig. 5). We found that ISG15 deficiency led to reduced levels of the negative regulator USP18 because of increased proteolysis due, at least in part, to SKP2-mediated ubiquitination, resulting in stronger responses to IFN-α/β and an ensuing amplification of IFN-α/β-induced responses. The observed enhancement of cellular responses to IFN-α/β provides an explanation for the lack of overt viral infection phenotypes in patients with ISG15 deficiency5. More importantly, human ISG15 is not only redundant for antiviral immunity in these patients, it is a key negative regulator of IFN-α/β. The role of ISG15 in stabilizing USP18 is essential and independent of conjugation. We have not tested whether ISGylated proteins might also stabilize USP18. The broad susceptibility to viral infections of ISG15-deficient mice2 but not humans21 may be due to different biochemical properties of murine versus human ISG15 and/or USP18, affecting their interaction potential. Simultaneously, the lack of free extracellular ISG15 resulted in lower IFN-γ production by lymphocytes, thereby underlying MSMD in these patients5.The phenotypic dichotomy of this monogenic disease, with both autoimmunity and MSMD, probably reflects the functional dichotomy of intracellular versus extracellular ISG15, respectively. There is currently no evidence that IFN-α/β-inducible human intracellular ISG15 exerts antiviral effects on documented viral infections via ISGylation. On the contrary, we show that IFN-α/β-inducible ISG15 is essential to negatively regulate IFN-α/β responses via USP18 stabilization, thereby preventing autoinflammatory consequences of uncontrolled IFN-α/β amplification.
METHODS
Exome capture, sequencing, alignments and variant calling
Exome sequencing was performed on two affected sisters and their unaffected mother. Libraries were prepared following the kit manufacturer’s protocol. In brief, we randomly fragmented 3 µg of purified genomic DNA with an ultrasonoscope (Covaris, Massachusetts, USA) to produce fragments of 150~200 base pairs (bp) in length. The shotgun library was then subjected to hybridization with the Agilent SureSelect Human All Exon 50Mb kit (Agilent Technologies, California, USA) for exome capture, followed by several cycles of amplification before quality control with a 2100 Bioanalyzer (Agilent Technologies) and quantification by RT–qPCR with the StepOnePlus Real-Time PCR System (Life Technologies, California, USA). The final library was sequenced on an Illumina Hiseq2000 (Illumina, California, USA) with 90-bp paired-end reads, following the Hiseq2000 protocol. We used BWA Aligner to align the sequences with the human genome reference sequence (hg18 build). Downstream processing was carried out with the Genome analysis toolkit (GATK) SAMtools and Picard Tools (http://picard.sourceforge.net). Substitution calls were made with a GATK Unified Genotyper, whereas insertion/deletion (indel) calls were made with a GATK Indel GenotyperV2. All calls with a read coverage ≤2× and a Phred-scaled SNP quality ≤20 were filtered out. All the variants were annotated with the GATK Genomic Annotator. For filtering purposes, we used the 1000 Genomes and EVS databases, and an in-house exome database containing data for over 1,000 individuals. For the comparative analysis of P1, P2, P5 and P6, we removed variants with less than six reads and a mapping quality of less than 40 (per GATK), and we used the dbSNP database in addition to the 1000 Genomes and EVS databases.
qPCR and microarray analysis
We assessed the expression of six known interferon-stimulated genes in whole blood. Total RNA was extracted from whole blood with the PAXgene (PreAnalytix) RNA isolation kit. RNA concentration was assessed with a spectrophotometer (FLUOstar Omega, Labtech). Quantitative reverse transcription PCR analysis was performed with the TaqMan Universal PCR Master Mix (Applied Biosystems) and cDNA derived from40 ng of total RNA. The relative abundance of target transcripts, measured with TaqMan probes for IFI27 (Hs01086370_m1), IFI44L (Hs00199115_m1), IFIT1 (Hs00356631_g1), ISG15 (Hs00192713_m1), RSAD2 (Hs01057264_m1), and SIGLEC1 (Hs00988063_m1), was normalized with respect to the expression of HPRT1 (Hs03929096_g1) and 18S (Hs999999001_s1) and assessed with Applied Biosystems StepOne Software v2.1 and Applied Biosystems Data Assist Software v3.01. Data for the patients are expressed relative to mean values for 23 normal controls. For PBMCs, total RNA was extracted with the RNeasy Mini kit (Qiagen). It was not possible to standardize the amount of RNA added to the cDNA synthesis reaction, but expression was nonetheless normalized with respect to the level of expression of HPRT1 (Hs03929096_g1) and 18S (Hs999999001_s1). For PBMCs and hTert fibroblasts, total RNA was extracted with the RNeasy Mini kit (Qiagen) and the standard protocol for hybridization to Affymetrix HTA 2.0 chips was performed. In microarray experiments, we used 1,000 IU ml−1 IFN-α2b as a stimulus. Affymetrix Expression Console and Transcriptome Analysis Console were used for analysis. Upregulation or downregulation was defined as significant if expression was twofold higher or lower for P1, P2 and P3 IFN-α2b-stimulated cells than for C1, C6 and C18 IFN-α2b-stimulated cells.
Cell lines and reagents
Human fibrosarcoma HLLR1-1.4 cells, which have been described elsewhere25, were cultured in Dulbecco’smodified Eagle’s medium(DMEM) (Gibco) supplemented with 10% fetal calf serum, hypoxanthine, thymidine and aminopterin (HAT) and 400 µg ml−1 G418 (Gibco). WISH amnion-derived epithelial cells, HEK293T cells, SV40-transformed and hTert-immortalized fibroblasts from controls and patients were cultured in DMEM supplemented with 10% fetal calf serum. EBV-transformed B cells from patient P1 and controls were cultured in RPMI medium supplemented with 10% fetal calf serum. Recombinant IFN-α2b was a gift from D. Gewert (Wellcome Foundation, Beckenham, Kent, UK; now at BioLauncher Ltd, Cambridge, UK) or was purchased from Schering; IFN-β was obtained from Biogen Idec, Cambridge, MA, USA. IFNs were purified to specific activities of >108 units per mg of protein. Cycloheximide (CHX) (Sigma) was used at a concentration of 20 µg ml−1.
Plasmids and transfection
HEK293T cells were transiently transfected with the FuGENE6 system (Roche Applied Science). The pMET7-USP18 expression vector has been described elsewhere27; pcDNA3+ His6-3×FlagISG15 (Fig. 4d, e and Extended Data Fig. 4d) was provided by J. M. Huibregtse (University of Texas at Austin, TX, USA), pBabe-3×Flag6His-ISG15 (BUG3354) and pBabe-3×Flag6His-ISG15(ΔGG) (BUG3355) (Fig.4c and Extended Data Fig. 4e) were derived from this plasmid; pcDNA3 Flag–SKP2 and HA–ubiquitin constructs were obtained from E. Bianchi (Institut Pasteur, Paris, France). In co-transfection experiments, empty vector was added to keep the total amount of DNA constant.
siRNA-mediated silencing
USP18, UBE1L, UbcH8, HERC5, SKP2 ON-TARGET plus SMART pools and a control siRNA (ON-TARGET plus non-targeting pool) were obtained from Dharmacon. Three individual ISG15-targeting siRNAs from Sigma were tested: ISG15#9, GGACAAAUGCGACGAACCU; ISG15#11, GCAGAUCA CCCAGAAGAUU; ISG15#12, GCAACGAAUUCCAGGUGUC. All efficiently targeted ISG15 transcripts. In all the experiments shown, ISG15#12 was used. Cells were transfected with 25 nM siRNA for 24 h in the presence of the Lipofectamine RNAi max reagent (Invitrogen), according to the manufacturer’s instructions. Cells were then stimulated with IFN.
Protein analyses
Cells were lysed in modified RIPA buffer (50 mM Tris/HClpH8, 200 mM NaCl, 1% Nonidet P40, 0.5% deoxycholate, 0.05% SDS, 2 mM EDTA), 1 mM orthovanadate and a protease inhibitor cocktail and subjected to western blotting. For co-immunoprecipitation, cells were lysed in 50 mM Tris, pH 6.8, 0.5% Nonidet P40, 200 mM NaCl, 10% glycerol, 1 mM EDTA and a protease inhibitor cocktail. For the analysis of USP18 ubiquitination (Fig. 4d and Extended Data Fig. 4d and 4e), cells were lysed in modified RIPA buffer and a protease inhibitor cocktail, and USP18 was immunoprecipitated from post-nuclear lysates for 2 h at 4 °C. It was then subjected to protein A capture and western blotting. The antibodies used were directed against STAT1 and STAT2 (Millipore), phospho-Tyr 701 STAT1 (Cell Signaling Technology), phospho-Tyr 689 STAT2 (Millipore), USP18 and AKT (Cell Signaling Technology), actin (Sigma), p27 (Santa Cruz Biotechnology), SKP2 (Invitrogen), HA (Santa Cruz Biotechnology), ISG15 (a gift from E. C. Borden, Cleveland Clinic, Cleveland, OH, USA or purchased from ABGENT, no. AP1150a), IFIT1 (a gift from G. Sen, Cleveland Clinic, Cleveland, OH, USA), OAS2 p69 (a gift from A. Hovanessian, Universitè Paris-Descartes, Paris, France), MxA (a gift from O. Haller, University of Freiburg, Freiburg, Germany) and ubiquitin (clone FK2 recognizing mono- and poly-ubiquitinated proteins, Enzo Life Sciences). An enhanced chemilum-inescence detection reagent was used for detection (Western Lightning, Perkin Elmer). Relative band intensities were measured with a Fuji ImageQuant LAS-4000.
Statistics
For larger samples there is a normal distribution, while for smaller samples (for example, n = 2) a distribution cannot be established. For larger samples there is a normal variance, while for smaller samples (for example, n = 2) variance cannot be established. Statistical methods used were defined in the legends of the figures where appropriate.
Autoantibody detection
Screening for a broad panel of IgG and IgA autoantibodies was performed with autoantibody arrays (University of Texas Southwestern Medical Center, Genomic and Microarray Core Facility), as previously described31. In brief, diluted serum samples were incubated in duplicate with the autoantigen array and the autoantibodies binding to the antigens were detected with Cy3 and Cy5 fluorescent labelled anti-Ig antibodies (IgG and IgA), by the generation of TIFF images. Genepix Pro 6.0 software was used to analyse the images. Net fluorescence intensities (defined as fluorescence intensity for the spot minus background fluorescence intensity) for duplicate spots were averaged. The signal-to-noise ratio (SNR) was used as a quantitative measurement of the ability to resolve true signal from background noise and SNR values of at least 3 were considered to differentiate a true signal from background noise.
Data were normalized as follows:immunoglobulin positive control (IgG or IgA) values across all samples were averaged and the positive control value for each sample was divided by the mean positive control value to generate a normalization factor for each sample. Each signal was then multiplied by the normalization factor for each block (sample).
Extended Data
Extended Data Figure 1. Mutations in ISG15-deficient individuals, allele characterization and serum IFN-α concentrations.

a, Sanger sequencing of ISG15 exon 2 from genomic DNA in kindred C, with the variants highlighted. b, The wild type (WT) and three mutant alleles (G55X, L114fs, E127X- ISG15) were inserted into an expression vector and used to transfect HEK293T cells. Other HEK293T cells were mock-transfected (mock) or left untransfected (not transf.). The cell lysates isolated were subjected to western blotting, with recombinant human (Rh)-ISG15 used as a control. c, d, Plasma samples from P1, P2, P3, P5, P6 and the mother and brother of P5/6 were used in cytopathic protection assays, to measure antiviral activity (c) and blocking antibodies against IFN-α were used to assess specificity (d) (experiment was performed one time).
Extended Data Figure 2. A form of ISG15 that cannot be conjugated rescues the phenotype of ISG15-deficient cells.

a, Lentiviral particles containing luciferase, wild-type (WT) ISG15-RFP or ISG15(ΔGG)-RFP genes were used to transduce hTert-immortalized fibroblasts from C1, a STAT1−/− subject, P1, P2 and P3. RFP-positive cells were obtained by sorting and were cultured for a few weeks. The cells were then treated with 1,000 IU of IFN-α2b for 12 h, washed with PBS and left to rest for 36 h, after which relative mRNA levels for IFIT1 were determined. b, The experimental setting described in a was used in the presence or absence of vehicle control, anti-ISG15 antibodies or control IgG for luciferase and wild-type ISG15–RFP-transduced C1 and P1 hTert-immortalized fibroblasts (showing representative experiments with technical replicates and s.e.m., out of 3 performed).
Extended Data Figure 3. Prolonged IFN signalling, low USP18, and high interferon-stimulated-gene-encoded protein levels in patient-derived cells and in ISG15-silenced human fibrosarcoma HLLR1-1.4 cells.

a, Left panels, SV40-immortalized fibroblasts from two controls (C10 and C12) and two ISG15-deficient patients (P1 and P2) were stimulated with IFN-β (500 pM) for 4 to 36 h. Cell lysates (30 µg) were analysed with the indicated antibodies. Right, EBV-transformed B cells from control (C3) and patient P1 were stimulated with IFN-β for 8 to 24 h. Cell lysates (30 µg) were analysed with the indicated antibodies. b, HLLR1-1.4 cells were transfected with control siRNA or ISG15 siRNA. One day post-transfection, IFN-β (500 pM) was added for various amounts of time. Cell lysates (30 µg) were analysed with the indicated antibodies (MxA and MX1 are used synonymously). c, WISH cells were stimulated and lysates analysed as described in b. d, HLLR1-1.4 cells were transfected with control siRNA, USP18 siRNA and UbcH8 (also known as UBE2E2) siRNA (left) or control siRNA, USP18 siRNA and HERC5 siRNA (right). One day post-transfection, cells were left untreated (naive) or were primed for 8 h with IFN-β (500 pM). Cells were washed and left to rest for 16 h before being pulsed for 30 min with 100pM IFN-α2 or IFN-β. Cell lysates (30 µg) were analysed with the indicated antibodies.
Extended Data Figure 4. ISG15 controls the stability of the USP18 protein, but not of other interferon-stimulated-gene products.

a, HLLR1-1.4 cells were transfected with either control siRNA or ISG15 siRNA. One day post-transfection, cells were stimulated with IFN-β (500 pM) for 6 h. Cycloheximide (CHX, 20 µgml−1) was then added for various time periods, from 30 min to 5 h. Cell lysates (30 µg) were analysed with the indicated antibodies. b, As in a, with additional controls, cells treated with IFN only. Several interferon-stimulated genes were analysed. c, hTert-immortalized fibroblasts from patient P1 transduced with lentiviral particles expressing RFP and luciferase and wild-type ISG15 (LV ISG15 WT) were stimulated with IFN-β (500 pM) for 6 h. CHX was then added for the indicated times. Cell lysates (15 µg) were analysed by western blotting. d, HEK293T cells were transfected with USP18, HA–ubiquitin and Flag–ISG15 as indicated. Two days later, cells were lysed in modified RIPA buffer, USP18 was immunoprecipitated (IP) and analysed with anti-USP18 antibodies. Left panels, cell lysates (30 µg) were analysed by western blot with the indicated antibodies. Right panels, the immunoprecipitates were gel separated and transferred onto a membrane. The membrane was cut into two parts above the 50 kDa marker, both of which were blotted with anti-USP18 antibodies. The top part was exposed for 2 min, the bottom part for 20 s. Asterisk indicates IgG heavy chain. e, HEK293T cells were transfected with 1 µg of the USP18 construct alone or with 1 µg of HA– ubiquitin, in the presence or absence of Flag-tagged ISG15, either wild type or a mutant form of ISG15 that cannot be conjugated as it lacks the two carboxyterminal glycine residues (Flag–ISG15(ΔGG)). Two days later, cells were lysed in modified RIPA buffer, USP18 was immunoprecipitated and analysed with anti-HA or anti-USP18 antibodies. Asterisk indicates IgG heavy chain. f, hTert-immortalized fibroblasts from patient P3 were transfected with control siRNA or SKP2 siRNA. We added IFN-β (500 pM) 24 h later and the cells were incubated for the indicated times. Cell lysates were analysed with the indicated antibodies and USP18 levels were determined as a function of actin levels.
Extended Data Figure 5. Autoantibody development in ISG15-deficient individuals.

a, b, Serum samples from ISG15-deficient, SLE and AGS patients were evaluated for the presence of IgG and IgA autoantibodies in a blinded experiment. Values for the negative control samples for each antigen were averaged and ratios of each sample to the mean for the negative controls plus 2 standard deviations were calculated, with values greater than 1 considered positive. A heat map of the ratio values was generated with MultiExperiment Viewer software (MeV, DFCI Boston, MA), with values coded as follows: 0, blue; 1, black; 5, yellow.
Extended Data Table 1.
Patients have normal titres of antibodies against many viral antigens
| Antibody responses | ||||||||
|---|---|---|---|---|---|---|---|---|
| HAV | EBV | Measles | VZV | CMV | HSV-1, HSV-2 |
Mumps | Influenza A |
|
| Reference value | (<49%) | α-VCA IgG | (<70) | (mUI/ml <150) | (<0.6) IgG | (<1.1) | (<50) | (<30) |
| P1 | NO | YES | NO | YES | YES | YES | N/A | YES |
| P2 | N/A | YES | NO | NO | YES | NO | NO | YES |
| P3 | YES | YES | NO | YES | YES | YES | YES | YES |
| P5 | NO | NO | NO | NO | YES | NO | NO | NO |
| P6 | NO | NO | YES | NO | YES | YES | NO | YES |
YES= antibodies present
NO= antibodies not present
N/A= not done
Serum samples from patients and, when available, immediate family members, were tested for the presence of antibodies against hepatitis A virus (HAV), EBV, measles, varicella zoster virus (VZV), cytomegalovirus (CMV), herpes simplex viruses 1 and 2 (HSV-1 and HSV-2), mumps and influenza A viruses. NA, not available; YES, antibodies present; NO, antibodies not present. α-VCA IgG denotes anti EBV capsid antibodies.
Extended Data Table 2.
Whole-exome sequencing results for patients with putative IBGC
| Whole Exome | ||||
|---|---|---|---|---|
| P5 | P6 | P5/6 Mother |
||
| Nonsense (stop-gained) | Total | 177 | 168 | 174 |
| Novel homozygous | 1* | 2* | 0 | |
| Novel heterozygous | 7 | 5 | 5 | |
| Readthrough (stop-lost) | Total | 78 | 77 | 74 |
| Novel homozygous | 0 | 0 | 0 | |
| Novel heterozygous | 1 | 1 | 1 | |
| Missense | Total | 11104 | 10967 | 11352 |
| Novel homozygous | 8 | 17 | 9 | |
| Novel heterozygous | 210 | 204 | 234 | |
| Silent | Total | 9848 | 9721 | 10065 |
| Novel homozygous | 6 | 9 | 1 | |
| Novel heterozygous | 97 | 92 | 99 | |
| Frameshift | Total | 178 | 168 | 178 |
| Novel homozygous | 0 | 0 | 0 | |
| Novel heterozygous | 4 | 3 | 4 | |
| Inframe | Total | 163 | 156 | 153 |
| Novel homozygous | 2 | 2 | 0 | |
| Novel heterozygous | 3 | 1 | 2 | |
| UTR | Total | 8508 | 8331 | 10131 |
| Novel homozygous | 15 | 22 | 29 | |
| Novel heterozygous | 117 | 127 | 137 | |
| Splice | Total | 3071 | 3067 | 3181 |
| Novel homozygous | 6 | 5 | 1 | |
| Novel heterozygous | 41 | 42 | 39 | |
| ncRNA | Total | 345 | 319 | 356 |
| Novel homozygous | 1 | 1 | 0 | |
| Novel heterozygous | 3 | 4 | 3 | |
Genomic DNA from the patients was used for massively parallel sequencing and the analysis (described in Methods) yielded the variants reported above. Asterisks denotes coding mutations reported in the main text. ncRNA, non-coding RNA; UTR, untranslated region.
Extended Data Table 3.
Homozygous variants of genes other than ISG15 present in P1, P2, P5 and P6
| P1 | P2 | P5 | P6 |
|---|---|---|---|
| SRP19 | C1orf101 | AIF1 | APOBEC4 |
| GAB4 | ALS2CR11 | C19orf55 | |
| MLL3 | C19orf55 | DEFB136 | |
| PIK3CB | HLA-DRB5 | KAZN | |
| THUMPD2 | LILRA3 | KDM4B | |
| ZC4H2 | LST1 | LRRFIP2 | |
| MUC16 | MGAM | ||
| PRDX2 | MUC16 | ||
| ZC3H12B | PADI4 | ||
| PLEKHN1 | |||
| PLXNB3 | |||
| PRDX2 | |||
| SMCR7 | |||
| TAS1R2 | |||
| TAS2R60 | |||
| TIAM2 |
Homozygous nonsense and missense variations from the four patients that were not present in the 1000 Genomes, EVS or dbSNP databases.
Extended Data Table 4.
ISG15-deficient hTert fibroblasts have enhanced responses to IFN-α
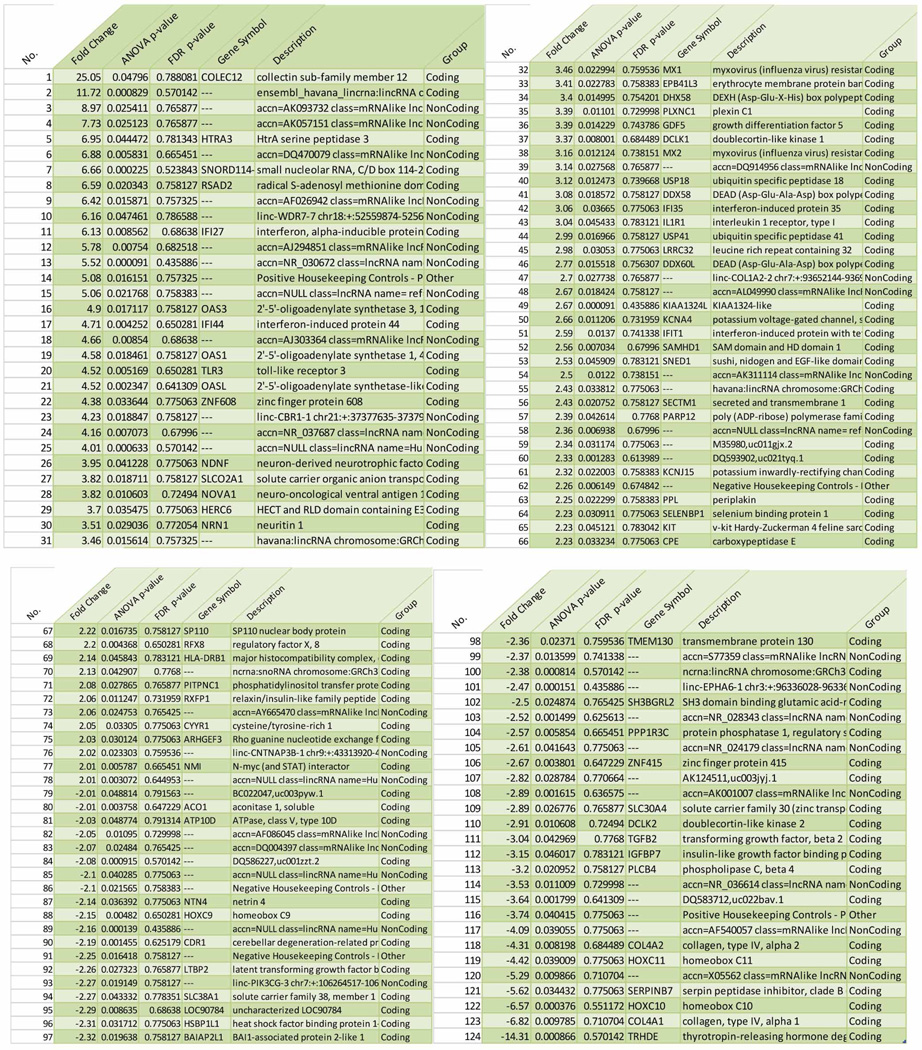 |
We treated hTert-immortalized fibroblasts from three controls and three patients with 1,000 IU IFN-α2b for 12 h. The cells were then washed three times with PBS and left to rest for an additional 36 h, after which we isolated mRNA for microarray analysis. We found that 124 transcripts were significantly different in patients, by a factor of more than two with respect to control cell lines, in statistical analyses based on ANOVA and false discovery rates.
Supplementary Material
Acknowledgements
The Laboratory of Human Genetics of Infectious Diseases is supported by grants from the French National Agency for Research (ANR), the EU grant HOMITB (HEALTH-F32008-200732), the St Giles Foundation, the National Center for Research Resources and the National Center for Advancing Sciences (NCATS), National Institutes of Health grant number 8UL1TR000043, the Rockefeller University, the National Institute of Allergy and Infectious Diseases grant number R37AI095983, Institut Merieux research grant and the Empire State StemCell fund through NYSDOH Contract #C023046 to Flow Cytometry Research Core at the Rockefeller University. The Cytokine Signaling Unit is supported by the Institut Pasteur, CNRS and INSERM. S.P. and G.U. received funding from the EU Seventh Framework Programme under grant agreement 223608. V.F.-N. was supported by the Ligue contre le Cancer. L.R. is a Human Frontier Science Program long-term fellow. L.D.N. was supported by the National Institute of Allergy and Infectious Diseases grant number 1PO1AI076210-01A1. Y.J.C. thanks the Manchester Biomedical Research Centre and the Greater Manchester Comprehensive Local Research Network, the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement 241779, and the European Research Council (GA 309449). A.G.-S. acknowledges NIAID grants U19AI083025 and P01AI090935 for support. We thank C. Daussy for technical assistance, E. Bianchi and F. Michel for discussions. We thank D. Zhang and the members of the Zhang laboratory for assistance, advice and discussions. This work was supported by Chinese National Natural Science Foundation grants (81000079, 81170165) to X.Z. D.B. is supported by the National Institute of Allergy and Infectious Diseases grant number R00AI106942-02.
Microarray data have been deposited in the Gene Expression Omnibus under accession number GSE60359; WES data have been deposited in the BioProject database under accession number PRJNA167660.
Footnotes
Online Content Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Supplementary Information is available in the online version of the paper.
Author Contributions D.B., X.Z., B.P.-B., V.F.-N., O.S., D.M., P.G., A.G.-S., L.A., P.L., L.D.N., S.B.-D., Y.J.C., J.-L.C. and S.P. wrote the manuscript. D.B., X.Z., B.P.-B., V.F.-N., S.D.S., C.Y., S.V., Z.L., I.T., G.I.R., C.C., N.M., S.A.M., Y.I., B.B., S.O., L.Z., X.W., H.J., W.L., T.H., D.L., T.M., B.W., D.Y., L.R., G.U., P.G., F.R., S.-Y.Z., E.J., J.B., A.G.-S., L.A., P.L., L.D.N., S.B.-D., Y.J.C., J.-L.C. and S.P. designed and/or performed experiments. M.L., J.-Y.L., Q.K.W., O.S., D.M., N.M., I.T. and S.A.M. took clinical care of the patients and provided advice.
The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
References
- 1.Jeon YJ, Yoo HM, Chung CH. ISG15and immune diseases. Biochim. Biophys. Acta. 2010;1802:485–496. doi: 10.1016/j.bbadis.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morales DJ, Lenschow DJ. The antiviral activities of ISG15. J. Mol. Biol. 2013;425:4995–5008. doi: 10.1016/j.jmb.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skaug B, Chen ZJ. Emerging role of ISG15 in antiviral immunity. Cell. 2010;143:187–190. doi: 10.1016/j.cell.2010.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang D, Zhang DE. Interferon-stimulated gene 15 and the protein ISGylation system. J. Interferon Cytokine Res. 2011;31:119–130. doi: 10.1089/jir.2010.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bogunovic D, et al. Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science. 2012;337:1684–1688. doi: 10.1126/science.1224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baechler EC, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl Acad. Sci.USA. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crow YJ. Aicardi–Goutières syndrome. Handb. Clin. Neurol. 2013;113:1629–1635. doi: 10.1016/B978-0-444-59565-2.00031-9. [DOI] [PubMed] [Google Scholar]
- 8.Crow YJ, Rehwinkel J. Aicardi–Goutières syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum. Mol. Genet. 2009;18:R130–R136. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rice GI, et al. Assessment of interferon-related biomarkers in Aicardi–Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case–control study. Lancet Neurol. 2013;12:1159–1169. doi: 10.1016/S1474-4422(13)70258-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malakhova OA, et al. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006;25:2358–2367. doi: 10.1038/sj.emboj.7601149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tokarz S, et al. The ISG15 isopeptidase UBP43 is regulated by proteolysis via the SCFSkp2 ubiquitin ligase. J. Biol. Chem. 2004;279:46424–46430. doi: 10.1074/jbc.M403189200. [DOI] [PubMed] [Google Scholar]
- 12.Kendall B, Cavanagh N. Intracranial calcification in paediatric computed tomography. Neuroradiology. 1986;28:324–330. doi: 10.1007/BF00333438. [DOI] [PubMed] [Google Scholar]
- 13.Livingston JH, Stivaros S, van der Knaap MS, Crow YJ. Recognizable phenotypes associated with intracranial calcification. Dev. Med. Child Neurol. 2013;55:46–57. doi: 10.1111/j.1469-8749.2012.04437.x. [DOI] [PubMed] [Google Scholar]
- 14.Manyam BV. What is and what is not ‘Fahr’s disease’. Parkinsonism Relat. Disord. 2005;11:73–80. doi: 10.1016/j.parkreldis.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 15.Wang C, et al. Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nature Genet. 2012;44:254–256. doi: 10.1038/ng.1077. [DOI] [PubMed] [Google Scholar]
- 16.Keller A, et al. Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nature Genet. 2013;45:1077–1082. doi: 10.1038/ng.2723. [DOI] [PubMed] [Google Scholar]
- 17.Nicolas G, et al. Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology. 2013;80:181–187. doi: 10.1212/WNL.0b013e31827ccf34. [DOI] [PubMed] [Google Scholar]
- 18.Briggs TA, et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nature Genet. 2011;43:127–131. doi: 10.1038/ng.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu. Rev. Immunol. 2002;20:581–620. doi: 10.1146/annurev.immunol.20.081501.125851. [DOI] [PubMed] [Google Scholar]
- 20.Casanova JL, Abel L. The genetic theory of infectious diseases: a brief history and selected illustrations. Annu. Rev. Genomics Hum. Genet. 2013;14:215–243. doi: 10.1146/annurev-genom-091212-153448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bogunovic D, Boisson-Dupuis S, Casanova JL. ISG15: leading a double life as a secreted molecule. Exp. Mol. Med. 2013;45:e18. doi: 10.1038/emm.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennett L, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raymond AA, Zariah AA, Samad SA, Chin CN, Kong NC. Brain calcification in patients with cerebral lupus. Lupus. 1996;5:123–128. doi: 10.1177/096120339600500207. [DOI] [PubMed] [Google Scholar]
- 24.Durfee LA, Lyon N, Seo K, Huibregtse JM. The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Mol. Cell. 2010;38:722–732. doi: 10.1016/j.molcel.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broering R, et al. The interferon stimulated gene 15 functions as a proviral factor for the hepatitis C virus and as a regulator of the IFN response. Gut. 2010;59:1111–1119. doi: 10.1136/gut.2009.195545. [DOI] [PubMed] [Google Scholar]
- 26.Chua PK, et al. Modulation of alpha interferon anti-hepatitis C virus activity by ISG15. J. Gen. Virol. 2009;90:2929–2939. doi: 10.1099/vir.0.013128-0. [DOI] [PubMed] [Google Scholar]
- 27.François-Newton V, et al. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon α response. PLoS ONE. 2011;6:e22200. doi: 10.1371/journal.pone.0022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Francois-Newton V, Livingstone M, Payelle-Brogard B, Uze G, Pellegrini S. USP18 establishes the transcriptional and anti-proliferative interferon α/β differential. Biochem. J. 2012;446:509–516. doi: 10.1042/BJ20120541. [DOI] [PubMed] [Google Scholar]
- 29.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and β-TrCP: tipping the scales of cancer. Nature Rev. Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hertzog PJ, Williams BR. Fine tuning type I interferon responses. Cytokine Growth Factor Rev. 2013;24:217–225. doi: 10.1016/j.cytogfr.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 31.Li QZ, et al. Protein array autoantibody profiles for insights into systemic lupus erythematosus and incomplete lupus syndromes. Clin. Exp. Immunol. 2007;147:60–70. doi: 10.1111/j.1365-2249.2006.03251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.