

Summary
Glucocorticoid (GC) effects are mediated by the glucocorticoid receptor (GR). Several studies demonstrated that a lower number of receptors per cell was associated with poor GC response. The regulation of GR expression is complex; the levels of GR can be autologously regulated by its ligand and also by transcriptional, post-transcriptional and post-translational mechanisms. Using three human myeloma cell lines, that parallel the development of GC resistance, this work describes the mechanism involved in downregulation of GR expression. The decreased expression was neither due to mutations in the GR gene nor due to methylation of the promoters. A gradual decrease in GR transcripts was seen during the development of resistance, the level of expression of exon 1–2 RNA fragments remained the same in sensitive and resistant cell lines but ChIP assay demonstrated that RNA polymerase II, detectable throughout exon 2 to 3 in the sensitive cell, was undetectable on exon 3 in the resistant variant suggesting lower or no transcription at this site. These studies demonstrated that downregulation of GR mRNA in resistant cell line involves a block to transcriptional elongation within intron B of the GR gene. This block may represent an important element in the regulation of GR expression.
Keywords: Glucocorticoid Receptor, multiple myeloma, transcription elongation block, glucocorticoid resistance
Introduction
Most of the glucocorticoid (GC) hormone effects are mediated by the glucocorticoid receptor (GR). The GR is a member of the nuclear hormone receptor super family of ligand-activated transcription factors and participates in numerous signaling pathways leading to altered gene expression in target cells and tissues (Adcock, et al 1999, Cairns, et al 1993, Drouin, et al 1987, Yamamoto and Alberts 1976). There is only one known GR gene, but several GR isoforms arose as a result of alternative splicing events (Yudt and Cidlowski 2002). In addition, the GR gene contains at least three promoters whose utilization gives rise to at least five separate transcripts containing different 5′-untranslated first exons (Breslin, et al 2001). A large number of studies about the relationship between GR expression and the clinical response to GC therapy found that a lower number of receptors per cell was associated with poor treatment response (Costlow, et al 1982, Iacobelli, et al 1987, Mastrangelo, et al 1980, Pui, et al 1984, Quddus, et al 1985). Thus, the level of expression of GR is an important determinant of the type and magnitude of the cellular response to the hormone (Vanderbilt, et al 1987). Consequentially, processes that regulate the expression of the human GR gene are important and must be tightly regulated.
The regulation of GR expression is complex and not totally understood. Transcriptional regulation from the three known GR promoter regions is complex and influenced by developmental and hormonal factors (Breslin, et al 2001, Kalinyak, et al 1987). In addition, the levels of GR can be autologous regulated by its ligand and transcriptional, post-transcriptional and post-translational mechanisms may be involved (Burnstein, et al 1991, Burnstein, et al 1994, Dong, et al 1988, Hoeck, et al 1989, McIntyre and Samuels 1985, Vedeckis, et al 1989). Thus, although GR expression is considered ubiquitous as GRs are expressed in almost all cells, the level of expression and receptor regulation vary considerably within and between tissues (Breslin and Vedeckis 1998).
Furthermore, although the primary control point in the regulation of gene expression is the frequency of transcription initiation which depends on the interaction of transcription factors with promoters or enhancer elements, regulation of gene expression at the level of transcript elongation is well established in prokaryotes (Platt 1986) and is emerging as an important control mechanism for eukaryotic gene expression. If transcriptional arrest occurs within the coding region of a gene, the arrested complex would block subsequently initiated RNA polymerases, thereby effectively repressing RNA synthesis from the affected gene. This type of mechanism has been implicated in the regulation of many genes such as MYC, MYB, FOS, ADA, ODC1, HIST3H3, TNF, HSPA4, etc. (Uptain, et al 1997).
Prior studies demonstrated that the lack of responsiveness to dexamethasone in MM.1R cells was coupled with significantly low levels of GRα (Chauhan, et al 2002, Chauhan, et al 2000, Moalli and Rosen 1994). In addition to GRα, our previous work in MM model systems with GC sensitive MM.1S and GC resistant cell lines demonstrated that GC resistant cell lines express low levels of all GR isoforms (GRβ, GR-P and GRγ) (Sanchez-Vega, et al 2006). In this study we show that downregulation of GR mRNA involves a block to transcriptional elongation within intron B of the GR gene. This block may represent an important element in the regulation of GR gene expression.
Materials and methods
Cell Lines
A myeloma cell line, MM.1, was established from the peripheral blood cells of a patient in the leukemic phase of MM. The patient treatment regimen included GCs (Goldman-Leikin, et al 1989). From this parental heterogeneous cell population, three separate cell lines that parallel the progression of the disease and development of GC resistance were established. GC administration induces apoptosis in a GC-sensitive subclone (MM.1S). A resistant subclone to GC-induced-apoptosis was also isolated (MM.1R). With increased duration of dexamethasone exposure in vitro, two distinct phenotypes emerge from MM.1R. The “early” form (MM.1RE) is a transient phenotype that arises immediately following selection of resistant cells from the parental MM.1 cell line that expresses the GR-A isoform. With further growth in the presence of dexamethasone for 4 to 6 months, the expression of the “late” phenotype (MM.1RL) is developed. MM.1RL is a stable resistant phenotype representative of patients in the later stages of the disease (Moalli, et al 1992). These three cell lines, designated MM.1S, MM.1RE, and MM.1RL, which can be distinguished based on their sensitivity to GCs, were a gift from Dr. Steven T. Rosen (Northwestern University, Chicago, IL). All cells were grown in RPMI-1640 (GIBCO) supplemented with 10% heat inactivated fetal bovine serum (FBS) (GIBCO, Invitrogen) in 5% carbon dioxide at 37°C. All cell lines are free of mycoplasma contamination as determined by Hoechst dye staining and PCR assays.
DNA and RNA Extraction
Cells were harvested when the cultures were growing in exponential phase. DNA and total RNA were extracted using QIAGEN DNA and RNeasy extraction kits (QIAGEN, Valencia, CA). Total RNA was treated with DNAse I to eliminate genomic DNA contamination. RNA and DNA were quantified spectrophotometrically; in addition, all RNA samples were also quantified with the Ribogreen kit (Molecular Probes, Eugene, OR).
Primers and Probes
Primers for sequencing of promoter A and for sequencing of exons 3, 4, 5, 6, 7 and 8 have been described before (Antonini, et al 2002, Breslin, et al 2001). The remaining primers for regular PCR and sequencing were designed with Primer3 software (Rozen and Skaletsky 2000) available on line3. Primers for real-time PCR quantification of promoters 1A1, 1A2, 1A3, 1B and 1C were identical to previous study (Pedersen and Vedeckis 2003). The rest of primers and probes used in the real-time PCR study were designed with Primer Express software (Applied Biosystems, Foster City, CA). The primers and probes were purchased from Sigma-Genosys (The Woodlands, TX). The sequences of all the primers and probes used in the present study are listed in Table I supplemental data.
Conventional PCR and Sequencing
Amplification of the different exons from DNA samples were done using Platinum® taq polymerase chain reaction (PCR) SuperMix (Invitrogen, San Diego, CA). Sequencing reactions were done with ABI PRISM® BigDye™ terminator v3.1 cycle sequencing chemistry (Applied Biosystems, Foster City, CA), and resolved in an ABI PRISM® 310 genetic analyzer (Applied Biosystems). The sequencing results were compared to the normal GR gene sequence in GeneBank accession number AY436590. Conventional PCR and sequencing reactions were carried on a MBS Satellite Thermal Cycler (Thermo Hybaid, Milford, CA).
Quantitative Real Time RT-PCR
The levels of GR mRNA expression were determined by One-Step real-time PCR with primers specific for GRα, GRβ, GRγ and GR-P localized on exons 8–9α, 8–9β, 3–4 and exon 7-intron G, respectively using TaqMan® one-step RT-PCR master mix (Applied Biosystems). The assays to quantify the different exon 1-containing transcripts were designed to amplify a RNA fragment spanning each specific exon 1 and exon 2. The 18S rRNA was amplified from the same samples to serve as an internal standard (Applied Biosystems). All reactions were carried out in an ABI PRISM® 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA) using standard cycling conditions for real-time assays. The efficiency of all real-time PCR systems was between 95% (slope of −3.45) and 107% (slope of −3.1).
Chromatin Immunoprecipitation Assay (ChIP)
ChIP assays were performed following the Upstate commercial protocol with some modifications (Upstate, Lake Placid, NY). Briefly, the histones and polymerase II were cross-linked to the DNA by incubation of 1 × 107 cells with 1% formaldehyde (final concentration) at room temperature for 10 min. The cells were washed with 1x PBS twice, and resuspended in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl [pH 8.1]). After lysis, the cell extracts were sonicated (Sonic Dismembrator 60, Fisher Scientific), the debris were pelleted and the supernatant was diluted 4-fold with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris HCl, pH 8.1, 167 mM NaCl). The proteins were incubated with 5 μg of antibody overnight at 4°C. The protein-DNA-antibody complexes were precipitated by with salmon sperm DNA/Protein A Agarose-50% Slurry and the beads were pelleted and washed once each with Low Salt Immune Complex Wash Buffer, (0.1% SDS, 1% Triton X-100, 2mM EDTA, 20mM Tris-HCl, pH 8.1, 150mM NaCl), High Salt Immune Complex Wash Buffer, (0.1% SDS, 1% Triton X-100, 2mM EDTA, 20mM Tris-HCl, pH 8.1, 500mM NaCl), LiCl Immune Complex Wash Buffer, (0.25M LiCl, 1% IGEPAL-CA630, 1% deoxycholic acid (sodium salt), 1mM EDTA, 10mM Tris, pH 8.1), and twice with TE Buffer, (10mM Tris-HCl, 1mM EDTA, pH 8.0). The DNA-protein complexes were obtained by extracting the beads with elution buffer (1% SDS, 0.1 M NaHCO3). The cross-linking of the DNA protein complexes were reversed with 5M NaCl at 65°C for 4 h. Samples were incubated with Proteinase K and the DNA was recovered by phenol/chloroform extraction and ethanol precipitation. 20 ng of DNA were used for PCR reactions.
Antibodies used in the assay were anti-trimethyl-histone H3 (Lys4), anti-acetyl-histone H4, anti-acetyl histone H3 (Lys9), anti-dimethyl histone H3 (Lys9), and anti-RNA polymerase II all from Upstate (Upstate, Lake Placid, NY). Primers for PCR are listed on Table II supplemental data.
Results
Mutation analysis of the GR gene
Mutations in the GR gene, albeit rare, have been detected in several GC resistance malignances (Bray and Cotton 2003). To identify possible mutations or polymorphisms in MM affecting the response to GCs, the GR gene was sequenced and compared between the three cell lines. Sequencing of the three GR promoters to check for possible mutations in some of the response elements located in them, showed no mutations that could lead to differences in the RNA expression levels between these cell lines. In addition, no alterations were found in any of the eight coding exons, exon-intron boundary regions, or 3′-UTR regions (including the AUUUA motifs in 3′UTR, that affect RNA stability) of the GR gene between the MM.1S, MM.1RE, and MM.1RL cell lines.
Methylation analysis of the GR promoters
In MM.1S, MM.1RE, and MM.1RL cell lines there is only GR expression from promoters B and C (Sanchez-Vega, et al 2006), these two promoters are located in a CpG island that makes them susceptible to a repression in RNA transcription by methylation. Therefore, the down-regulation in GR expression in resistant cell lines could be due to methylation in these promoters. To test this hypothesis, MM.1S and MM.1RL cell lines were treated with 5 μM of the demethylating agent 5-aza-deoxycytidine (5-aza-dCyd) for six days and the levels of GR expression were measured by real-time PCR. As shown in Figure 1, 5-aza-dCyd treatment does not induce changes in GR expression. This concentration and time of treatment have been shown to result in hypomethylation.
Figure 1. Relative expression of GRα in response to 5 μM 5-aza-deoxycytidine (5-aza-dCyd) treatment in MM.1S and MM.1RL cell lines.

Expression values are calculated with respect to GRα expression at time 0.
Analysis of GR transcripts containing exon 2
As published before (Sanchez-Vega, et al 2006), because the number of mRNA fragments containing exons 1 and 2 was similar in these cell lines, however the transcripts containing the rest of the exons were dramatically reduced in resistant cells, we wanted to determine the exact point where this discrepancy arises. To start with, we analyzed if the exon 3 is expressed at the same level as the exon 2 a real-time PCR assay was done using a forward primer located on exon 2 and a reverse primer located on exon 3.
As shown in Table I, the number of RNA fragments containing exon 3 is lower than the number of exon 2-containing transcripts, indicating that the point of discrepancy is between these two exons. Furthermore, compare to promoter C, for each specific cell line, exon 2–3 expression was 100-fold, 1,000-fold and 10,000 fold lower in sensitive, early resistant and late resistant cell lines, respectively. Taken together, these data suggest that the dramatic decline in RNA transcripts is due to an alteration in transcription over intron B.
Table I.
Number of exons 1C-2, 1B-2 and 2–3 containing transcripts expressed in MM.1S, MM.1RE and MM.1RL cell lines per microgram of total RNA.
| Exons 1B-2 | Exons 1C-2 | Exons 2–3 | |
|---|---|---|---|
| MM.1S | 5.8·105 | 4.8·106 | 4.4·104 |
| MM.1RE | 2.1·105 | 1.6·106 | 2.2·103 |
| MM.1RL | 1.3·105 | 8.3·105 | 9.3·101 |
To determine if some of the intron B sequences remain in the transcripts containing the exon 2, RNA samples corresponding to the three cell lines were amplified using different sets of primers located on intron B. When intron segments were amplified from the RNA samples, we are able to detect amplification of intron B sequences as far as 2,000 bp downstream from the end of exon 2 but not at 20,000 bp downstream of this exon in MM.1S, MM.1RE, and MM.1RL cell lines (Lanes 2 through 12, right panel, Figure 2). This amplification corresponded to RNA transcripts containing sequences of intron B and not to DNA amplification because the amplification of the same RNA samples without a reverse transcription step did not amplify any fragment (Lanes 8 through 13, right panel, Figure 2). To verify that the PCR system works properly, DNA samples were amplified for both regions located at 2,000 and 20,000 bp 3′ from the end of exon 2 (Left panel, Figure 2).
Figure 2. A) Scheme representing the location of primers used to amplify intron B sequences.
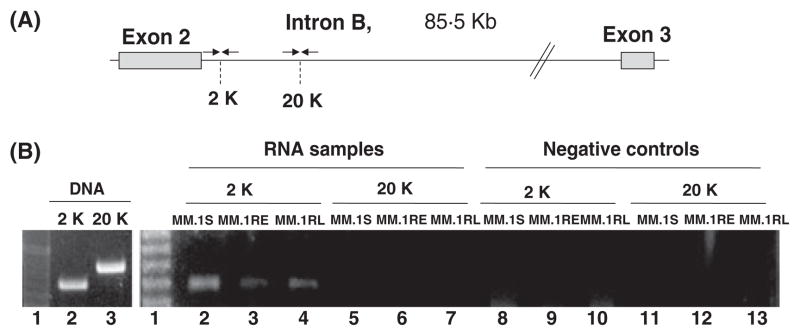
Arrows represents the primers, and 2K and 20K, specify the nucleotide position on kilo base pairs on intron B for the amplified fragments. B) Amplification of intron B sequences from RNA in MM.1S, MM.1RE, and MM.1RL cell lines. Left Panel), Reaction controls. First lane in gel corresponds to a molecular size marker. Lanes 2 and 3 correspond to the amplification of two DNA segments around the positions 2,000 and 20,000 pair bases of intron B, respectively. Right Panel), RT-PCR assay of intron B sequences. First lane in gel corresponds to a molecular size marker. Lanes 2 through 7 correspond to the amplification of RNA samples from MM.1S, MM.1Re, and MM.1RL cell lines Lanes 8 through 13 correspond to the same samples amplified without the reverse transcriptase. 2K and 20K, specify the nucleotide position on kilo base pairs on intron B amplified in the assay.
To establish more precisely the length of the intronic sequence retained in the GR transcripts, we used more sets of primers localized in intron B that amplify fragments of RNA located at 4,000, 8,000, and 10,000 bp downstream from the end of exon 2. Again, when intron segments were amplified from the RNA samples, we were able to detect amplification of intron B sequences as far as 4,000 bp all cell lines (Lanes 2 through 4, Figure 3), and at 10,000 bp in MM.1S and MM.1RL. Interestingly we were not able to detect any amplification of intron B sequences further than 4,000 bp in the MM.1RL cell line.
Figure 3. A) Scheme representing the location of primers used to amplify intron B sequences.
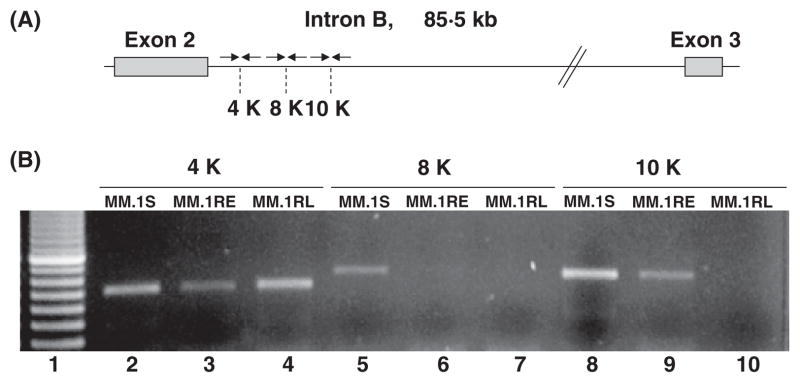
Arrows represents the primers, and 4K, 8K and 10K, specify the nucleotide position on kilo base pairs on intron B for the amplified fragments. B) Amplification of intron B sequences from RNA in MM.1S, MM.1RE, and MM.1RL cell lines. RT-PCR assay of intron B sequences. First lane in gel corresponds to a molecular size marker. Lanes 2 through 10 correspond to the amplification of RNA samples from MM.1S, MM.1RE, and MM.1RL. 4K, 8K and 10K, specify the nucleotide position on on kilo base pairs intron B amplified in the assay.
Analysis of intron B DNA sequences in sensitive and resistant cell lines
To determine if the discrepancy we found in the intron B sequences at the level of RNA is also found at the DNA level, the same sequences were amplify from DNA samples.
Surprisingly, when DNA samples from MM.1S and MM.1RL are amplified we were not able to get any amplification product from the intron B region that goes from 8,000 bp to 12,000 bp downstream of the end of exon 2 (Figure 4). To find out if this lack of amplification is due to a deletion of the intronic sequences in the GR gene in the resistant cell line, we perform a long-range PCR with the forward primer located in the region of 4 Kb and the reverse primer located in the region of 20 Kb. As expected for the sensitive cell line, and in agreement with the real-time PCR data, a PCR product of about 16,000 bp was detected, this PCR fragment was almost undetectable in the early resistant cell line, and not detectable in the late resistant cell line (Figure 5). In addition, no shorter fragments corresponding to a deletion in this region were detected.
Figure 4. A) Scheme representing the location of primers used to amplify intron B sequences.
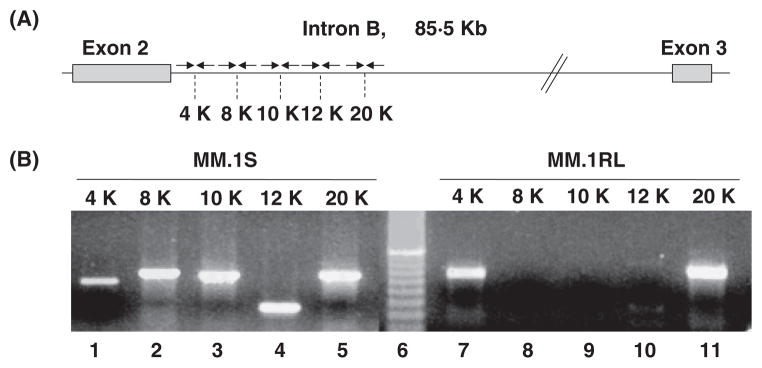
Arrows represents the primers, and 4K, 8K, 10K, 12K and 20K, specify the nucleotide position on kilo base pairs on intron B for the amplified fragments. B) Amplification of intron B sequences from DNA in MM.1S and MM.1RL cell lines. PCR assay of intron B sequences. Sixth lane in gel corresponds to a molecular size marker. Lanes 1 through 5 correspond to the amplification of DNA samples from MM.1S, and lanes 7 through 11 to the amplification of DNA samples from MM.1RL. 4K, 8K, 10K, 12K and 20K, specify the nucleotide position on kilo base pairs on intron B amplified in the assay.
Figure 5. A) Scheme representing the location of primers used to amplify intron B sequences.

Arrows represents the primers located at 4K (forward primer), and 20K (reverse primer), used to amplify a fragment of 16,000 bp on intron B. B) Amplification of intron B sequences from DNA in MM.1S, MM.1RE, and MM.1RL cell lines. First lane in gel corresponds to a molecular size marker. Lanes 2 through 4 correspond to the amplification of a 16,000 bp fragment of intron B from nucleotide 4,000 to nucleotide 20,000, from MM.1S, MM.1RE, and MM.1RL.
Analysis of chromatin structure of intron B in sensitive and resistant cell lines
One possible explanation to the lack of amplification of the 4 – 20 Kb region could be a different chromatin reorganization of this region between sensitive and resistant cell lines. To test this hypothesis we performed a chromatin immunoprecipitation assay of this region using antibodies specific for histone modifications and for the RNA polymerase II (Pol II).
An enrichment of Pol II was consistently observed across the entire transcribed region in MM.1S cells. In contrast, Pol II was only detected in the region comprising exon 2 through 4 Kb downstream to this exon, and barely from 20 Kb upstream to exon 3 through exon 3 in MM.1RL cell line. Confirming a block on transcription elongation located in intron B (Figure 6).
Figure 6. Chromatin immunoprecipitation (ChIP) assays at GR locus testing histone modifications in MM.1S and MM.1RL cell lines.
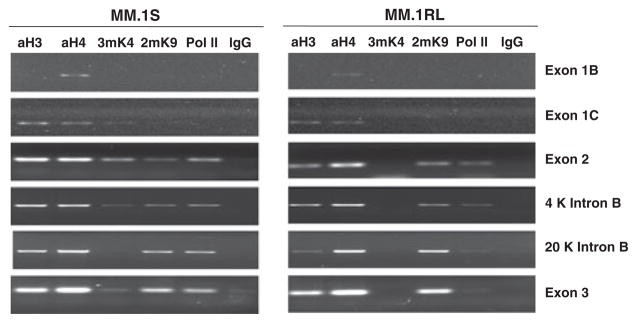
Immunoprecipitations were done with anti-acetyl-histone H3 (aH3), anti-acetyl histone H4 (aH4), anti-trimethyl histone H3 at Lys4 (3mK4), anti-dimethyl histone H3 at Lys9 (2mK9), anti-RNA polymerase II (Pol II), or immunoglobulin G (IgG) as a negative control. Right panel corresponds to MM.1RL and Left panel corresponds to MM.1S. The different rows show the histone modifications from exon 1C to exon 3. 4 Kb and 20 Kb, specify the nucleotide position on intron B downstream exon 2.
Besides, as histone H3 trimethylated on Lys4 (3mK4) is linked to transcriptionally active regions (Schneider, et al 2004), we next determined whether the methylated status of this histone in intron B is different in sensitive versus resistant cell line using antibodies specific for the lys4 trimethylated form of H3. In contrast to MM.1RL where 3mK4 is not detected, this modification is observed in exons 1C, 2 and 3 and to a lesser extent throughtout intron B in the sensitive cell line (Figure 6).
Histone H3 trimethylated on Lys9 (3mK9) was linked to active as well to silent genomic regions, suggesting a different role of this modification in different tissues or developmental phases (Vakoc, et al 2006). In order to test if this modification was involved in the transcriptional block found in the resistant cell we check its distribution in both cell lines. No difference in enrichment of this modified form of H3 was detected in the promoter or intronic regions between the sensitive and resistant cell lines (Figure 6).
Acetylation of lysines in the N termini of core histones is closely linked with transcriptional activation. Conversely, histone deacetylase complexes targeted to methylated promoters maintain these residues in a hypoacetylated state, inhibiting transcription on the process (Eden, et al 1998). ChIP analysis using antibodies against acetylated H3 and H4, revealed a small difference in enrichment of this modification between the two cell lines. Again, the MM.1RL cell line shows a lower degree of acetylation than the MM.1S cell line.
Discussion
In most cells tested to date, the GR is down-regulated in response to hormone treatment and the cells remain viable (Burnstein, et al 1991, Kalinyak, et al 1987, Rosewicz, et al 1988). This reduction in cellular receptor levels leads to insensitivity to subsequent hormone administrations.
Previous work involving comparative analysis of gene profiles of GC-sensitive (MM1.S) versus GC-resistant (MM1.RE and MM1.RL) myeloma cells demonstrated significantly lower levels of GR transcripts and protein expression in resistant cells (Sanchez-Vega, et al 2006). Levels of GR transcripts were comparable with the expression of total GR protein. Development of resistance correlated with an overall reduction in GR mRNA levels. The GR alpha is the predominant isoform in the sensitive cell line (MM1.S) decreasing in expression in the early resistant cells (MM1.RE) and virtually undetectable in late resistant cells (MM1.RL). The fact that overexpression of GR alpha in the resistant cell line restored sensitivity to GC further underscores the importance of this isoform of GR in these myeloma cells. Current work, which focused on the mechanisms for the gradual decrease in global GR transcripts in resistant cell lines, revealed a few points. First, the decrease of GR expression in the resistant cell line could be due to a mutation in a regulatory signal located on the promoter of the gene. Nevertheless, we sequenced the three promoters and the coding regions and we did not found any mutation that could be responsible for the differences in expression between these cell lines (data not shown).
Second, the expression of GR transcripts could be repressed by hypermethylation of GR promoters. Hence, we analyzed the likelihood of epigenetic changes in the promoters of the GR gene. However, treatment with the hypomethylating agent 5-aza-dCyd, does not change the levels of expression of full length GR mRNA (Figure 1). In addition, although a gradual decrease in full-length GR transcripts is seen during the development of resistance, the level of expression of exon 1-exon 2 RNA fragments remains the same in sensitive and resistant cell lines (Table I). This observation rules out the possibility of promoter methylation as a mechanism for the decline in GR mRNA expression in the resistant variants of the MM cell line, and suggests that an inhibition in the initiation of transcription may not be the cause for the overall reduction of GR mRNAs in resistant cell lines.
Third, the stability of the GR transcripts is partly dependent on the half-life of these transcripts, which is based on the AU-rich elements (AREs). A polymorphism composed of an A to G mutation in an ATTTA motif located in the 3′ UTR of exon 9β of the GR gene is associated with rheumatoid arthritis (Derijk, et al 2001). These motifs are found in AU-rich elements (AREs) in the 3′ UTR of short-lived mRNAs of cytokines, transcription factors and proto-oncogenes, and are often destabilizing elements for RNAs (Chen and Shyu 1995). There are four and ten AUUUA motifs in GRβ and GRα mRNAs, respectively (Schaaf and Cidlowski 2002). Because, this polymorphism in an AUUUA motif of the GRβ mRNA results in increased stability of GRβ mRNA and a small increase in GRβ protein expression, we determined the presence of any mutations that could alter the expression of the GR isoforms in our model system. There were no mutations in any of the AUUUA motifs located in both exons 9. These observations and the fact that GR mRNA has a half-life of 1–6 hours and that we are not able to detect GR transcripts in the late resistant cell line at any point but consistently obtain high levels of expression of exon 1-exon 2 fragments, rules out the possibility of a faster RNA degradation in the resistant cell line.
Fourth, as shown in the ChIP assay we can detect high levels of polymerase II throughout exon 2 to exon 3 in the sensitive cell line and although we can detect some pol II in exon 2 in the resistant cell line, this protein was almost undetectable in exon 3. In addition, elongating RNA polymerase II recruits histone methyltransferases to methylate lysines 4 and 9 of histone H3 in nucleosomes in the body of actively transcribed genes (Eissenberg and Shilatifard 2006). In agreement with this observation the ChIP assay shows a chromatin structure related to a more actively transcribed gene in GR sensitive cells than in GR resistant cells. As with other genes regulated by transcription elongation blocks, Intron B is a large exon comprising 85 Kb, however only the sequences located between 8,000 and 15,000 pb downstream of exon 2 seems to be implicated in this block.
In conclusion, all these observations point to a block on the transcriptional elongation in the second intron (Intron B) as a mechanism in the development of resistance. In this work we propose a different mechanism than the ones described until today, involved in GR mRNA regulation.
Transcript elongation is a dynamic process that does not occur at a constant rate. Throughout the elongation phase, RNA polymerase can encounter blocks to elongation from which there are three outcomes. Many paused complexes, after some period, continue chain elongation. A proportion terminates transcription, and the remainder converts to an arrested state, in which the ternary complex is intact but incapable of further transcription in the absence of accessory factors (Kerppola and Kane 1991). Few pause, arrest, or termination signals are 100% efficient. The mechanism by which sequences might block transcription is unknown, although eukaryotic DNA sequences representing transcription blocks have been identified, they appear to be characterized more by diversity than by a common theme. (Bentley and Groudine 1988, Chen, et al 1991, Connelly and Manley 1989, Dedrick, et al 1987, Keene, et al 1999, Kerppola and Kane 1988, Maa, et al 1990, Mechti, et al 1991, Meulia, et al 1992, Reines, et al 1987, Shor, et al 1995). Thus, more experiments are needed in order to find out the mechanism involved in the transcription elongation regulation of the GR gene.
Our current study used cell line as a model system which was derived from plasma cells obtained from patient with MM who was treated with dexamethasone. Later on, GC resistant variant cell lines were created by in vitro selection with dexamethasone. Recent work in primary plasma cells obtained from patients with myeloma suggested that baseline GR expression level directly correlates with the clinical outcome in 351 newly-diagnosed myeloma patients who were treated with GC-containing regimens (Ma, et al 2008). Patients with low levels of GR expression (14% of the population) were associated with significantly (p = <0.01) worse 5-year event-free survival (20% vs 52%) and overall survival (34% vs 68%) compared to those with high levels of GR expression (86% of population). It would be important to determine if the mechanism for decline in GR transcripts is similar in these primary plasma cells as in the cell lines being reported by us in the current study.
Supplementary Material
Abbreviations List
- MM
Multiple Myeloma
- GR
Glucocorticoid receptor
- GCs
Glucocorticoids
- DBD
DNA-binding domain
- LBD
ligand-binding domain
- PCR
polymerase chain reaction
- RT-PCR
reverse transcription-polymerase chain reaction
- PMSF
phenyl methyl sulforyl fluoride
- FBS
fetal bovine serum
- PBS
phosphate buffered saline
- FITC
fluorescein isothiocyanate
- PI
propidium iodide
- bp
base pairs
- WT
wild type
Footnotes
Supported by Grant CA85915 from the National Cancer Institute.
References
- Adcock IM, Nasuhara Y, Stevens DA, Barnes PJ. Ligand-induced differentiation of glucocorticoid receptor (GR) trans-repression and transactivation: preferential targetting of NF-kappaB and lack of I-kappaB involvement. Br J Pharmacol. 1999;127:1003–1011. doi: 10.1038/sj.bjp.0702613. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Antonini SR, Latronico AC, Elias LL, Cukiert A, Machado HR, Liberman B, Mendonca BB, Moreira AC, Castro M. Glucocorticoid receptor gene polymorphisms in ACTH-secreting pituitary tumours. Clin Endocrinol (Oxf) 2002;57:657–662. doi: 10.1046/j.1365-2265.2002.01639.x. [DOI] [PubMed] [Google Scholar]
- Bentley DL, Groudine M. Sequence requirements for premature termination of transcription in the human c-myc gene. Cell. 1988;53:245–256. doi: 10.1016/0092-8674(88)90386-8. [DOI] [PubMed] [Google Scholar]
- Bray PJ, Cotton RG. Variations of the human glucocorticoid receptor gene (NR3C1): pathological and in vitro mutations and polymorphisms. Hum Mutat. 2003;21:557–568. doi: 10.1002/humu.10213. [DOI] [PubMed] [Google Scholar]
- Breslin MB, Geng CD, Vedeckis WV. Multiple promoters exist in the human GR gene, one of which is activated by glucocorticoids. Mol Endocrinol. 2001;15:1381–1395. doi: 10.1210/mend.15.8.0696. [DOI] [PubMed] [Google Scholar]
- Breslin MB, Vedeckis WV. The human glucocorticoid receptor promoter upstream sequences contain binding sites for the ubiquitous transcription factor, Yin Yang 1. J Steroid Biochem Mol Biol. 1998;67:369–381. doi: 10.1016/s0960-0760(98)00138-1. [DOI] [PubMed] [Google Scholar]
- Burnstein KL, Bellingham DL, Jewell CM, Powell-Oliver FE, Cidlowski JA. Autoregulation of glucocorticoid receptor gene expression. Steroids. 1991;56:52–58. doi: 10.1016/0039-128x(91)90124-e. [DOI] [PubMed] [Google Scholar]
- Burnstein KL, Jewell CM, Sar M, Cidlowski JA. Intragenic sequences of the human glucocorticoid receptor complementary DNA mediate hormone-inducible receptor messenger RNA down-regulation through multiple mechanisms. Mol Endocrinol. 1994;8:1764–1773. doi: 10.1210/mend.8.12.7708063. [DOI] [PubMed] [Google Scholar]
- Cairns C, Cairns W, Okret S. Inhibition of gene expression by steroid hormone receptors via a negative glucocorticoid response element: evidence for the involvement of DNA-binding and agonistic effects of the antiglucocorticoid/antiprogestin RU486. DNA Cell Biol. 1993;12:695–702. doi: 10.1089/dna.1993.12.695. [DOI] [PubMed] [Google Scholar]
- Chauhan D, Auclair D, Robinson EK, Hideshima T, Li G, Podar K, Gupta D, Richardson P, Schlossman RL, Krett N, Chen LB, Munshi NC, Anderson KC. Identification of genes regulated by dexamethasone in multiple myeloma cells using oligonucleotide arrays. Oncogene. 2002;21:1346–1358. doi: 10.1038/sj.onc.1205205. [DOI] [PubMed] [Google Scholar]
- Chauhan D, Pandey P, Hideshima T, Treon S, Raje N, Davies FE, Shima Y, Tai YT, Rosen S, Avraham S, Kharbanda S, Anderson KC. SHP2 mediates the protective effect of interleukin-6 against dexamethasone-induced apoptosis in multiple myeloma cells. J Biol Chem. 2000;275:27845–27850. doi: 10.1074/jbc.M003428200. [DOI] [PubMed] [Google Scholar]
- Chen CY, Shyu AB. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- Chen Z, Innis JW, Sun MH, Wright DA, Kellems RE. Sequence requirements for transcriptional arrest in exon 1 of the human adenosine deaminase gene. Mol Cell Biol. 1991;11:6248–6256. doi: 10.1128/mcb.11.12.6248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly S, Manley JL. RNA polymerase II transcription termination is mediated specifically by protein binding to a CCAAT box sequence. Mol Cell Biol. 1989;9:5254–5259. doi: 10.1128/mcb.9.11.5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costlow ME, Pui CH, Dahl GV. Glucocorticoid receptors in childhood acute lymphocytic leukemia. Cancer Res. 1982;42:4801–4806. [PubMed] [Google Scholar]
- Dedrick RL, Kane CM, Chamberlin MJ. Purified RNA polymerase II recognizes specific termination sites during transcription in vitro. J Biol Chem. 1987;262:9098–9108. [PubMed] [Google Scholar]
- Derijk RH, Schaaf MJ, Turner G, Datson NA, Vreugdenhil E, Cidlowski J, de Kloet ER, Emery P, Sternberg EM, Detera-Wadleigh SD. A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor beta-isoform mRNA is associated with rheumatoid arthritis. J Rheumatol. 2001;28:2383–2388. [PubMed] [Google Scholar]
- Dong Y, Poellinger L, Gustafsson JA, Okret S. Regulation of glucocorticoid receptor expression: evidence for transcriptional and posttranslational mechanisms. Mol Endocrinol. 1988;2:1256–1264. doi: 10.1210/mend-2-12-1256. [DOI] [PubMed] [Google Scholar]
- Drouin J, Charron J, Gagner JP, Jeannotte L, Nemer M, Plante RK, Wrange O. Pro-opiomelanocortin gene: a model for negative regulation of transcription by glucocorticoids. J Cell Biochem. 1987;35:293–304. doi: 10.1002/jcb.240350404. [DOI] [PubMed] [Google Scholar]
- Eden S, Hashimshony T, Keshet I, Cedar H, Thorne AW. DNA methylation models histone acetylation. Nature. 1998;394:842. doi: 10.1038/29680. [DOI] [PubMed] [Google Scholar]
- Eissenberg JC, Shilatifard A. Leaving a mark: the many footprints of the elongating RNA polymerase II. Curr Opin Genet Dev. 2006;16:184–190. doi: 10.1016/j.gde.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Goldman-Leikin RE, Salwen HR, Herst CV, Variakojis D, Bian ML, Le Beau MM, Selvanayagan P, Marder R, Anderson R, Weitzman S, et al. Characterization of a novel myeloma cell line, MM.1. J Lab Clin Med. 1989;113:335–345. [PubMed] [Google Scholar]
- Hoeck W, Rusconi S, Groner B. Down-regulation and phosphorylation of glucocorticoid receptors in cultured cells. Investigations with a monospecific antiserum against a bacterially expressed receptor fragment. J Biol Chem. 1989;264:14396–14402. [PubMed] [Google Scholar]
- Iacobelli S, Marchetti P, De Rossi G, Mandelli F, Gentiloni N. Glucocorticoid receptors predict response to combination chemotherapy in patients with acute lymphoblastic leukemia. Oncology. 1987;44:13–16. doi: 10.1159/000226435. [DOI] [PubMed] [Google Scholar]
- Kalinyak JE, Dorin RI, Hoffman AR, Perlman AJ. Tissue-specific regulation of glucocorticoid receptor mRNA by dexamethasone. J Biol Chem. 1987;262:10441–10444. [PubMed] [Google Scholar]
- Keene RG, Mueller A, Landick R, London L. Transcriptional pause, arrest and termination sites for RNA polymerase II in mammalian N- and c-myc genes. Nucleic Acids Res. 1999;27:3173–3182. doi: 10.1093/nar/27.15.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola TK, Kane CM. Intrinsic sites of transcription termination and pausing in the c-myc gene. Mol Cell Biol. 1988;8:4389–4394. doi: 10.1128/mcb.8.10.4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola TK, Kane CM. RNA polymerase: regulation of transcript elongation and termination. Faseb J. 1991;5:2833–2842. doi: 10.1096/fasebj.5.13.1916107. [DOI] [PubMed] [Google Scholar]
- Ma S, Zhan F, Ma C, Tessel M, Zhang L, Rajkumar V, Singhal S, Barlogie B, Shaughnessy JD, Jr, Krett N, Rosen ST. Glucocorticoid receptor expression correlates with the clinical outcome in myeloma patients treated with glucocorticoid-containing regimens. Blood. 2008 in press. [Google Scholar]
- Maa MC, Chinsky JM, Ramamurthy V, Martin BD, Kellems RE. Identification of transcription stop sites at the 5′ and 3′ ends of the murine adenosine deaminase gene. J Biol Chem. 1990;265:12513–12519. [PubMed] [Google Scholar]
- Mastrangelo R, Malandrino R, Riccardi R, Longo P, Ranelletti FO, Iacobelli S. Clinical implications of glucocorticoid receptor studies in childhood acute lymphoblastic leukemia. Blood. 1980;56:1036–1040. [PubMed] [Google Scholar]
- McIntyre WR, Samuels HH. Triamcinolone acetonide regulates glucocorticoid-receptor levels by decreasing the half-life of the activated nuclear-receptor form. J Biol Chem. 1985;260:418–427. [PubMed] [Google Scholar]
- Mechti N, Piechaczyk M, Blanchard JM, Jeanteur P, Lebleu B. Sequence requirements for premature transcription arrest within the first intron of the mouse c-fos gene. Mol Cell Biol. 1991;11:2832–2841. doi: 10.1128/mcb.11.5.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulia T, Krumm A, Spencer C, Groudine M. Sequences in the human c-myc P2 promoter affect the elongation and premature termination of transcripts initiated from the upstream P1 promoter. Mol Cell Biol. 1992;12:4590–4600. doi: 10.1128/mcb.12.10.4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moalli PA, Pillay S, Weiner D, Leikin R, Rosen ST. A mechanism of resistance to glucocorticoids in multiple myeloma: transient expression of a truncated glucocorticoid receptor mRNA. Blood. 1992;79:213–222. [PubMed] [Google Scholar]
- Moalli PA, Rosen ST. Glucocorticoid receptors and resistance to glucocorticoids in hematologic malignancies. Leuk Lymphoma. 1994;15:363–374. doi: 10.3109/10428199409049738. [DOI] [PubMed] [Google Scholar]
- Pedersen KB, Vedeckis WV. Quantification and glucocorticoid regulation of glucocorticoid receptor transcripts in two human leukemic cell lines. Biochemistry. 2003;42:10978–10990. doi: 10.1021/bi034651u. [DOI] [PubMed] [Google Scholar]
- Platt T. Transcription termination and the regulation of gene expression. Annu Rev Biochem. 1986;55:339–372. doi: 10.1146/annurev.bi.55.070186.002011. [DOI] [PubMed] [Google Scholar]
- Pui CH, Dahl GV, Rivera G, Murphy SB, Costlow ME. The relationship of blast cell glucocorticoid receptor levels to response to single-agent steroid trial and remission response in children with acute lymphoblastic leukemia. Leuk Res. 1984;8:579–585. doi: 10.1016/0145-2126(84)90006-7. [DOI] [PubMed] [Google Scholar]
- Quddus FF, Leventhal BG, Boyett JM, Pullen DJ, Crist WM, Borowitz MJ. Glucocorticoid receptors in immunological subtypes of childhood acute lymphocytic leukemia cells: a Pediatric Oncology Group Study. Cancer Res. 1985;45:6482–6486. [PubMed] [Google Scholar]
- Reines D, Wells D, Chamberlin MJ, Kane CM. Identification of intrinsic termination sites in vitro for RNA polymerase II within eukaryotic gene sequences. J Mol Biol. 1987;196:299–312. doi: 10.1016/0022-2836(87)90691-7. [DOI] [PubMed] [Google Scholar]
- Rosewicz S, McDonald AR, Maddux BA, Goldfine ID, Miesfeld RL, Logsdon CD. Mechanism of glucocorticoid receptor down-regulation by glucocorticoids. J Biol Chem. 1988;263:2581–2584. [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vega B, Krett N, Rosen ST, Gandhi V. Glucocorticoid receptor transcriptional isoforms and resistance in multiple myeloma cells. Mol Cancer Ther. 2006;5:3062–3070. doi: 10.1158/1535-7163.MCT-06-0344. [DOI] [PubMed] [Google Scholar]
- Schaaf MJ, Cidlowski JA. AUUUA motifs in the 3′UTR of human glucocorticoid receptor alpha and beta mRNA destabilize mRNA and decrease receptor protein expression. Steroids. 2002;67:627–636. doi: 10.1016/s0039-128x(02)00015-6. [DOI] [PubMed] [Google Scholar]
- Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6:73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- Shor J, Ben-Asher E, Aloni Y. Transcription elongation of the murine ornithine decarboxylase (ODC) gene is regulated in vitro at two downstream elements by different attenuation mechanisms. Oncogene. 1995;10:1587–1596. [PubMed] [Google Scholar]
- Uptain SM, Kane CM, Chamberlin MJ. Basic mechanisms of transcript elongation and its regulation. Annu Rev Biochem. 1997;66:117–172. doi: 10.1146/annurev.biochem.66.1.117. [DOI] [PubMed] [Google Scholar]
- Vakoc CR, Sachdeva MM, Wang H, Blobel GA. Profile of histone lysine methylation across transcribed mammalian chromatin. Mol Cell Biol. 2006;26:9185–9195. doi: 10.1128/MCB.01529-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderbilt JN, Miesfeld R, Maler BA, Yamamoto KR. Intracellular receptor concentration limits glucocorticoid-dependent enhancer activity. Mol Endocrinol. 1987;1:68–74. doi: 10.1210/mend-1-1-68. [DOI] [PubMed] [Google Scholar]
- Vedeckis WV, Ali M, Allen HR. Regulation of glucocorticoid receptor protein and mRNA levels. Cancer Res. 1989;49:2295s–2302s. [PubMed] [Google Scholar]
- Yamamoto KR, Alberts BM. Steroid receptors: elements for modulation of eukaryotic transcription. Annu Rev Biochem. 1976;45:721–746. doi: 10.1146/annurev.bi.45.070176.003445. [DOI] [PubMed] [Google Scholar]
- Yudt MR, Cidlowski JA. The glucocorticoid receptor: coding a diversity of proteins and responses through a single gene. Mol Endocrinol. 2002;16:1719–1726. doi: 10.1210/me.2002-0106. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.