

Abstract
The RCA (Roseobacter clade affiliated) cluster, with an internal 16S rRNA gene sequence similarity of >98%, is the largest cluster of the marine Roseobacter clade and most abundant in temperate to (sub)polar oceans, constituting up to 35% of total bacterioplankton. The genome analysis of the first described species of the RCA cluster, Planktomarina temperata RCA23, revealed that this phylogenetic lineage is deeply branching within the Roseobacter clade. It shares not >65.7% of homologous genes with any other organism of this clade. The genome is the smallest of all closed genomes of the Roseobacter clade, exhibits various features of genome streamlining and encompasses genes for aerobic anoxygenic photosynthesis (AAP) and CO oxidation. In order to assess the biogeochemical significance of the RCA cluster we investigated a phytoplankton spring bloom in the North Sea. This cluster constituted 5.1% of the total, but 10–31% (mean 18.5%) of the active bacterioplankton. A metatranscriptomic analysis showed that the genome of P. temperata RCA23 was transcribed to 94% in the bloom with some variations during day and night. The genome of P. temperata RCA23 was also retrieved to 84% from metagenomic data sets from a Norwegian fjord and to 82% from stations of the Global Ocean Sampling expedition in the northwestern Atlantic. In this region, up to 6.5% of the total reads mapped on the genome of P. temperata RCA23. This abundant taxon appears to be a major player in ocean biogeochemistry.
Introduction
Our understanding of the role of abundant individual taxa in ocean biogeochemistry is hampered by the fact that so far very few of such taxa have been isolated and are available to genomic and postgenomic analyses (Yooseph et al., 2010). It is extremely difficult to obtain representative isolates of the major players and only novel approaches made this effort more successful (Giovannoni and Stingl, 2007). However, still today detailed information on the role of abundant individual taxa in oceanic cycling of matter is available only for Prochlorococcus and for Cand. Pelagibacter ubique of the SAR11 clade. Pelagibacter ubique of the SAR11 clade (Giovannoni et al., 2005; Tripp et al., 2008; Sowell et al., 2009; Thompson et al., 2011). Isolates of these taxa are available, their genomes have been sequenced and thus form a basis for postgenomic studies and relating metagenomic and, more importantly, metatranscriptomic and metaproteomic information to individual taxa.
The Roseobacter and SAR11 clades are the most prominent subdivisions of Alphaproteobacteria in the ocean's near surface waters (Giovannoni and Stingl, 2005). The RCA (Roseobacter clade affiliated) cluster with an internal sequence similarity of the 16S rRNA gene of at least 98%, constitutes up to 35% of total bacterioplankton and is most abundant in temperate to (sub)polar oceans, but absent in tropical and subtropical regions (Selje et al., 2004; Giebel et al., 2009; Giebel et al., 2011). It is divided into a subcluster with sequences from temperate regions and one with sequences of subpolar and polar origin (Giebel et al., 2011). Because of the high abundance and distinct biogeography of the RCA cluster, there is great interest to elucidate its functional role and biogeochemical significance. However, no information on the genomic data and features of organisms of this important cluster are yet available because RCA organisms defy isolation and cultivation greatly.
An isolate of this cluster, retrieved from the southern North Sea, became recently available and was characterized as type species of the first described species of the genus Planktomarina, Planktomarina temperata RCA23 (Giebel et al., 2013). In the North Sea, P. temperata RCA23 represents the most abundant ribotype of the RCA cluster that comprises persistently between 2% and 20% of total bacterioplankton (Selje et al., 2004; Giebel et al., 2011; Teeling et al., 2012) and is a major representative of the Roseobacter clade in the active bacterioplankton (Wemheuer et al., 2014). Based on these observations we hypothesized that the genome of this organism exhibits distinct features of its adaptation to life in the nutrient-poor pelagic environment with respect to genome organization and metabolic potential. Further, we hypothesized that the genome of P. temperata RCA23 is well represented in metagenomic and metatranscriptomic data in the North Sea because of its high abundance and activity. Therefore, we sequenced the genome of P. temperata RCA23 to elucidate its metabolic potential. In addition, we assessed its significance and functional role during a phytoplankton spring bloom in the southern North Sea by applying a metatranscriptomic approach. Finally, metagenomic data sets of pelagic marine systems were mined for the presence, abundance and genomic features of this organism.
Materials and methods
Origin and growth of P. temperata RCA23
P. temperata RCA23 was originally isolated from a water sample collected in the southern North Sea (Giebel et al., 2011, 2013). It was grown in liquid culture of autoclaved sea water amended with marine broth (40% of peptone and yeast extract; Giebel et al., 2013) to an optical density of 0.2. Biomass was harvested by 20 min centrifugation (Beckman JA10, Krefeld, Germany) at 7500 r.p.m. and 4 °C and DNA was extracted using the MasterPure DNA Purification Kit (Epicentre, Madison, Wisconsin, USA) according to the manufacturer's protocol.
Sequence determination, gene annotation and phylogenetic analyses
To sequence the genome of P. temperata RCA23 a pyrosequencing run was performed using a Roche GS-FLX 454 sequencer (Branford, CT, USA) with Titanium chemistry. All sequencing steps were performed according to the manufacturer's protocols and recommendations. In total 411 932 reads were generated and assembled to 78 contigs bigger than 500 bp with a 26-fold coverage. Furthermore, 576 fosmid Sanger-sequences were added to the data set to identify the contig order. Gap closure and polishing were carried out using the Staden software package (Staden, 1996) and PCR-based techniques on genomic DNA. Open reading frames were identified using YACOP (Tech and Merkl, 2003) and GLIMMER (Delcher et al., 2007). The open reading frame finding was inspected manually with Artemis and open reading frames were corrected by checking the GC frame plot, ribosomal binding site and blast hits against the NCBI nr database.
Orthologous protein sequences were identified using reciprocal BLASTp-analysis combined with global alignments based on the Needleman–Wunsch algorithm. Only hits with e-values <1e−20 were considered and additionally filtered, based on sequence identity cutoffs of the respective global alignments. A cutoff value of 30% sequence identity was chosen to identify orthologs. For multilocus sequence analysis, protein sequences of 162 genes, for which one ortholog but no paralog was found in every comparison strain, were concatenated. The multilocus sequence analysis tree was constructed using ARB v5.1 (Ludwig et al., 2004) and the evolutionary history was inferred using the neighbor-joining method and recovered reproducibly with the maximum-likelihood method (Figure 1).
Figure 1.

Neighbor-joining tree based on multilocus sequence analysis (MLSA) of genome-sequenced organisms of the Roseobacter clade. The tree was constructed using ARB v5.1 and is based on a similar tree of Newton et al. (2010) but includes additional genome sequences and an extended gene set. Filled circles indicate nodes also recovered reproducibly with maximum-likelihood calculation. Numbers at the nodes are bootstrap values (only >50% are shown) from 1000 replicates. Subclades of the Roseobacter clade are marked by different colors. (T) indicates type strains. Escherichia coli K12 MG1655 was used as outgroup.
Study area, sample collection and chlorophyll
The significance of the RCA cluster was investigated during a phytoplankton spring bloom in the southern North Sea. Samples were collected at 11 stations at 2 m depth between 25 and 31 May 2010 on board RV Heincke by 4 l Niskin bottles mounted on a CTD rosette (Sea-Bird, Bellevue, WA, USA). For pyrosequencing, metagenomic and metatranscriptomic analyses, 50 l of sea water were prefiltered through a 10-μm nylon net and a filter sandwich consisting of a precombusted glass fiber and 3-μm polycarbonate filter (47 mm diameter). Bacterioplankton was harvested from a prefiltered 1-liter sample on a filter sandwich consisting of a glass fiber and 0.2-μm polycarbonate filter (47 mm). One filter sandwich was used for RNA extraction. For gene expression analysis and metagenome sequencing, at least four filter sandwiches were subjected to RNA and DNA extraction. Chlorophyll a concentrations were determined as described (Giebel et al., 2011).
Enumeration of bacteria, CARD-FISH and BrdU-FISH
Bacterial cell numbers were determined by flow cytometry after staining of subsamples with SybrGreen I (Sigma-Aldrich, Munich, Germany), preserved with glutardialdehyde (final concentration 1%) and stored at −80 °C until further analysis as described Giebel et al. (2011). The abundance of RCA cells was determined by catalyzed reporter deposition fluorescence in situ hybridization (CARD-FISH) and the proportion of DNA-synthesizing RCA cells by incorporation of BrdU (4 h incubation) according to Pernthaler et al. (2002) and applying an RCA-specific probe set (RCA996, C Beardsley and I Bakenhus, unpublished data). Cells were counted via epifluorescence microscopy using × 1000 magnification and suitable filter sets for DAPI-stained total cells, Alexa488-stained RCA cells, and Cy3-stained (Perkin Elmer, Waltham, MA, USA) BrdU-positive cells, respectively.
16S rRNA gene amplicons, metagenomics and transcriptomics
Environmental DNA and RNA were coextracted and the composition of the active bacterial community was assessed by 16S rRNA PCRs as described before Wemheuer et al. (2014). For metagenomic and metatranscriptomic analyses DNA and cDNA were sequenced on an Illumina/Solexa GAIIx system (San Diego, CA, USA). In total, 54 334 282 paired-end sequences of 100 bp were generated for the metagenomic and 78 042 122 single-read sequences of 75–100 bp for the metatranscriptomic data sets, respectively (Supplementary Table S1). The sequences were quality trimmed and the Illumina adapter sequences were removed with Trimmomatic v0.30 (Bolger et al., 2014) using the following parameters: adapter:2:40:15 leading:3 trailing:3 slidingwindow:4:15 minlen:50. Reads derived from ribosomal RNA gene fragments were filtered with SortMeRNA (Kopylova et al., 2012). The remaining sequences were mapped with Bowtie 2 (Langmead and Salzberg, 2012) using the implemented end-to-end mode, which requires that the entire read align from one end to the other.
To compare the RNA-Seq results, the read counts were normalized to remove biases like the length of the transcript and the sequencing depth of a sample. We used the Nucleotide activity Per Kilobase of exon model per million Mapped reads (NPKM), a derivate of RPKM (reads per kilo base per million), as a normalized read count value (Wiegand et al., 2013).
Data deposition
The genome sequence of P. temperata RCA23 has been deposited at the NCBI GenBank database with the accession number CP003984. The metagenomic and metatranscriptomic data sets have been deposited at MG-Rast with the accession numbers: 4548721.3 (Station 3 gDNA), 4550305.3 (Station 3 cDNA), 4548722.3 (Station 9 gDNA), 4550303.3 (Station 9 cDNA), 4548723.3 (Station 13 gDNA) and 4550304.3 (Station 13 cDNA).
Results and discussion
Genomic features of P. temperata RCA23
P. temperata RCA23 is deeply branching within the Roseobacter clade, not affiliated to any of the known subclades, as shown by multilocus sequence analysis (Figure 1). The genome of P. temperata RCA23 encompasses 3.29 Mbp and carries 3101 genes of which 3054 encode proteins (Supplementary Table S2). It is the smallest of all closed and the third smallest of all genomes of the Roseobacter clade and carries the second lowest number of genes of all organisms of this clade (Figure 2a and Supplementary Table S2). The only organism of this clade with a significantly smaller genome is Rhodobacterales bacterium HTCC2255 (Luo et al., 2013). Further, partial genomes of two other organisms of the Roseobacter clade with estimated genome sizes of <3 Mbp were recently reported from sequence analyses of single amplified genomes retrieved from coastal and oceanic surface waters (Swan et al., 2013; Luo et al., 2014). Members of the Roseobacter clade with a genome size of <3.8 Mbp are exclusively of pelagic origin (Figure 2a and Supplementary Table S2). The mean genome sizes of the pelagic members of this clade is 4.06 Mbp and 0.5 Mbp smaller than that of members associated to other organisms, surfaces or sediments (Figure 2b and Supplementary Table S3). The relatively small genome of P. temperata RCA23 and other pelagic members of the Roseobacter clade and the reduction in genomic traits is consistent with genomic streamlining and adaptation to nutrient-poor pelagic marine ecosystems (see below; Giovannoni et al., 2005; Luo et al., 2013; Swan et al., 2013). The genomic difference between both groups of the Roseobacter clade obviously reflects the divergent evolutionary history of this clade with genomic streamlining of at least several pelagic members and gained genomic content by the associated members (Luo et al., 2013). The much larger genome size of P. temperata RCA23 and other pelagic members of the Roseobacter clade, however, harbour genomes much larger than highly streamlined marine pelagic bacteria such as Cand. P. ubique of the SAR11 clade. This feature points out that, besides streamlining, distinct aspects of their life history and adaptation to their niche in the marine pelagic realm were also important in shaping the evolution of these pelagic members of the Roseobacter clade (Giovannoni et al., 2014). These authors have identified a low number of σ-factors as a feature to distinguish between streamlined and non-streamlined genomes of bacteria with a more complex life style. The genome of P. temperata RCA23 harbors six σ-factors, a value much lower than that of other members of the Roseobacter clade and in the same range as that of many typical streamlined genomes of a size of <2 Mb (Giovannoni et al., 2014).
Figure 2.

Genomic traits of P. temperata RCA23 in comparison to other organisms of the Roseobacter clade. Relation between genome size and number of genes (a). Box–Whisker plot of the GC content (b), percentage coding genes (c) and CDS (coding DNA sequences) count (d) of 14 members of the Roseobacter clade associated to other organisms, surfaces or sediments and of 24 members with a pelagic life style listed in Supplementary Table S2. Data include 12 closed and 26 draft genomes. Blue circle and +: P. temperata RCA23. The boxes show the median (solid line), mean (dashed line), the 25th and 75th percentile and the whiskers the 10th and 90th percentiles. •: Outlayers. For further details see Supplementary Tables S2 and S3.
The GC content of 54% of P. temperata RCA23 is lower than that of all associated (mean=60.07±3.58%, N=14) and the fifth lowest of the 24 pelagic members of the Roseobacter clade included in this analysis (mean=59.28±6.82% Supplementary Tables S2 and S3). However it is substantially higher than the genomic GC content of many other marine bacteria, ranging below 40%, such as Rhodobacterales bacterium HTCC2255 (Supplementary Table S2), Cand. P. ubique and pelagic bacteria subjected to single cell genome sequencing (Swan et al., 2013; Luo et al., 2014). It has been speculated that a reduced genomic GC content may be an adaptation to nitrogen limitation in marine systems (Swan et al., 2013), but clear-cut evidence is still missing, also because it is still unclear whether pelagic heterotrophic bacteria are carbon- or nitrogen-limited. The percentage of coding DNA of P. temperata RCA23 is very similar to the mean of all pelagic members of the Roseobacter clade which, however, is significantly higher than the mean of the organisms of this clade associated to other organisms, surfaces or sediment (Figure 2c and Supplementary Table S3).
The genome of P. temperata RCA23 shares not >65.7% of homologous genes (2009) with other members of the Roseobacter clade (Supplementary Tables S2 and S4). It harbors no prophage, which is consistent with the fact that prophage induction using mitomycin C and UV light was unsuccessful (H-A Giebel, unpublished data). Further, the genome carries no plasmid, no complete GTA (gene transfer agent) cluster (Supplementary Table S4) and no CRISPR (clustered regularly interspaced small palindromic repeats). CRISPR are uncommon in the genomes of most organisms of the Roseobacter clade and were so far detected in only two members of this clade, Dinoroseobacter shibae and Maritimibacter alkaliphilus HTCC2654. The lack of plasmids in the genome of P. temperata RCA23 and other pelagic members of this clade with a genome size of <3.5 Mb (Supplementary Table S2) appears to be an adaptation to the pelagic life style and exhibits another feature of streamlining not considered yet in other analyses. In contrast, plasmids are common genomic elements of all members of the Roseobacter clade associated to other organisms, surfaces and sediments and constitute between 2% and 33% of the genomic content (Supplementary Table S2; Pradella et al., 2010).
GTAs have been detected in Alphaproteobacteria and all genomes of the Roseobacter clade except in Rhodobacterales bacteria HTCC2083 and HTCC2255 (Zhao et al., 2009; Newton et al., 2010). GTA-related gene transfer was suggested as a potential adaptation mechanism of these bacteria to maintain the metabolic flexibility in the dynamic marine environment (Biers et al., 2008). The fact that the genome of P. temperata RCA23 encodes only three putative GTA-related genes (c18030, c18040 and c18050; Supplementary Table S4) implies that this mode of gene transfer was discarded during adaptation to the pelagic life style, presumably because of little benefits by this type of genetic exchange in the nutrient-poor pelagic environment. Hence, the mentioned missing genomic features, the absence of plasmids and GTA in the genome of P. temperata RCA23 indicate a less diversified genomic organization as compared to other organisms of the Roseobacter clade and suggests reduced genetic exchange with other members of this clade.
In comparison to typical groups of other Roseobacter clade organisms (Newton et al., 2010) 10 genomic islands (GIs), harboring 22.6% of all genes, are present in the genome of P. temperata RCA23 (Supplementary Figure S1). This bacterium encodes genes for chemotaxis, possesses a monotrichous flagellum and is motile (Table 1; Giebel et al., 2013). Phylogenetic analysis of the flagella synthesis cluster present in GI 3 shows that the flagellar gene sets of the Roseobacter clade are divided into two distinct groups (Supplementary Figure S2). The flagella genes of the majority of the Roseobacter clade organisms fall into group I, but those of P. temperata RCA23 into group II. This group contains relatively few sequences of Roseobacter clade members but additionally includes strains of the SAR116 clade and of Rhodobacter sphaeroides. The existence of these two groups of flagella gene clusters obviously reflects the evolutionary history of the Roseobacter clade with substantial lateral gene transfer (Luo et al., 2013). GI 5 harbours the genes for the group I CO dehydrogenase (coxI; Supplementary Table S4). The existence of the coxI gene cluster enables members of the Roseobacter clade to oxidize CO (Cunliffe, 2011).
Table 1. Comparison of selected biosynthetic/catabolic genes and pathways of representative members of the Roseobacter clade.
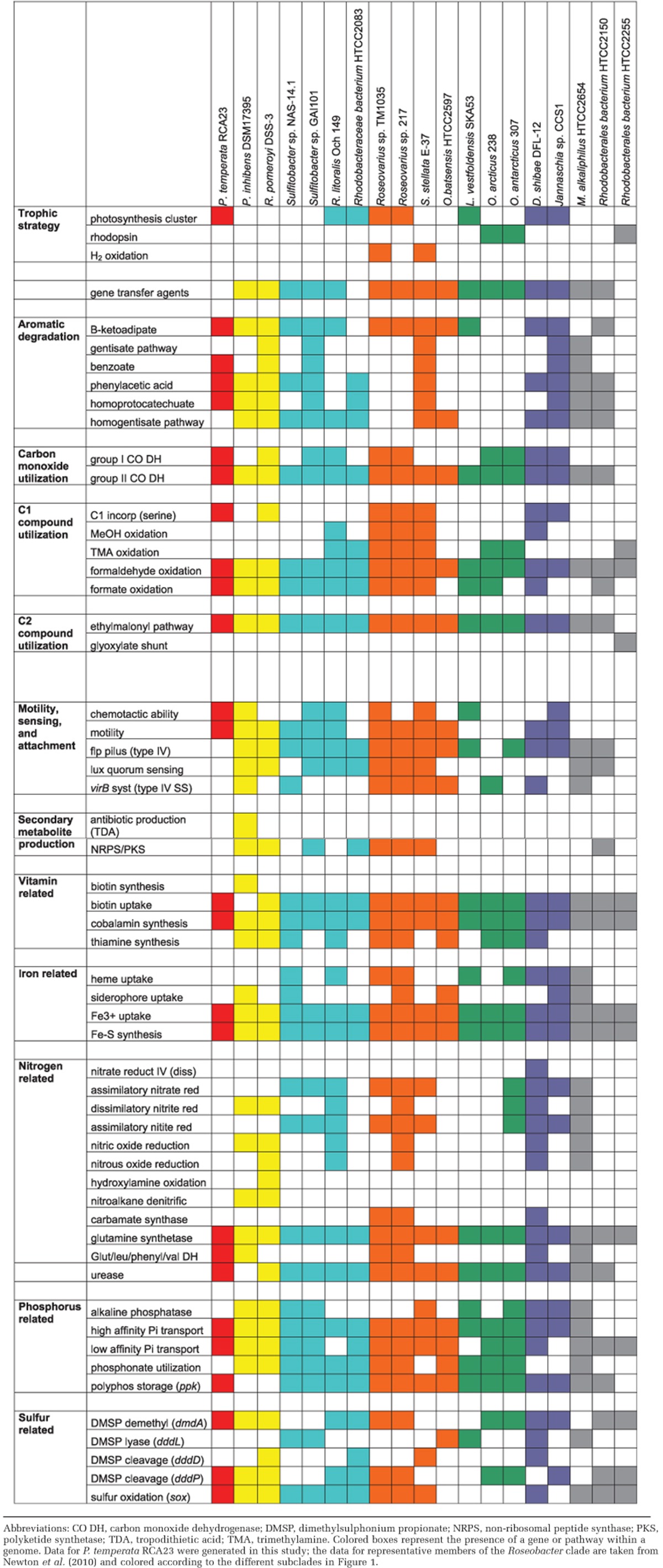
The genome of P. temperata RCA23 encodes all basic metabolic functions. Carbohydrates are metabolized via the Entner–Doudoroff pathway that appears typical for the Roseobacter clade and also for the SAR11 clade (Giovannoni et al., 2005; Fuerch et al., 2009). Dimethylsulphonium propionate can be degraded via the demethylation and the cleavage pathway (Table 1 and Supplementary Table S4) and enhances growth of P. temperata RCA23 when added additionally to the medium (Giebel et al., 2013). This organism is able to carry out AAP (Giebel et al., 2013). The organization of the photosynthetic operon, however, differs from that of other members of the Roseobacter clade (Supplementary Figure S3 and Supplementary Table S4). In accordance to many other Roseobacter clade organisms, nitrogen can only be taken up in reduced form as the genome harbors no genes encoding enzymes for reducing nitrate and nitrite, but gene clusters encoding enzymes for uptake of ammonium, amino acids, putrescine/spermidine and to utilize urea (Table 1 and Supplementary Table S4). These genomic features are in line with growth tests of P. temperata RCA23 (Giebel et al., 2013). Whether oxidized sulfur compounds can be reduced is unclear because a putative gene cluster for assimilatory sulfate reduction exists (c14410-c14360), but lacks the typical cysH gene known from other organisms of the Roseobacter clade. Instead a smaller variant harboring a phosphoadenosine phosphosulfate reductase domain was found (c14380).
The genome of P. temperata RCA23 encodes 287 transport proteins, a number lower than the mean of the pelagic members of the Roseobacter clade (Supplementary Figure S4A and B; Supplementary Tables S2 and S3). A total of 208 transport proteins of P. temperata RCA23 belongs to the ATP-binding cassette (ABC) family (Supplementary Figure S4B and C; Supplementary Table S2). This number is lower than the mean of the pelagic members of the Roseobacter clade, whereas the proportion of ABC transporters of P. temperata RCA23 (72.5%) is very close to and the number of ABC transporters per Mb of P. temperata RCA23 (63.2) identical to the mean of all pelagic members (Supplementary Table S2). Interestingly, the other two deeply branching pelagic members of the Roseobacter clade, Rhodobacterales bacteria HTCC2255 and HTCC 2150, have the lowest numbers of ABC transporters per Mb of all roseobacters, 47.6 and 50.2, values close to that of Cand. P. ubique, 51.1 (Supplementary Table S2). The mean of the absolute numbers of ABC transporters of the pelagic members is significantly lower than that of the associated Roseobacter clade members (Student's t-test; P=0.014; Supplementary Table S3), presumably a result of the different evolutionary history of both groups of this clade with respect to gene acquisition and loss (Luo et al., 2013). A high proportion of ABC transporters, which exhibit high substrate affinities, was interpreted as an adaptation of pelagic bacteria to the nutrient-poor oceanic environment (Giovannoni et al., 2005; Lauro et al., 2009). As all members of the Roseobacter clade exhibit a rather high percentage of ABC transporters, this feature may not be an adaptation to the nutrient-poor oceanic environment. It may also reflect the generally highly dynamic nutrient supply to members of this clade, considered as opportunistic marine bacteria and being able to adapt to a variety of nutrient conditions (Brinkhoff et al., 2008; Teeling et al., 2012; Luo et al., 2013). The number of tripartite ATP-independent periplasmic transporters of P. temperata RCA23 is lower than the mean of the pelagic as well as associated members of the Roseobacter clade (Figure 3d and Supplementary Table S3). The great majority of tripartite ATP-independent periplasmic transporters of P. temperata RCA23 mediates uptake of dicarboxylic acids (Supplementary Table S4). Physiological tests showed that P. temperata RCA23 is able to grow on a large variety of organic substrates including amino acids, monosaccharides and short-chain fatty acids (Giebel et al., 2013). This high versatility appears to reflect the high number of transporter proteins.
Figure 3.

(a) Bathymetry of the southern North Sea (German Bight) and sampling stations in May 2010. Arrows indicate stations where the samples for the metagenomic and transcriptomic analyses were collected. (b) Chlorophyll a, bacterial abundance, salinity and temperature at 2 m depth. (c) Percentages of the RCA cluster as detected by CARD-FISH with an RCA-specific probe, of DNA-synthesizing RCA cells (BrdU positive) and of active RCA (cDNA). Samples in (b) and (c) are ordered as inside and outside the bloom except station 1, which was a separate bloom in the southernmost area. *: Missing data.
Even though quite a few genomic features are similar to those of other members of the Roseobacter clade, the genome of P. temperata RCA23 is distinct in lacking genes encoding the Flp pilus (type IV) for attachment, the VirB system for discharge of genetic material and protein, and genes encoding enzymes for quorum sensing and synthesizing other secondary metabolites (Table 1). The only other organism of the Roseobacter clade also missing the genes encoding attachment properties is Rhodobacterales bacterium HTCC2255 (Table 1). In Octadecabacter arcticus these genes are present but fragmented into three partial clusters and not arranged homologously to the other organisms of this clade (Table 1; Vollmers et al., 2013). The lacking feature for attachment in P. temperata RCA23 is consistent with the observation that the RCA cluster has not been detected in the fraction of particle-associated bacteria in the North Sea (Giebel et al., 2011) and that growth on agar plates is weak and unreliable (Giebel et al., 2013). This lacking attachment ability, however, is in contrast to the life style of another RCA cluster isolate, strain LE17, retrieved from Californian coastal waters that lives associated with a dinoflagellate (Mayali et al., 2008).
In order to distinguish between genomic features of copiotrophic and oligotrophic bacteria Lauro et al. (2009) compared Photobacterium angustum S14 (copiotrophic; genome size 5.10 Mbp), and Sphingopyxis alaskensis RB 2256 (oligotrophic; genome size 3.37 Mbp). These authors found that categories of clusters of orthologous groups (COG) for motility (N), transcription (K), defense mechanisms (V) and signal transduction (T) constitute lower and COG categories for lipid transport and metabolism (I) and secondary metabolites, biosynthesis, transport and catabolism (Q) higher fractions in the oligotrophic as compared to the copiotrophic bacterium. The fractions of COG categories K, V and T in the genome of P. temperata RCA23 constitute even smaller fractions and COG categories I and Q higher fractions than in S. alaskensis (Supplementary Table S5), confirming the finding by Lauro et al. (2009) and emphasizing that P. temperata RCA23 is a truly oligotrophic bacterium in these respects. Interestingly, COG category I (lipid transport and metabolism) of the pelagic members of the Roseobacter clade constitutes a significantly higher fraction relative to the associated members (Supplementary Table S5) and P. temperata RCA23 has the third highest fraction of this COG category of all pelagic roseobacters (Supplementary Table S5). Genes affiliated to this COG category include short-chain fatty acid dehydrogenases, hydratases, dehydratases and acetyltransferases (Supplementary Table S4). The COG category of cell motility (N) of P. temperata RCA23 has a fraction not as low as that of S. alaskensis, suggesting that this feature is of adaptive significance for the life style of P. temperata RCA23 in the pelagic environment. The respective means of the associated and pelagic groups of the Roseobacter clade do not exhibit such a clear-cut difference between these COG categories and thus may reflect that members of this clade in general are more adapted to nutrient-poor environments than P. angustum (Supplementary Table S5).
Occurrence of the RCA cluster and P. temperata RCA23 in the North Sea
In order to assess the significance of the RCA cluster and P. temperata RCA23 among the active microbial players in the southern North Sea, we investigated bacterioplankton abundance, composition and activity in and outside a diatom-dominated phytoplankton spring bloom in the German Bight (Figure 3). The RCA cluster dominated the Roseobacter clade by >90% (Wemheuer et al., 2014), constituted 3.0–6.5% (mean=5.1±1.2%) of total bacterioplankton and the active bacterioplankton community to even higher proportions, to 10–31.3% (mean=18.5±7.7%), as determined by the cDNA derived from the 16S rRNA (Figure 3c; Wemheuer et al., 2014). Between 11.9% and 49.6% (mean=34.0±10.7%) of the RCA cells were actively dividing as determined by incorporation of bromodeoxyuridine (BrdU; Figure 3c).
One sample collected outside (station 3) and two samples collected inside the bloom, during day (1100 hours; station 13) and at night (0330 hours; station 9), were subjected to a metagenomic and metatranscriptomic analysis. The genome of P. temperata RCA23 was retrieved from the metagenomes to >96% and accounted for 7.8–15.4% of total reads (Table 2; Figure 4). It was also retrieved from the metatranscriptomes, to 17.3% outside and to at least 93.4% inside the bloom (Table 2; Figure 4). Inside the bloom it accounted for 6.7% of total metatranscriptomic reads during night and for 8% during day (Table 2). These data, in line with the other above-mentioned data, show that P. temperata RCA23 was an abundant and in the bloom also highly active member of the bacterioplankton in the southern North Sea.
Table 2. Days of sampling, mapped reads, percentage of genomic coverage and percentage of total reads of P. temperata RCA23, Cand. Pelagibacter ubique HTCC1062 of the SAR11 clade and of Gammaproteobacterium HTCC2207 of the SAR92 cluster of the metagenome (DNA) and metatranscriptome (RNA) retrieved at stations outside (non-bloom) and inside the phytoplankton spring bloom during day (1100 hours) and night (0330 hours).
| Station | Target | No. of reads | Organism (lineage) | Mapped reads | Percentage of genome coverage | Percentage of total reads |
|---|---|---|---|---|---|---|
| 3 | ||||||
| Non-bloom | DNA | 23 381 812 | P. temperata RCA23 | 1 819 024 | 97.1 | 7.8 |
| 26 May 2010 | Cand. P. ubique HTCC1062 (SAR11) | 407 611 | 92.7 | 1.7 | ||
| HTCC2207 (SAR92) | 50 361 | 44.6 | 0.2 | |||
| RNA | 2 223 804 | P. temperata RCA23 | 19 435 | 17.3 | 0.9 | |
| Cand. P. ubique HTCC1062 (SAR11) | 16 947 | 17.9 | 0.8 | |||
| HTCC2207 (SAR92) | 4021 | 2.6 | 0.2 | |||
| 9 | ||||||
| bloom, night | DNA | 12 671 944 | P. temperata RCA23 | 1 783 951 | 96.4 | 14.1 |
| 29 May 2010 | Cand. P. ubique HTCC1062 (SAR11) | 112 728 | 89.0 | 0.9 | ||
| HTCC2207 (SAR92) | 45 926 | 50.2 | 0.4 | |||
| RNA | 8 101 537 | P. temperata RCA23 | 543 596 | 93.4 | 6.7 | |
| Cand. P. ubique HTCC1062 (SAR11) | 35 853 | 42.6 | 0.4 | |||
| HTCC2207 (SAR92) | 35 853 | 34.1 | 0.3 | |||
| 13 | ||||||
| bloom, day | DNA | 16 156 091 | P. temperata RCA23 | 2 480 608 | 96.0 | 15.4 |
| 29 May 2010 | Cand. P. ubique HTCC1062 (SAR11) | 23 806 | 66.3 | 0.1 | ||
| HTCC2207 (SAR92) | 238 054 | 95.5 | 1.5 | |||
| RNA | 15 349 574 | P. temperata RCA23 | 1 222 858 | 94.6 | 8.0 | |
| Cand. P. ubique HTCC1062 (SAR11) | 27 026 | 40.9 | 0.2 | |||
| HTCC2207 (SAR92) | 179 429 | 89.1 | 1.2 | |||
| GOS02-13 | DNA | 1 083 836 | P. temperata RCA23 | 12 254 | 81.9 | 1.1 |
| Norwegian fjord | DNA | 863 687 | P. temperata RCA23 | 54 895 | 83.9 | 6.4 |
| RNA | 256 537 | 4176 | 23.7 | 1.6 | ||
For exact locations of the stations see Figure 3a.
Figure 4.

Circular plot of metagenome and transcriptome read mappings onto the genome of P. temperata RCA23. Read counts from the Bowtie 2 mappings are shown. From innermost to outer circle: GC content, CDS counts, stations 3, 9 and 13 (DNA and cDNA), unmappable regions and genomic islands.
For comparison and to validate our genome mapping approach, we carried out similar genomic mappings in the metagenomes and transcriptomes for organisms of the SAR11 clade and SAR92 cluster, phylogenetic lineages also abundant at these stations (Wemheuer et al., 2014). The genomes of Cand. P. ubique HTCC1062 of the SAR11 clade and of the Gammaproteobacterium HTCC2207 of the SAR92 cluster were retrieved to 92.7% and 44.6% in the metagenome outside the bloom, and to 50.2% and 95.6% inside the bloom, with some differences between day and night (Table 2). Both genomes accounted for not >1.7% of total metagenomic reads. The genome of Cand. P. ubique HTCC1062 had already been retrieved to 96% from metagenomic data of a North Sea phytoplankton spring bloom, but only from the pooled data of all combined samples (Teeling et al., 2012). We retrieved the genomes of Cand. P. ubique HTCC1062 and of Gammaproteobacterium HTCC2207 also from metatranscriptomic reads, in the bloom sample collected during the day to 40.9% and 89.1%, respectively, and at night to 42.6% and 34.1%, respectively. These data show little variations between day and night for the transcriptome of Cand. P. ubique HTCC1062, but a much lower fraction of the genome of HTCC2207 transcribed at night. The transcriptomic reads of both organisms accounted for not >1.2% of total metatranscriptomic reads, indicating that these organisms were far less abundant and active than P. temperata RCA23.
The fact that at least 93% of the genome of P. temperata RCA23 were transcribed in the bloom is remarkable because it reflects that the population of this abundant organism was highly active and used basically the entire potential of its metabolic properties. The largest fraction of the remaining unmapped regions refers to hypothetical proteins mostly located in GIs (Figure 4). To the best of our knowledge this is the first report of such a high fraction of the genome of an abundant marine bacterium transcribed and retrieved from a metatranscriptome. A similarly high genome coverage in a metatranscriptome analysis was recently found in a sediment-derived microbial community degrading hexadecane under anoxic conditions. The genome of Smithella spec., largely dominating this community, was transcribed to 94% (Embree et al., 2014). Transcribed genomic fractions of 52–95% have been reported from pure culture experiments with various bacteria such as Helicobacter pylori (Sharma et al., 2010), Bacillus anthracis (Passalacqua et al., 2009), Bacillus subtilis (Nicolas et al., 2012) and Prochlorococcus MED4 (Wang et al., 2014). Hence, the high fraction of the genome of P. temperata RCA23 transcribed is in line with these reports but may indicate that the ambient population of this organism consisted of cells in different metabolic stages with respect to growth and substrate utilization.
A more detailed analysis of the transcriptomic patterns of P. temperata RCA23 shows that transcripts of COG category E (amino-acid transport and metabolism) exhibited the highest NPKM-normalized reads of all COG categories, followed by COG category R (general function) and J (translation, ribosomal structure and biogenesis) (Table 3, Supplementary Table S6). The high values of normalized transcripts of amino-acid and carbohydrate transport and metabolism (categories E and G) appear to reflect the significance of these substrates for growth of P. temperata RCA23, and is consistent with the broad substrate spectrum of this organism (Giebel et al., 2013). Pronounced differences between the day and night transcriptome occurred in COG categories for translation, ribosomal structure and biogenesis (category J) and for cell motility, but not for chemotaxis (both category N), with higher fractions at night than at day (Supplementary Table S6). Further, the genes c29660, c29670 and c29760, encoding the light-harvesting protein B-870 beta chain PufB and alpha chain PufA and cytochrome c551, respectively, were highly overexpressed at night (Supplementary Figure S5 and Supplementary Table S6). This observation is in line with the well-known fact that bacteriochlorophyll a is only produced during night in AAP bacteria (Wagner-Döbler and Biebl, 2006). Genes encoding small heat-shock protein IbpA (c1240, c20690), RNA polymerase σ-32 factors RpoH1 and RpoH2 (c9960, c21310), chaperonin GroEL (c20740), GroS (c20750) and chaperone protein DnaK (c30090) were also highly overexpressed at night. The small heat-shock protein DnaK and chaperonin GroS, GroEL are important in preventing stress-induced aggregation of various proteins in the cytosol of bacteria (Mogk et al., 2003; Stolyar et al., 2007; Ting et al., 2010). Hence, P. temperata RCA23 obviously undergoes some stress during night. So far we have no clue about a more specific type of stress that upregulates the transcription of these genes during night.
Table 3. NPKM-normalized reads in the COG categories with type of metabolism of the genome of P. temperata RCA23 retrieved in the metatranscriptome at stations 9 and 13 in the German Bight of the North Sea.
| COG category | Type of metabolism | Station 9 (night) | Station 13 (day) |
|---|---|---|---|
| NPKM | NPKM | ||
| B | Chromatin structure and dynamics | 661 | 954 |
| C | Energy production and conversion | 61408 | 57017 |
| D | Cell cycle control, cell division and chromosome partitioning | 7190 | 7533 |
| E | Amino acid transport and metabolism | 101802 | 107628 |
| F | Nucleotide transport and metabolism | 24455 | 26003 |
| G | Carbohydrate transport and metabolism | 59872 | 59448 |
| H | Coenzyme transport and metabolism | 36820 | 42486 |
| I | Lipid transport and metabolism | 45455 | 49575 |
| J | Translation, ribosomal structure and biogenesis | 71558 | 59835 |
| K | Transcription | 44710 | 41837 |
| L | Replication, recombination and repair | 36761 | 39159 |
| M | Cell wall/membrane/envelope biogenesis | 37299 | 39404 |
| N | Cell motility | 8425 | 4975 |
| O | Post-translational modification, protein turnover and chaperones | 50598 | 45863 |
| P | Inorganic ion transport and metabolism | 32016 | 32078 |
| Q | Secondary metabolites biosynthesis, transport and catabolism | 16285 | 16089 |
| R | General function prediction only | 84440 | 90432 |
| S | Function unknown | 68057 | 68212 |
| T | Signal transduction mechanisms | 20797 | 19875 |
| U | Intracellular trafficking, secretion and vesicular transport | 7431 | 7254 |
| V | Defense mechanisms | 8258 | 8799 |
Abbreviations: COG, clusters of orthologous groups; NPKM, Nucleotide activity Per Kilobase of exon model per million Mapped reads.
As a conclusion from this analysis the population of P. temperata RCA23 undergoes intense metabolic reconstruction during night including protein synthesis and stress response, but also enhances the synthesis of flagella proteins. This intense metabolic reconstruction may be a result of the different modes of energy conservation of this AAP bacterium during day and night. It has been shown that Roseobacter litoralis, another AAP bacterium of the Roseobacter clade, changes its proteome drastically, mainly by downregulating protein synthesis, when shifted from dark to light conditions (Zong and Zhao, 2012). Hence the mode of energy conservation appears to have a pronounced effect on the global regulation of metabolic networks in AAP bacteria of the Roseobacter clade, and presumably beyond, with implications of their diurnal participation in turnover of organic matter in marine ecosystems.
Global and biogeochemical significance of the RCA cluster and P. temperata RCA23
Because of the known global distribution of the RCA cluster (Selje et al., 2004) we examined publicly available metagenomic data sets for the presence of genomic features of P. temperata RCA23. The photosynthetic operon was present in an environmental bacterial artificial chromosome clone derived from Californian coastal waters (Supplementary Figure S3; Béjà et al., 2002; Yutin and Béjà, 2005) and in metagenomic data sets from a Norwegian fjord, the western English Channel, the western coastal Atlantic visited during the Global Ocean Sampling expedition (GOS), and in the eastern (Monterey Bay) and southwestern Pacific in Australian waters (Rusch et al., 2007; Yutin et al., 2007; Gilbert et al., 2008, 2010; Thomas et al., 2010; Rich et al., 2011).
The genome of P. temperata RCA23 was retrieved from metagenomic and metatranscriptomic data of a Norwegian fjord to 83.9% and 23.7%, respectively (Gilbert et al., 2008; Table 2). At the GOS stations, the genome of P. temperata RCA23 was retrieved from the metagenomic data to 81.9% (Table 2) and accounted for 0.7% to 6.5% of the mapped reads (Supplementary Figure S6B). This finding, together with the observation that the structure of the photosynthetic operon of the AAP bacteria at these stations was similar to that of P. temperata RCA23 (see above), is consistent with the assumption that this organism constituted largely the AAP communities at these stations. In the GOS and the Norwegian fjord data sets, 80% and 81%, respectively, of the genes of P. temperata RCA23 showed a coverage of ⩾80%. In contrast, 62.5% of the genes of the GIs were not mapped or showed a low coverage (Supplementary Table S7).
Conclusion
This study, on the basis of the sequenced genome of P. temperata RCA23 and the North Sea metagenome and metatranscriptome, sheds new light on the significance of the RCA cluster for biogeochemical processes in marine pelagic systems, considering the high abundances of this organism and the entire RCA cluster in temperate to polar oceans (Selje et al., 2004; West et al., 2008; Giebel et al., 2009, 2011; Wemheuer et al., 2014). It appears to carry out a life style well adapted to the nutrient- and energy-poor marine pelagic realm. The entire population of this organism, and possibly even single cells, simultaneously transcribe almost the entire genome during phytoplankton bloom situations and convert complementary energy by harvesting light. This versatile physiology together with the wealth of high-affinity transporter proteins, despite the absence of distinct genomic and metabolic traits typical for the Roseobacter clade, and a streamlined genome relative to other Roseobacter clade organisms, helps to explain the success of this abundant taxon in temperate and presumably also (sub)polar marine systems. Our findings illustrate how important it is to dissect the role of individual taxa for better understanding their participation in processing organic and inorganic matter in pelagic marine systems.
Acknowledgments
We thank Renate Gahl-Janssen for growing P. temperata RCA23 and harvesting biomass for DNA extraction, Melissa Büngener for technical assistance in sequence analyses, the crew of RV Heincke and M Wolterink for helping with sample collection and processing during the North Sea cruise and the prophage induction experiment. This work was supported by grants from the Volkswagen Foundation in Lower Saxony, Germany, and by TRR51 of Deutsche Forschungsgemeinschaft.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary Material
References
- Béjà O, Suzuki MT, Heidelberg JF, Nelson WC, Preston CM, Hamada T, et al. Unsuspected diversity among marine aerobic anoxygenic phototrophs. Nature. 2002;415:630–633. doi: 10.1038/415630a. [DOI] [PubMed] [Google Scholar]
- Biers EJ, Wang K, Pennington C, Belas R, Chen F, Moran MA. Occurrence and expression of gene transfer agent (GTA) genes in marine bacterioplankton. Appl Environ Microbiol. 2008;74:2933–2939. doi: 10.1128/AEM.02129-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.2014Trimmomatic: a flexible trimmer for Illumina sequence data Bioinformaticse-pub ahead of print 28 April 2014doi: 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed]
- Brinkhoff T, Giebel HA, Simon M. Diversity, ecology, and genomics of the Roseobacter clade: a short overview. Arch Microbiol. 2008;189:531–539. doi: 10.1007/s00203-008-0353-y. [DOI] [PubMed] [Google Scholar]
- Cunliffe M. Correlating carbon monoxide oxidation with cox genes in the abundant Marine Roseobacter Clade. ISME J. 2011;5:685–691. doi: 10.1038/ismej.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embree M, Nagarajan H, Movahedi N, Chitsaz H, Zengler K. Single-cell genome and metatranscriptome sequencing reveal metabolic interactions of an alkane-degrading methanogenic community. ISME J. 2014;8:757–767. doi: 10.1038/ismej.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuerch T, Preusse M, Tomasch J, Zech H, Wagner-Döbler I, Rabus R, et al. Metabolic fluxes in the central carbon metabolism of Dinoroseobacter shibae and Phaeobacter gallaeciensis, two members of the marine Roseobacter clade. BMC Microbiol. 2009;9:209. doi: 10.1186/1471-2180-9-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebel HA, Brinkhoff T, Zwisler W, Selje N, Simon M. Distribution of Roseobacter RCA and SAR11 lineages and distinct bacterial communities from the subtropics to the Southern Ocean. Environ Microbiol. 2009;11:2164–2178. doi: 10.1111/j.1462-2920.2009.01942.x. [DOI] [PubMed] [Google Scholar]
- Giebel HA, Kalhoefer D, Lemke A, Thole S, Gahl-Janssen R, Simon M, et al. Distribution of Roseobacter RCA and SAR11 lineages in the North Sea and characteristics of an abundant RCA isolate. ISME J. 2011;5:8–19. doi: 10.1038/ismej.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebel HA, Kalhoefer D, Gahl-Janssen R, Choo YJ, Lee K, Cho JC, et al. Planktomarina temperata gen. nov., sp. nov., belonging to the globally distributed RCA cluster of the marine Roseobacter clade, isolated from the German Wadden Sea. Int J Syst Evol Microbiol. 2013;63:4207–4217. doi: 10.1099/ijs.0.053249-0. [DOI] [PubMed] [Google Scholar]
- Gilbert JA, Field D, Huang Y, Edwards R, Weizhong L, Gilna R, et al. Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities. PLoS One. 2008;3:e3042. doi: 10.1371/journal.pone.0003042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JA, Field D, Swift P, Thomas S, Cummings D, Temperton B, et al. The taxonomic and functional diversity of microbes at a temperate coastal site: a ‘multi-omic' study of seasonal and diel temporal variation. PLoS One. 2010;5:e15545. doi: 10.1371/journal.pone.0015545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, Baptista D, et al. Genome streamlining in a cosmopolitan oceanic bacterium. Science. 2005;309:1242–1245. doi: 10.1126/science.1114057. [DOI] [PubMed] [Google Scholar]
- Giovannoni SJ, Stingl U. The importance of culturing bacterioplankton in the ‘omics' age. Nat Rev Microbiol. 2007;5:820–826. doi: 10.1038/nrmicro1752. [DOI] [PubMed] [Google Scholar]
- Giovannoni SJ, Stingl U. Molecular diversity and ecology of microbial plankton. Nature. 2005;437:343–348. doi: 10.1038/nature04158. [DOI] [PubMed] [Google Scholar]
- Giovannoni SJ, Cameron Thrash J, Temperton B. Implications of streamlining theory for microbial ecology. ISME J. 2014;8:1553–1565. doi: 10.1038/ismej.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopylova E, Noé L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–3217. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg S. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauro FM, McDougald D, Thomas T, Williams TJ, Egan S, Rice S, et al. The genomic basis of trophic strategy in marine bacteria. Proc Natl Acad Sci USA. 2009;106:15527–15533. doi: 10.1073/pnas.0903507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Csu M, Hughes A, Moran MA. Evolution of divergent life history strategies in marine Alphaproteobacteria. mbio. 2013;4:e00373-13. doi: 10.1128/mBio.00373-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Swan BK, Stepanauskas R, Hughes A, Moran MA. Evolutionary analysis of a streamlined lineage of surface ocean Roseobacters. ISME J. 2014;8:1428–1439. doi: 10.1038/ismej.2013.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayali X, Franks PS, Azam F. Cultivation and ecosystem role of a marine Roseobacter clade-affiliated cluster bacterium. Appl Environ Microbiol. 2008;74:2595–2603. doi: 10.1128/AEM.02191-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A, Deuerling E, Vorderwülbecke S, Vierling E, Bukau B. Small heat shock proteins, ClpB and the DnaK system form a functional triade in reversing protein aggregation. Mol Microbiol. 2003;50:585–595. doi: 10.1046/j.1365-2958.2003.03710.x. [DOI] [PubMed] [Google Scholar]
- Newton RJ, Griffin LE, Bowles KM, Meile C, Gifford S, Givens CE, et al. Genome characteristics of a generalist marine bacterial lineage. ISME J. 2010;4:784–798. doi: 10.1038/ismej.2009.150. [DOI] [PubMed] [Google Scholar]
- Nicolas P, Mäder U, Dervyn E, Rochat T, Léduc A, et al. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science. 2012;335:1103–1106. doi: 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- Passalacqua KD, Varadarajan A, Ondov BD, Okou DT, Zwick ME, Bergman NH. Structure and complexity of a bacterial transcriptome. J Bacteriol. 2009;191:3203–3211. doi: 10.1128/JB.00122-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernthaler A, Pernthaler J, Schattenhofer M, Amann R. Identification of DNA-synthesizing bacterial cells in coastal North Sea plankton. Appl Environ Microbiol. 2002;68:5728–5736. doi: 10.1128/AEM.68.11.5728-5736.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradella S, Päuker O, Petersen J. Genome organization of the marine Roseobacter clade member Marinovum algicola. Arch Microbiol. 2010;192:115–126. doi: 10.1007/s00203-009-0535-2. [DOI] [PubMed] [Google Scholar]
- Rich VI, Pham VD, Eppley JM, Shi Y, DeLong EF. Time-series analyses of Monterey Bay coastal microbial picoplankton using a ‘genome proxy' microarray. Environ Microbiol. 2011;13:116–134. doi: 10.1111/j.1462-2920.2010.02314.x. [DOI] [PubMed] [Google Scholar]
- Rusch DB, Halpern A, Sutton G, Heidelberg KB, Williamson S, Yooseph S, et al. The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol. 2007;5:e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selje N, Simon M, Brinkhoff T. A newly discovered Roseobacter cluster in temperate and polar oceans. Nature. 2004;427:445–448. doi: 10.1038/nature02272. [DOI] [PubMed] [Google Scholar]
- Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;460:250–254. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- Sowell SM, Wilhelm LJ, Norbeck AD, Lipton LS, Nicora CD, Barofsky DF, et al. Transport functions dominate the SAR11 metaproteome at low-nutrient extremes in the Sargasso Sea. ISME J. 2009;3:93–105. doi: 10.1038/ismej.2008.83. [DOI] [PubMed] [Google Scholar]
- Staden R. The Staden sequence analysis package. Mol Biotech. 1996;5:233–241. doi: 10.1007/BF02900361. [DOI] [PubMed] [Google Scholar]
- Stolyar S, He Q, Joachimiak MP, He ZL, Yang ZK, Borglin SE, et al. Response of Desulfovibrio vulgaris to alkaline stress. J Bacteriol. 2007;189:8944–8952. doi: 10.1128/JB.00284-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan BK, Tupper B, Sczyrba A, Lauro FM, Martinez-Garcia M, González JM, et al. Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc Natl Acad Sci USA. 2013;110:11463–11468. doi: 10.1073/pnas.1304246110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tech M, Merkl R. YACOP: Enhanced gene prediction obtained by a combination of existing methods. In Silico Biology. 2003;3:441–451. [PubMed] [Google Scholar]
- Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science. 2012;336:608–611. doi: 10.1126/science.1218344. [DOI] [PubMed] [Google Scholar]
- Thomas T, Rusch D, DeMaere MZ, Yung PY, Lewis M, Halpern A, et al. Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME J. 2010;4:1557–1567. doi: 10.1038/ismej.2010.74. [DOI] [PubMed] [Google Scholar]
- Thompson AW, Huang K, Saito MA, Chisholm SW. Transcriptome response of high- and low-light-adapted Prochlorococcus strains to changing iron availability. ISME J. 2011;5:1580–1594. doi: 10.1038/ismej.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L, Williams TJ, Cowley MJ, Lauro FM, Guilhaus M, Raftery MJ, et al. Cold adaptation in the marine bacterium, Sphingopyxis alaskensis, assessed using quantitative proteomics. Environ Microbiol. 2010;12:2658–2676. doi: 10.1111/j.1462-2920.2010.02235.x. [DOI] [PubMed] [Google Scholar]
- Tripp HJ, Kitner JB, Schwalbach MS, Dacey JWH, Wilhelm LJ, Giovannoni SJ. SAR11 marine bacteria require exogenous reduced sulphur for growth. Nature. 2008;452:741–744. doi: 10.1038/nature06776. [DOI] [PubMed] [Google Scholar]
- Vollmers J, Voget S, Dietrich S, Gollnow K, Smits M, Meyer K, et al. Poles apart: extreme genome plasticity and a new xanthorhodopsin-like gene family in the genomes of Octadecabacter arcticus 238 and Octadecabacter antarcticus 307. PLoS One. 2013;8:e63422. doi: 10.1371/journal.pone.0063422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner-Döbler I, Biebl H. Environmental biology of the marine Roseobacter lineage. Ann Rev Microbiol. 2006;60:255–280. doi: 10.1146/annurev.micro.60.080805.142115. [DOI] [PubMed] [Google Scholar]
- Wang B, Lu L, Lv H, Jiang H, Qu G, Tian C, et al. The transcriptome landscape of Prochlorococcus MED4 and the factors for stabilizing the core genome. BMC Microbiol. 2014;14:11. doi: 10.1186/1471-2180-14-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemheuer B, Güllert S, Billerbeck S, Giebel H-A, Voget S, Simon M, et al. Impact of a phytoplankton bloom on the diversity of the active bacterial community in the southern North Sea as revealed by metatranscriptomic approaches. FEMS Microbiol Ecol. 2014;87:378–389. doi: 10.1111/1574-6941.12230. [DOI] [PubMed] [Google Scholar]
- West NJ, Obernosterer I, Zemb Lebaron P. Major differences of bacterial diversity and activity inside and outside of a natural iron-fertilized phytoplankton bloom in the Southern Ocean. Environ Microbiol. 2008;10:738–756. doi: 10.1111/j.1462-2920.2007.01497.x. [DOI] [PubMed] [Google Scholar]
- Wiegand S, Dietrich S, Hertel R, Bongaerts J, Evers S, Volland S, et al. RNA-Seq of Bacillus licheniformis: active regulatory RNA features expressed within a productive fermentation. BMC Genomics. 2013;14:667. doi: 10.1186/1471-2164-14-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yooseph S, Nealson KH, Rusch DB, McCrown JP, Dupont CL, Kim M, et al. Genomic and functional adaptation in surface ocean planktonic prokaryotes. Nature. 2010;468:60–67. doi: 10.1038/nature09530. [DOI] [PubMed] [Google Scholar]
- Yutin N, Béjà O. Putative novel photosynthetic reaction centre organizations in marine aerobic anoxygenic photosynthetic bacteria: insights from metagenomics and environmental genomics. Environ Microbiol. 2005;7:2027–2033. doi: 10.1111/j.1462-2920.2005.00843.x. [DOI] [PubMed] [Google Scholar]
- Yutin N, Suzuki MT, Teeling H, Weber M, Venter JC, Rusch DB, et al. Assessing diversity and biogeography of aerobic anoxygenic phototrophic bacteria in surface waters of the Atlantic and Pacific Oceans using the Global Ocean Sampling expedition metagenomes. Environ Microbiol. 2007;9:1464–1475. doi: 10.1111/j.1462-2920.2007.01265.x. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Wang K, Budinoff C, Buchan A, Lang A, Jiao N, et al. Gene transfer agent (GTA) genes reveal diverse and dynamic Roseobacter and Rhodobacter populations in the Chesapeake Bay. ISME J. 2009;3:364–373. doi: 10.1038/ismej.2008.115. [DOI] [PubMed] [Google Scholar]
- Zong R, Zhao N. Proteomic responses of Roseobacter litoralis OCh149 to starvation and light regimen. Microbes Environ. 2012;27:430–442. doi: 10.1264/jsme2.ME12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.