

Background: It was unknown whether human β-arrestin 2 could be SUMOylated.
Results: Human β-arrestin 2 is SUMOylated on Lys-295. SUMOylation attenuates β-arrestin 2 inhibition of IL-1R/TRAF6 signaling.
Conclusion: SUMOylation attenuates human β-arrestin 2 inhibition of TRAF6 and IL-1R signaling.
Significance: We show SUMOylation as a novel mechanism in regulation of β-arrestin 2-mediated IL-1R-TRAF6 signaling.
Keywords: Arrestin, Cell Signaling, IL-1, Small Ubiquitin-like Modifier (SUMO), Ubiquitylation (Ubiquitination), Arrestin Cell Signaling
Abstract
β-Arrestin 2 as an adaptor plays a role in the regulation of receptor desensitization, trafficking, and signaling. Bovine β-arrestin 2 has been shown to be SUMOylated on the lysine 400 residue, which links it to the endocytosis of the β2-adrenergic receptor. Here we identify a major SUMOylation site, lysine 295, on human β-arrestin 2. SUMOylation on this site attenuates β-arrestin 2 binding to TRAF6, then enhances TRAF6 oligomerization and autoubiquitination, and consequently leads to the increase of TRAF6-mediated NF-κB/AP-1 activation. We further determine SENP1 as a specific de-SUMOylation protease that can reverse the SUMOylation of β-arrestin 2-mediated processes. Our study reveals SUMOylation as a novel mechanism in the regulation of β-arrestin 2-mediated IL-1R/TRAF6 signaling.
Introduction
IL-1 signaling is a key player in the regulation of inflammatory processes. IL-1 stimulates IL-1R and downstream signaling molecules and subsequently activates the transcription factors NF-κB and AP-1, which control the expression of key immunoregulatory genes (1–3). TRAF6 is a critical mediator for the Toll-like/interleukin-1 receptor superfamily (4, 5). As a RING domain containing E3 ubiquitin ligase, TRAF6 is recruited to the receptor complexes and forms oligomers upon signaling activation and then leads to Lys-63-linked polyubiquitination of itself and downstream signaling molecules (6–8). Lys-63 ubiquitin-conjugated TRAF6 recruits TAB2 and activates the TAB2-associated TAK1 kinase, which subsequently phosphorylates and activates IκB kinases. IκB kinase then phosphorylates IκBα, leading to degradation of IκBα and, consequently activation of NF-κB. In addition, TAK1 can also activate the JNK and p38 MAPK family members, then triggering AP-1 activation (8–10). Both of oligomerization and autoubiquitination are critical for TRAF6 activity toward downstream targets to mediate IL-1β- or LPS-induced NF-κB/AP-1 activation.
β-Arrestin 2 (also known as arrestin 3), along with β-arrestin 1 (arrestin 2) and visual rod (arrestin 1) and cone (arrestin 4) arrestins, is part of a small family of cytosolic adaptor proteins functioning as crucial adaptors/mediators in the regulation of a variety of cell surface receptor desensitization, trafficking, and signaling activities (11–13). β-Arrestins have been shown to be modified by phosphorylation, ubiquitination, and nitrosylation (14–16). These modifications have linked β-arrestins to G protein-coupled receptor (GPCR)3 internalization, trafficking and signal transduction (13). Additionally, β-arrestin 2 directly interacts with TRAF6 after TLR/IL-1R activation, leading to prevention of autoubiquitination of TRAF6 and activation of NF-κB and AP-1 (17).
Recently, bovine β-arrestin 2 has been reported as a SUMOylated protein. The SUMOylation site Lys-400 locates within the C tail of bovine β-arrestin 2, which mediates GPCR endocytosis, and the SUMOylation of bovine β-arrestin 2 has been shown to promote β-arrestin 2-mediated β2-adrenergic receptor internalization (18). Here we also show human/murine β-arrestin 2 as a SUMOylated target. However, lysine 295, not lysine 400, has been identified as a major SUMO-conjugated site on human β-arrestin 2. We further found that SUMOylation decreases β-arrestin 2 inhibition of TRAF6 oligomerization and autoubiquitination and, consequently, promotes TRAF6-mediated NF-κB/AP-1 activation. Our study reveals SUMOylation as a novel mechanism in the regulation of β-arrestin 2-mediated IL-1R/TRAF6 signaling.
EXPERIMENTAL PROCEDURES
Plasmids and Antibodies
The plasmids β-arrestin 2-HA, FLAG-TRAF6, and His-ubiquitin were provided by Dr. Ping Wang (Institute of Biomedical and Science, East China Normal University, China). FLAG-SUMO1, FLAG-SENP1, HA-SUMO1, RGS-SENP1, and RGS-SENP1-mu, a SENP1 catalytic mutant, have been described previously (19–21). β-Arrestin 2 point mutants (K295R and K400R) were generated using site-directed mutagenesis. SENP1-GFP, SENP1mu-GFP, and GFP-TRAF6 were generated using standard cloning procedures and PCR-based mutagenesis (Vazyme Biotech Co., Ltd). Antibodies against FLAG M2 and HA were from Sigma; GFP from Eptomics; RGS-his from Qiagen; and β-arrestin 2, IκBα, SUMO1, ERK, and p-ERK from CST.
Immunoprecipitation and Immunoblotting
Transfected cells were lysed in radioimmune precipitation assay buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 0.1% SDS, 1% sodium deoxycholate, and a mixture of protease inhibitors) and cleared by centrifugation. Cleared cell lysates were incubated with 10 μl of anti-FLAG M2-agarose affinity gel (Sigma) or 10 μl of anti-HA-agarose affinity gel (Sigma) for 2 h. To perform an endogenous assay, MEF cells were lysed in ice-cold radioimmune precipitation assay buffer. Cleared cell lysates were incubated with anti-SUMO-1 antibody (1:200) and 15–20 μl of protein A/G beads (Santa Cruz Biotechnology) for 4 h at 4 °C. After extensive washing, beads were boiled at 100 °C for 10 min. Proteins were resolved by SDS-PAGE and transferred onto PVDF membranes (Millipore), followed by immunoblotting using corresponding antibodies according to the instructions of the manufacturer. Immunoblots were analyzed using the LAS-4000 system (Fujifilm).
Ubiquitination Assay
HEK293T cells were transfected with His-ubiquitin and FLAG-TRAF6. The transfected cells were lysed by denatured buffer (6 m guanidine-HCl, 0.1 m Na2HPO4/NaH2PO4, and 10 mm imidazole), followed by Talon bead purification. The ubiquitination was detected by Western blot analysis.
Luciferase Assay
NF-κB- or AP-1-dependent firefly luciferase plasmids were transiently transfected into HEK293T cells with Renilla luciferase plasmids and others. The cells were harvested 36 h after transfection, and luciferase assays were performed using the Dual-Luciferase reporter assay system (Promega). The relative luciferase activity was normalized on the basis of the Renilla luciferase activity. Data represent three independent experiments performed in duplicate.
Stable Cell Line
β-Arrestin 2 WT or β-arrestin 2 K295R mutant (K295R) lentiviral plasmids were transfected into HEK293T cells with lentivirus packaging vectors by calcium phosphate-DNA coprecipitation method. Viral supernatants were collected 48 h after transfection. MCF-7 cells were infected by lentiviral supernatant in the presence of 10 μg/ml Polybrene for 12 h. 48–72 h later, the cells were sorted for stable cell lines by flow cytometry.
RNA Isolation and Real-time RT-PCR
Total RNA was isolated from cells by using Tripure isolation reagent (Roche). For mRNA analysis, an aliquot containing 2 μg of total RNA was reverse-transcribed using the cDNA synthesis kit (Takara). Real-time PCR was performed using SYBR Green PCR master mix (Applied Biosystems) and detected by the ABI Prism 7500 sequence detection system (Applied Biosystems). The primers for real-time RT-PCR were as follows: GAPDH, 5′-GAGCTGAACGGGAAGCTCACTG-3′ (sense) and 5′-TGGTGCTCAGTGTAGCCCAGGA-3′ (antisense); TNFα, 5′-CCCTCTGGCCCAGGCAGTCA-3′ (sense) and 5′-ATGGGTGGAGGGGCAGCCTT-3′ (antisense).
ELISA Assay
After serum starvation for 12 h, MCF-7 cells were cultured for 24 h with recombinant human IL-1β (20 ng/ml) (Bioworld Technology). The concentration of TNFα in culture supernatants was determined with a human-specific ELISA kit (ExCell Bio), followed by analysis with a SYNERGY microplate reader (BioTek).
RESULTS
SUMO Conjugates Human β-Arrestin 2 on Lys-295
Wyatt et al. (18) have reported previously that bovine β-arrestin 2 is conjugated by SUMO on residue Lys-400. However, when aligning the bovine, human, and murine β-arrestin 2 sequences, we noticed that the Lys-400 residue in human/murine β-arrestin 2 is not in a conserved SUMO consensus motif, ΨKXE (Fig. 1A), which raised the question of whether human/murine as well as bovine β-arrestin 2 could be SUMOylated. To test this, we performed an in vivo SUMOylation assay in HEK293T cells by cotransfecting human β-arrestin 2-HA and FLAG-SUMO1. As shown in Fig. 1, B and C, the SUMOylated β-arrestin 2 band was readily detected in cells transfected with β-arrestin 2-HA and FLAG-SUMO1. We also observed that endogenous human/mouse β-arrestin 2 was modified by endogenous SUMO1 in HEK293T cells and MEFs (Fig. 1D and Fig. 3C). These results showed that human/murine β-arrestin 2 could be also conjugated by SUMO similarly as bovine β-arrestin 2.
FIGURE 1.

SUMO conjugates human β-arrestin 2 on Lys-295. A, the alignment of bovine/human/murine β-arrestin 2 sequences sowing the SUMO consensus motif. B and C, human β-arrestin 2 is SUMOylated in vivo. HEK293T cells were transfected with the indicated plasmids, and the transfected cells were harvested 36 h after transfection and immunoprecipitated with anti-FLAG M2-agarose beads or anti-HA beads. The immunoprecipitates (IP) and the original whole-cell lysates (WCL) were analyzed by immunoblotting (IB) with anti-HA or anti-FLAG antibodies. D, HEK293T cells lysates were immunoprecipitated with anti-β-arrestin 2 antibody or control IgG. The immunoprecipitates and the whole-cell lysates were analyzed by immunoblotting with anti-SUMO1 or anti-β-arrestin 2 antibodies. E, Lys-295 is the major SUMOylation site of human β-arrestin 2. HEK293T cells were transfected with the indicated plasmids, and the transfected cell lysates were immunoprecipitated with anti-FLAG M2-agarose beads, followed by Western blot analysis by using anti-ΗΑ or anti-FLAG antibodies.
FIGURE 3.

SENP1 de-SUMOylates β-arrestin 2. A, SENP1 binds to β-arrestin 2. HEK293T cells were transfected with the indicated plasmids, and the transfected cell lysates were immunoprecipitated (IP) with anti-FLAG M2-agarose beads, followed by WB analysis with anti-ΗΑ or anti-FLAG antibodies. IB, immunoblot; WCL, whole-cell lysate. B, SENP1 deconjugates SUMOylated β-arrestin 2 in vivo. HEK293T cells were transfected with the indicated plasmids, and the transfected cell lysates were immunoprecipitated with anti-FLAG M2-agarose beads. The immunoprecipitates and the whole-cell lysates were analyzed by immunoblotting with anti-β-arrestin 2, anti-FLAG, or anti-RGS-his antibodies. MU, mutant. C, SUMOylated β-arrestin 2 is accumulated in Senp1−/− MEFs. MEF cell lysates were immunoprecipitated with anti-SUMO1 antibody or control IgG. The immunoprecipitates and cell lysates were analyzed by immunoblotting with anti-β-arrestin 2 or anti-SUMO1 antibodies. D, SENP1 enhances β-arrestin 2 inhibition of TRAF6 autoubiquitination. The indicated plasmids were transfected into HEK293T cells. The cell lysates were purified by Talon beads. The precipitates and cell lysates were immunoblotted using anti-FLAG, anti-HA, or anti-GFP antibodies. E, SENP1 enhances β-arrestin 2 inhibition of TRAF6-mediated NF-κB and AP-1 activation. The indicated plasmids were cotransfected into HEK293T cells with an NF-κB- or AP-1-dependent firefly luciferase reporter and a Renilla luciferase reporter. The relative luciferase activity was measured 36 h after transfection and normalized on the basis of Renilla luciferase activity. *, p < 0.05; **, p < 0.01. F, FLAG-TRAF6-transfected wild-type and Senp1−/− MEFs were treated with IL-1β or left untreated before harvest. The cell lysates were immunoprecipitated with anti-FLAG M2-agarose beads, followed by WB analysis with anti-ubiquitin or anti-TRAF6 antibodies. G, β-arrestin 2 WT- or β-arrestin 2 K295R-MCF-7 stable cells were transfected with HA-TRAF6 and si-SENP1 or si-NS oligonucleotides. The cells were treated with IL-1β before harvest, the cell lysates were immunoprecipitated with anti-HA-agarose beads, and the immunoprecipitates and the cell lysates were analyzed by immunoblotting with anti-ubiquitin, anti-HA, or anti-FLAG antibodies. H, wild-type and Senp1−/− MEFs were treated with IL-1β for the indicated time before harvest, followed by WB analysis with the indicated antibodies.
We mutated Lys-400 on human β-arrestin 2 to test whether this residue was a SUMO conjugation site, as bovine β-arrestin 2. As shown in Fig. 1E, mutating the Lys-400 residue had no significant effect on human β-arrestin 2 SUMOylation. However, when mutating Lys-295, a lysine residue located in a conserved SUMO consensus motif, LKHE (ΨKXE), human β-arrestin 2 SUMOylation was significantly abolished, suggesting that Lys-295, but not Lys-400, on human β-arrestin 2 is the major residue to accept SUMO.
SUMOylation Attenuates Human β-Arrestin 2 Inhibition of TRAF6 Activation
Lys-400 resides within the C tail of bovine β-arrestin 2, which mediates GPCR endocytosis. SUMOylation on this site has been shown to promote β-arrestin 2-mediated β2-adrenergic receptor internalization (18). However, Lys-295 on human β-arrestin 2 locates in the TRAF6-binding domain. β-Arrestin 2 binding to TRAF6 has been shown to inhibit autoubiquitination of TRAF6 and activation of NF-κB and AP-1 responding to TLR-IL-1R signaling (17). We therefore proposed that SUMOylation on Lys-295 might regulate β-arrestin 2 interacting with TRAF6 and then modulate IL-1R/TRAF6 signaling. We first tested whether β-arrestin 2 SUMOylation is related to IL-1β stimulation. We showed that IL-1β induced human β-arrestin 2 SUMOylation (Fig. 2A). We then determined whether SUMOylation of human β-arrestin 2 could affect TRAF6 ubiquitination. As shown in Fig. 2B, the expression of β-arrestin 2 markedly reduced TRAF6 ubiquitination (lane 3 versus lane 2). Interestingly, the expression of SUMO1/Ubc9 could almost restore TRAF6 ubiquitination (Fig. 2B, lane 4 versus lane 3) but had no effect on β-arrestin 2 K295R-mutant (Fig. 2B, lane 5 versus lane 4). Mutating Lys-400 on human β-arrestin 2 did not affect TRAF6 ubiquitination (Fig. 2B, lane 6 versus lanes 3 and 4), suggesting that SUMOylation on Lys-295 reduces β-arrestin 2 inhibition of TRAF6 ubiquitination. We further tested whether SUMOylation of β-arrestin 2 had a similar effect on TRAF6-mediated NF-κB and AP-1 activation as on TRAF6 ubiquitination. HEK293T cells were transfected with an NF-κB- or AP-1-dependent luciferase reporter plus TRAF6 and with or without SUMO1/Ubc9. A luciferase assay from these transfected cells showed that SUMOylation on Lys-295 also attenuates β-arrestin 2 inhibition of NF-κB and AP-1 activation (Fig. 2C).
FIGURE 2.

SUMOylation attenuates human β-arrestin 2 inhibition of TRAF6 activation. A, SUMOylation of β-arrestin 2 is induced by IL-1β. HEK293T cells were transfected with FLAG-SUMO1 and β-arrestin 2-HA. The transfected cells were treated with IL-1β (20 ng/ml) for the indicated time before harvest. The cell lysates were immunoprecipitated (IP) with anti-FLAG M2-agarose beads and analyzed by immunoblotting (IB) with anti-β-arrestin 2, anti-p-IKKβ, anti-IKKβ, or anti-IκBα antibodies. IKKβ phosphorylation and IκBα degradation were used as indicators for the validity of IL-1β stimuli. The relative gray scale determination was analyzed using ImageJ software. WCL, whole-cell lysate. B, SUMOylation reduces β-arrestin 2 inhibition of TRAF6 autoubiquitination. The indicated plasmids were transfected into HEK293T cells. The Talon bead precipitates and cell lysates were analyzed by immunoblotting with anti-FLAG, anti-HA, anti-His, or anti-Myc antibodies. Ub, ubiquitin. C, SUMOylation attenuates β-arrestin 2 inhibition of TRAF6-mediated NF-κB and AP-1 activation. An NF-κB- or AP-1-dependent firefly luciferase reporter and a Renilla luciferase reporter plus the indicated plasmids were cotransfected into HEK293T cells. The relative luciferase activity was measured 36 h after transfection and normalized on the basis of Renilla luciferase activity. *, p < 0.05; **, p < 0.01. D, β-arrestin 2 WT-, β-arrestin 2 K295R-, or β-arrestin 2 K400R-MCF-7 stable cells were transfected with HA-TRAF6 or not as indicated. The cells were treated with IL-1β. The cell lysates were immunoprecipitated with anti-HA-agarose beads, and the immunoprecipitates and the cell lysates were analyzed by immunoblotting with anti-ubiquitin, anti-HA, or anti-β-arrestin 2 antibodies. E, β-arrestin 2 WT-, β-arrestin 2 K295R-, or β-arrestin 2 K400R-MCF-7 stable cells were transfected with an NF-κB- or AP-1-dependent firefly luciferase reporter and a Renilla luciferase reporter. The relative luciferase activity was measured 36 h after transfection and normalized on the basis of Renilla luciferase activity. *, p < 0.05; **, p < 0.01.
We generated human β-arrestin 2 wild-type, K295R-mutant, or K400R-mutant stably transfected MCF-7, a relatively low endogenous β-arrestin 2 cell line (data not shown), to further determine the role of SUMOylation in β-arrestin 2-mediated IL-1R signaling. As shown in Fig. 2D, IL-1β-induced TRAF6 ubiquitination was significantly lower in β-arrestin 2 (K295R)-MCF-7 cells than in β-arrestin 2 (WT)-MCF-7 or β-arrestin 2 (K400R)-MCF-7 cells. Similarly, IL-1β-induced NF-κB or AP-1 activation was also significantly lower in β-arrestin 2 (K295R)-MCF-7 cells than that in β-arrestin 2 (WT)-MCF-7 or β-arrestin 2 (K400R)-MCF-7 cells (Fig. 2E). These results suggest that SUMOylation on Lys-295 could attenuate β-arrestin 2 inhibition of TRAF6 activation and TRAF6-mediated IL-1R signaling.
SENP1 De-SUMOylates β-Arrestin 2
We observed that SUMO-specific protease 1 (SENP1) could bind to β-arrestin 2 (Fig. 3A). Therefore, we reasoned that SENP1 could be a deconjugation protease for SUMOylated β-arrestin 2. To test this possibility, we performed a de-SUMOylation assay in the transfected cells. As shown in Fig. 3B, the expression of SENP1 wild-type but not the SENP1 catalytic mutant made the SUMOylated β-arrestin 2 band disappear. We further confirmed SENP1 as a specific de-SUMOylation protease of β-arrestin 2 by observing the accumulation of the SUMOylated β-arrestin 2 in Senp1−/− MEFs (Fig. 3C).
Because SUMOylation reduces β-arrestin 2 inhibition of TRAF6 autoubiquitination and TRAF6-mediated NF-κB/AP-1 activation (Fig. 2), we speculated that SENP1 could enhance human β-arrestin 2 inhibition of TRAF6 ubiquitination and activation through de-SUMOylation. To test this, we performed an ubiquitination assay in HEK293T cells that were cotransfected with FLAG-TRAF6 and His-Ubiquitin plus β-arrestin 2-HA in the presence of SENP1-GFP wild-type or the catalytic mutant SENP1mu-GFP. As shown in Fig. 3D, the coexpression of SENP1 wild-type, not the catalytic mutant, and β-arrestin 2 decreased TRAF6 ubiquitination more than expression of β-arrestin 2 alone. Interestingly, SENP1-reduced TRAF6 ubiquitination depended on β-arrestin 2 because SENP1 action on TRAF6 ubiquitination could not be detected without the coexpression of β-arrestin 2. We also assessed the effect of SENP1 on TRAF6-mediated NF-κB and AP-1 activation by using an NF-κB- or AP-1-dependent luciferase assay. Similarly, SENP1 wild-type, not the catalytic mutant, can enhance β-arrestin 2 inhibition of TRAF6-mediated NF-κB or AP-1 activation (Fig. 3E).
We further determined the role of SENP1 in IL-1R signaling by using Senp1−/− MEFs. As shown in Fig. 3F, IL-1β induced more TRAF6 ubiquitination in Senp1−/− MEFs than that in wild-type MEFs. We also observed that IL-1β-induced more TRAF6 ubiquitination in SENP1 knockdown β-arrestin 2 stably transfected MCF-7 cells (Fig. 3G). More interestingly, there was no effect on β-arrestin 2 (K295R)-MCF-7 cells (Fig. 3G, lane 2 versus lane 4), indicating that SENP1 negative regulation of TRAF6 ubiquitination is dependent on de-SUMOylation of β-arrestin 2. Similarly, IL-1β induced more IκBα phosphorylation and ERK phosphorylation in Senp1−/− MEFs than that in wild-type MEFs (Fig. 3H). These data suggest that SENP1 enhances β-arrestin 2 inhibition of TRAF6-mediated signaling through de-SUMOylation of β-arrestin 2.
SUMOylation Decreases β-Arrestin 2 Binding to TRAF6
β-Arrestin 2 has been reported to directly interact with TRAF6 after TLR-IL-1R activation, leading to prevention of TRAF6 oligomerization and autoubiquitination (17). Because SUMOylation on Lys-295 attenuates β-arrestin 2 inhibition of TRAF6 autoubiquitination and activation (Fig. 2), we reasoned that SUMOylation might disrupt β-arrestin 2-TRAF6 interaction, which reduces β-arrestin 2 inhibition of TRAF6. To test this, we showed that the coexpression of SUMO1/Ubc9, which promotes β-arrestin 2 SUMOylation, could decrease both β-arrestin 2 wild-type and β-arrestin 2 K400R-mutant binding to TRAF6 (Fig. 4A, lanes 3 and 5 versus lane 2) but has no effect on β-arrestin 2 K295R-mutant (Fig. 4A, lane 4 versus lane 2). Furthermore, we assessed β-arrestin 2-TRAF6 interaction in wild-type and Senp1−/− MEFs. As shown in Fig. 4B, TRAF6 was able to bind to β-arrestin 2 in Senp1−/− MEFs much less than that in wild-type MEFs. These results suggest that SUMOylation on Lys-295 could attenuate β-arrestin 2 binding to TRAF6.
FIGURE 4.

SUMOylation decreases β-arrestin 2 binding to TRAF6. A, HEK293T cells were transfected with the indicated plasmids. The cell lysates were immunoprecipitated (IP) with anti-HA-agarose beads. The immunoprecipitates and cell lysates were analyzed by WB with anti-FLAG, anti-HA, or anti-Myc antibodies. IB, immunoprecipitation; WCL, whole-cell lysate. B, Senp1−/− MEFs or wild-type MEF cell lysates were immunoprecipitated with anti-β-arrestin 2 antibody or control IgG. Precipitates and cell lysates were analyzed by WB with anti-β-arrestin 2 or anti-TRAF6 antibodies. C, GFP-TRAF6 and FLAG-TRAF6 were cotransfected into HEK293T cells with β-arrestin 2-HA wild-type or K295R-mutant, and the transfected cells were harvested 24 h after transfection. The cell lysates were immunoprecipitated with anti-FLAG M2-agarose beads. The precipitates and cell lysates were analyzed by WB with anti-GFP, anti-FLAG, or anti-HA antibodies.
Because oligomerization is necessary for TRAF6 autoubiquitination (8), we reasoned that SUMOylation of β-arrestin 2 would enhance TRAF6 oligomerization by attenuating β-arrestin 2 interaction with TRAF6. To test this, we cotransfected differently tagged TRAF6 into HEK293T cells. The coexpression of β-arrestin 2 reduced the association of GFP-TRAF6 with FLAG-TRAF6 (Fig. 4C, lane 3 versus lane 2). Interestingly, the β-arrestin 2 K295R-mutant blocked TRAF6 oligomerization more efficiently than β-arrestin 2 wild-type did (Fig. 4C, lane 4 versus lane 3), suggesting that SUMOylation of β-arrestin 2 could enhance TRAF6 oligomerization through disrupting the interaction between β-arrestin 2 and TRAF6.
Deficiency in SUMOylation Enhances β-Arrestin 2 Inhibition of IL-1β-induced TNFα Expression
To determine the significance of β-arrestin 2 SUMOylation in vivo, we compared TNFα expression in β-arrestin 2 (WT)-MCF7 and β-arrestin 2 (K295R)-MCF7 cells. As shown in Fig. 5A, the expression of β-arrestin 2 wild-type reduced IL-1β-induced TNFα mRNA expression. However, the expression of β-arrestin 2 K295R-mutant showed more of a reduction of the TNFα mRNA level than β-arrestin 2 wild-type. Furthermore, the ELISA assay confirmed that IL-1β-treated β-arrestin 2 (K295R)-MCF7 cells produced much less TNFα in culture medium than β-arrestin 2 (WT)-MCF7 cells (Fig. 5B). Taken together, these results indicate that SUMOylation attenuates β-arrestin 2 inhibition of IL-1β-induced TNFα expression.
FIGURE 5.
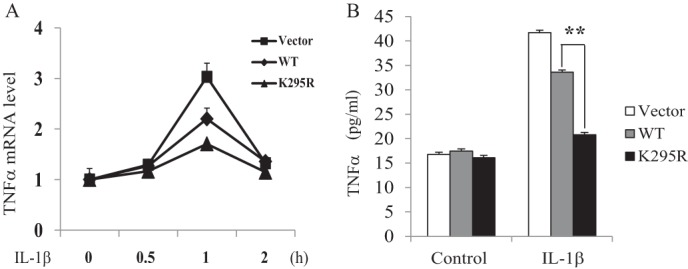
Deficiency in SUMOylation enhances β-arrestin 2 inhibition of IL-1β-induced TNFα expression. A, quantitative real-time RT-PCR analysis showing the relative TNFα mRNA level in β-arrestin 2 WT- or β-arrestin 2 K295R-MCF-7 stable cell lines treated with IL-1β for the indicated time. The results are representative of three experiments. B, ELISA analysis showing the TNFα concentration in supernatants of β-arrestin 2 WT- or β-arrestin 2 K295R-MCF-7 stable cell lines after IL-1β stimulation. The results are representative of three experiments. **, p < 0.01.
DISCUSSION
In this study, we identify Lys-295 as a major SUMOylation site on human β-arrestin 2. Because this site locates in the TRAF6-binding domain, SUMOylation can attenuate β-arrestin 2 binding to TRAF6. Therefore, SUMOylation of β-arrestin 2 can enhance TRAF6 oligomerization and autoubiquitination and, consequently, activate TRAF6-mediated IL-1R signaling. We also found SENP1 as a specific de-SUMOylation protease of β-arrestin 2 in these processes to enhance β-arrestin 2 inhibition of TRAF6 activation. These data reveal SUMOylation as a novel mechanism to attenuate β-arrestin 2 inhibition of TRAF6 and IL-1R signaling.
As an important adaptor, β-arrestins are widely involved in receptors, especially GPCR family, desensitization, trafficking, signaling, and regulating a growing list of cellular processes such as chemotaxis, apoptosis, inflammatory processes, and metastasis (13, 22). More recently, studies have shown that β-arrestins function as scaffold proteins for many signaling molecules in the cytoplasm and nucleus, thereby regulating gene expression and cellular responses (23). Besides traditional GPCR signaling, β-arrestins participate in the regulation of other signaling pathways, including TLR/IL-1R signaling and Wnt/β-catenin signaling (17, 24–26). In TLR/IL-1R signaling, β-arrestin 2 has been reported to directly bind to TRAF6 and then reduce TRAF6 autoubiquitination and activation, leading to a negative regulation of TLR/IL-1R signaling. Here we further find β-arrestin 2 SUMOylation, as a negative mechanism, to attenuate β-arrestin 2 inhibition of TRAF6 activation and then promote TRAF6 oligomerization and autoubiquitination.
SUMOylation is an essential posttranslational modification with critical roles in regulating protein functions such as localization, activity, and protein-protein interaction (27–29). Typically, SUMO-modified proteins contain a SUMO consensus motif defined as ΨKXE, where Ψ is a large hydrophobic residue and X represents any amino acid (30). Bovine β-arrestin 2 has been shown to be SUMOylated. SUMOylation of bovine β-arrestin 2 does not influence the affinity of receptor interaction but is important for AP-2 interaction and AP-2-mediated receptor internalization (18). However, the Lys-400 residue in the carboxyl-terminal region of human β-arrestin 2 is not a major SUMOylation site because mutation of this site did not significantly affect its SUMOylation status. We identified Lys-295 as a major SUMOylation site on human β-arrestin 2, although there might be other potential SUMOylation sites. This site locates in the TRAF6-binding domain of human β-arrestin 2. Therefore, we observed that SUMOylation of human β-arrestin 2 plays a role in the regulation of TRAF6 activation and TRAF6-mediated signaling. It would be interesting to study whether it is evolution-related that the different SUMOylation sites exist on bovine and human/murine β-arrestin 2. In summary, our study reveals that SUMOylation modulates human β-arrestin 2-mediated inhibition of TRAF6 and TRAF6-mediated IL-1R signaling (Fig. 6).
FIGURE 6.
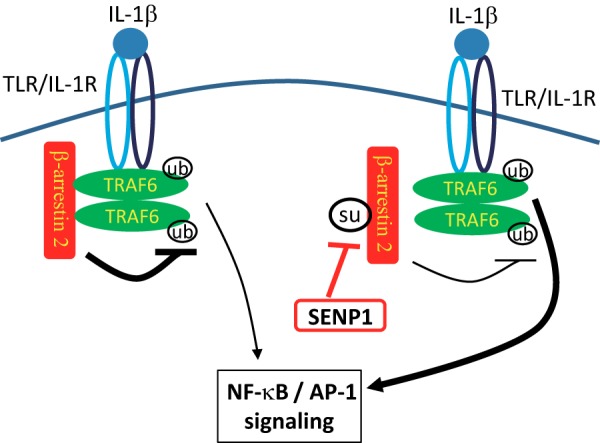
Working model for the regulation of IL-1R-TRAF6 signaling by β-arrestin 2 SUMOylation.
Acknowledgments
We thank Dr. Ping Wang and Dr. Yong Li for plasmids and reagents. We also thank Yong Zuo, Yanqiong Zou, and other members of the Cheng laboratory for assistance.
This work was supported by National Basic Research Program of China (973 Program) Grant 2013CB910902 (to J. C.) and National Natural Science Foundation of China Grants 91019021 (to J. C.) and 81302296 (to W. M.).
- GPCR
- G protein-coupled receptor
- SUMO
- small ubiquitin-like modifier
- MEF
- mouse embryonic fibroblast
- WB
- Western blot.
REFERENCES
- 1. Akira S. (2003) Toll-like receptor signaling. J. Biol. Chem. 278, 38105–38108 [DOI] [PubMed] [Google Scholar]
- 2. Kawai T., Akira S. (2005) Toll-like receptor downstream signaling. Arthritis Res. Ther. 7, 12–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lomaga M. A., Yeh W. C., Sarosi I., Duncan G. S., Furlonger C., Ho A., Morony S., Capparelli C., Van G., Kaufman S., van der Heiden A., Itie A., Wakeham A., Khoo W., Sasaki T., Cao Z., Penninger J. M., Paige C. J., Lacey D. L., Dunstan C. R., Boyle W. J., Goeddel D. V., Mak T. W. (1999) TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 13, 1015–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bradley J. R., Pober J. S. (2001) Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene 20, 6482–6491 [DOI] [PubMed] [Google Scholar]
- 5. Chung J. Y., Park Y. C., Ye H., Wu H. (2002) All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J. Cell Sci. 115, 679–688 [DOI] [PubMed] [Google Scholar]
- 6. Sun L., Deng L., Ea C. K., Xia Z. P., Chen Z. J. (2004) The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 14, 289–301 [DOI] [PubMed] [Google Scholar]
- 7. Yang W. L., Wang J., Chan C. H., Lee S. W., Campos A. D., Lamothe B., Hur L., Grabiner B. C., Lin X., Darnay B. G., Lin H. K. (2009) The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325, 1134–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang C., Deng L., Hong M., Akkaraju G. R., Inoue J., Chen Z. J. (2001) TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351 [DOI] [PubMed] [Google Scholar]
- 9. Scherer D. C., Brockman J. A., Chen Z., Maniatis T., Ballard D. W. (1995) Signal-induced degradation of I κ B α requires site-specific ubiquitination. Proc. Natl. Acad. Sci. U.S.A. 92, 11259–11263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deng L., Wang C., Spencer E., Yang L., Braun A., You J., Slaughter C., Pickart C., Chen Z. J. (2000) Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103, 351–361 [DOI] [PubMed] [Google Scholar]
- 11. Smith W. C., Gurevich E. V., Dugger D. R., Vishnivetskiy S. A., Shelamer C. L., McDowell J. H., Gurevich V. V. (2000) Cloning and functional characterization of salamander rod and cone arrestins. Invest. Ophthalmol. Vis. Sci. 41, 2445–2455 [PubMed] [Google Scholar]
- 12. Lefkowitz R. J. (2004) Historical review: a brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol. Sci. 25, 413–422 [DOI] [PubMed] [Google Scholar]
- 13. Shenoy S. K., Xiao K., Venkataramanan V., Snyder P. M., Freedman N. J., Weissman A. M. (2008) Nedd4 mediates agonist-dependent ubiquitination, lysosomal targeting, and degradation of the β2-adrenergic receptor. J. Biol. Chem. 283, 22166–22176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin F. T., Chen W., Shenoy S., Cong M., Exum S. T., Lefkowitz R. J. (2002) Phosphorylation of β-arrestin2 regulates its function in internalization of beta(2)-adrenergic receptors. Biochemistry 41, 10692–10699 [DOI] [PubMed] [Google Scholar]
- 15. Ozawa K., Whalen E. J., Nelson C. D., Mu Y., Hess D. T., Lefkowitz R. J., Stamler J. S. (2008) S-nitrosylation of β-arrestin regulates β-adrenergic receptor trafficking. Mol. Cell 31, 395–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shenoy S. K., McDonald P. H., Kohout T. A., Lefkowitz R. J. (2001) Regulation of receptor fate by ubiquitination of activated β 2-adrenergic receptor and β-arrestin. Science 294, 1307–1313 [DOI] [PubMed] [Google Scholar]
- 17. Wang Y., Tang Y., Teng L., Wu Y., Zhao X., Pei G. (2006) Association of β-arrestin and TRAF6 negatively regulates Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol. 7, 139–147 [DOI] [PubMed] [Google Scholar]
- 18. Wyatt D., Malik R., Vesecky A. C., Marchese A. (2011) Small ubiquitin-like modifier modification of arrestin-3 regulates receptor trafficking. J. Biol. Chem. 286, 3884–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cheng J., Kang X., Zhang S., Yeh E. T. (2007) SUMO-specific protease 1 is essential for stabilization of HIF1α during hypoxia. Cell 131, 584–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kamitani T., Nguyen H. P., Kito K., Fukuda-Kamitani T., Yeh E. T. (1998) Covalent modification of PML by the sentrin family of ubiquitin-like proteins. J. Biol. Chem. 273, 3117–3120 [DOI] [PubMed] [Google Scholar]
- 21. Gong L., Millas S., Maul G. G., Yeh E. T. (2000) Differential regulation of sentrinized proteins by a novel sentrin-specific protease. J. Biol. Chem. 275, 3355–3359 [DOI] [PubMed] [Google Scholar]
- 22. Whalen E. J., Rajagopal S., Lefkowitz R. J. (2011) Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol. Med. 17, 126–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. DeWire S. M., Ahn S., Lefkowitz R. J., Shenoy S. K. (2007) β-Arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 [DOI] [PubMed] [Google Scholar]
- 24. Kovacs J. J., Hara M. R., Davenport C. L., Kim J., Lefkowitz R. J. (2009) Arrestin development: emerging roles for β-arrestins in developmental signaling pathways. Dev. Cell 17, 443–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bonnans C., Flacelière M., Grillet F., Dantec C., Desvignes J. P., Pannequin J., Severac D., Dubois E., Bibeau F., Escriou V., Crespy P., Journot L., Hollande F., Joubert D. (2012) Essential requirement for β-arrestin2 in mouse intestinal tumors with elevated Wnt signaling. Proc. Natl. Acad. Sci. U.S.A. 109, 3047–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dickey B. F., Walker J. K., Hanania N. A., Bond R. A. (2010) β-Adrenoceptor inverse agonists in asthma. Curr. Opin. Pharmacol. 10, 254–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Creton S., Jentsch S. (2010) SnapShot: the SUMO system. Cell 143, 848–848.e1 [DOI] [PubMed] [Google Scholar]
- 28. Geiss-Friedlander R., Melchior F. (2007) Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 8, 947–956 [DOI] [PubMed] [Google Scholar]
- 29. Hay R. T. (2005) SUMO: a history of modification. Mol. Cell 18, 1–12 [DOI] [PubMed] [Google Scholar]
- 30. Rodriguez M. S., Dargemont C., Hay R. T. (2001) SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem. 276, 12654–12659 [DOI] [PubMed] [Google Scholar]