

Background: A traceable form of PKCδ is needed to investigate PKCδ functions.
Results: Analog-specific PKCδ could utilize N6-(benzyl)-ATP to phosphorylate PKCδ substrates and was specifically inhibited by PP1 analogs.
Conclusion: Binding to the catalytic domain glycine-rich loop, Lys-378, Glu-428, Leu-430, and Phe-633 is the likely mechanism by which N6-(benzyl)-ATP and PP1 analogs interact with analog-specific PKCδ.
Significance: This study provides tools to investigate PKCδ-mediated pathways.
Keywords: ATP, Chemical Biology, Protein Kinase C (PKC), Protein Phosphorylation, Stroke
Abstract
To better study the role of PKCδ in normal function and disease, we developed an ATP analog-specific (AS) PKCδ that is sensitive to specific kinase inhibitors and can be used to identify PKCδ substrates. AS PKCδ showed nearly 200 times higher affinity (Km) and 150 times higher efficiency (kcat/Km) than wild type (WT) PKCδ toward N6-(benzyl)-ATP. AS PKCδ was uniquely inhibited by 1-(tert-butyl)-3-(1-naphthyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (1NA-PP1) and 1-(tert-butyl)-3-(2-methylbenzyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (2MB-PP1) but not by other 4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP1) analogs tested, whereas WT PKCδ was insensitive to all PP1 analogs. To understand the mechanisms for specificity and affinity of these analogs, we created in silico WT and AS PKCδ homology models based on the crystal structure of PKCι. N6-(Benzyl)-ATP and ATP showed similar positioning within the purine binding pocket of AS PKCδ, whereas N6-(benzyl)-ATP was displaced from the pocket of WT PKCδ and was unable to interact with the glycine-rich loop that is required for phosphoryl transfer. The adenine rings of 1NA-PP1 and 2MB-PP1 matched the adenine ring of ATP when docked in AS PKCδ, and this interaction prevented the potential interaction of ATP with Lys-378, Glu-428, Leu-430, and Phe-633 residues. 1NA-PP1 failed to effectively dock within WT PKCδ. Other PP1 analogs failed to interact with either AS PKCδ or WT PKCδ. These results provide a structural basis for the ability of AS PKCδ to efficiently and specifically utilize N6-(benzyl)-ATP as a phosphate donor and for its selective inhibition by 1NA-PP1 and 2MB-PP1. Such homology modeling could prove useful in designing molecules to target PKCδ and other kinases to understand their function in cell signaling and to identify unique substrates.
Introduction
PKC is a family of 10 serine-threonine kinases that regulate a broad spectrum of cellular functions (1, 2). In general, PKC isozymes contain a regulatory domain in the amino-terminal region, followed by a flexible hinge region and a conserved catalytic domain in the carboxyl-terminal tail. The catalytic domain is composed of two lobes. The amino-terminal lobe contains a glycine-rich loop of the consensus sequence GXGXXGX and an invariant Lys that positions ATP for phosphoryl transfer. The carboxyl-terminal lobe contains an activation loop that binds protein substrates for catalysis. The sequence linking these two lobes also contributes to ATP binding and contains a gatekeeper amino acid residue that limits the size of a hydrophobic region within the ATP binding pocket and confers selectivity for binding nucleotides and small molecule inhibitors (1, 2). In PKC isozymes, this gatekeeper is a large hydrophobic residue, either Met or Ile.
Based on their amino-terminal structures and sensitivities to Ca2+ and diacylglycerol, PKCs are classified into conventional PKCs (α, βI, βII, and γ), novel PKCs (δ, ϵ, η, and θ), and atypical PKCs (ζ and λ/ι). Of interest to our laboratory is PKCδ, a member of the novel PKC subfamily, which we found to regulate behavioral responses to ethanol (3) as well as promote reperfusion injury after cerebral ischemia (4). To understand the molecular and cellular actions of PKCδ in physiological and pathophysiological states, it would be desirable to generate a form of PKCδ that can be specifically inhibited and can be used to identify PKCδ substrates for mapping downstream signaling pathways.
A chemical-genetics approach has been developed to identify immediate phosphorylation substrates of kinases and to study results of kinase inhibition by selective, cell-permeable, small molecule inhibitors (5, 6). This approach targets the structurally conserved ATP-binding pocket within all kinases to generate mutant alleles that can utilize specific ATP analogs in addition to ATP. The mutation creates a “cavity” by replacing a bulky gatekeeper with a smaller residue (alanine or glycine) in the ATP-binding pocket. The engineered “cavity” is located where the N6 amine of ATP usually sits, and thus allows for binding of structurally modified ATP analogs with bulky substitutions attached at the N6 position. Only the analog-specific (AS)3 kinase, and not the WT kinase, can efficiently use N6-substituted ATP analogs as phosphate donors. Therefore, only unique substrates of the AS kinase are labeled by the ATP analogs.
To further facilitate the identification and purification of substrates, an affinity tagging strategy was developed (7). First, an AS kinase mutant is used to thiophosphorylate the substrates with N6-(benzyl)-ATPγS. The thiophosphate group is then alkylated by para-nitrobenzyl mesylate to create thiophosphate ester epitopes that can be recognized by specific antibodies. The tagged substrates can be isolated by immunoprecipitation or immunoaffinity purification. The approach has been successfully used to identify direct substrates of several kinases, including JNK (8), v-Src (9), ERK2 (10), CDK1 (11), Raf-1 (12), CDK7 (13), and PKCϵ (14, 15).
AS kinase mutations are designed to be functionally silent with respect to kinase activity and substrate specificity. The engineered AS kinases are also uniquely sensitive to novel kinase inhibitors, such as analogs of PP1 (16). Based on this approach, we generated an AS PKCδ that can be regulated by specific ATP and PP1 analogs (3, 17). Here we studied specific interactions of these analogs with residues in the ATP binding pocket in silico. Our study provides valuable insights for the rational design of molecules to regulate and study PKCδ signaling.
EXPERIMENTAL PROCEDURES
Expression and Purification of WT and AS PKCδ
COS-7 and Neuro2A cells were transfected using the SuperFect Transfection Reagent (Qiagen) with amino-terminal FLAG epitope-tagged rat WT or AS PKCδ (M425A) in pcDNA3 (17). The corresponding gatekeeper for human, rat, and mouse PKCδ is Met-427, Met-425, and Met-425, respectively. After culture for 3 days, cells were washed once with PBS and lysed in buffer containing 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, phosphatase inhibitor mixture 1, and cOmpleteTM protease inhibitor mixture (Roche Applied Science). After 20 min of incubation at 4 °C, the lysates were centrifuged at 12,000 × g for 10 min. The abundance of WT and AS PKCδ in the lysate was determined by Western blotting using anti-PKCδ antibodies (BD Biosciences). To purify WT and AS PKCδ, the supernatants were incubated with anti-FLAG M2 antibody-conjugated agarose (Sigma-Aldrich) at 4 °C for 3 h. The agarose beads were washed three times with the lysis buffer. WT and AS PKCδ were eluted using a storage buffer containing FLAG peptide (20 mm HEPES, pH 7.4, 0.1 mm EGTA, 25% glycerol, 0.03% Triton X-100, 150 ng/μl FLAG peptide) and stored at −80 °C until use. The concentrations of WT and AS PKCδ were determined by ELISA using recombinant PKCδ prepared in SF9 cells (PanVera) as a standard.
Detection of PKCδ Substrates by in Vitro Kinase Assays
Substrates were phosphorylated in vitro by the mixed micelle PKC kinase assay described by Bell (18). FLAG-tagged WT or AS PKCδ (0.312 ng) were incubated in 80 μl of kinase buffer containing 20 mm HEPES (pH 7.4), 0.1 mm EGTA, 0.03% Triton X-100, 10 mm MgCl2, 48 μg of phosphatidylserine (Avanti), 100 nm phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich), and 200 nm histone 3. The reactions were started by the addition of 20 μl of ATP solution containing 250 μm ATP and 10 μCi of [γ-32P]ATP or N6-(benzyl)-[γ-32P]ATP. The inhibitor staurosporine (Calbiochem), bisindolylmaleimide I (Calbiochem), 1NA-PP1, 1NM-PP1, or 2NM-PP1 was added at 1 μm to inhibit PKCδ activity. ATP and PP1 analogs were gifts from K. Shokat (University of California, San Francisco). The reactions were incubated at 27 °C for 30 min and stopped by adding 25 μl of 5× SDS-PAGE sample buffer and heating the mixture at 90 °C for 5 min. Proteins were separated on 4–12% NuPage® gels (Invitrogen) and stained using Coomassie Blue or silver. Phosphorylated substrates were detected by autoradiography.
To detect substrates in neutrophils, we isolated neutrophils from bone marrow by Percoll density gradient centrifugation (4, 19). Neutrophils were lysed by freeze/thaw treatment in modified radioimmune precipitation buffer containing 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 5 mm EDTA, 5 mm EGTA, phosphatase inhibitor mixtures I and II (Sigma-Aldrich), and cOmpleteTM protease inhibitor mixtures (Roche Applied Science) and centrifuged at 20,000 × g for 15 min at 4 °C. The supernatant (100 μg) was incubated in 60 μl of PKC reaction buffer containing 20 mm HEPES, pH 7.4, 0.1 mm EGTA, 0.03% Triton X-100, and 10 mm MgCl2 at 27 °C for 30 min with 1 mm GTP, 100 ng of AS PKCδ purified from transfected COS-7 cells, 200 μm N6-(benzyl)-ATPγS, and 1 μm PMA. The thiophosphate groups on substrate proteins in the supernatant were then alkylated with 2.5 mm para-nitrobenzyl mesylate (Epitomics Inc.) for 2 h at room temperature. The reaction was stopped by the addition of SDS-PAGE sample buffer and analyzed by gel electrophoresis and silver staining. Thiophosphate esters on putative substrate proteins were detected by Western blotting using rabbit primary antibodies (1:15,000 dilution, Epitomics Inc.) and HRP-conjugated secondary antibodies (Jackson ImmunoResearch).
Kinetic Analysis of AS PKCδ with ATP Analogs and PP1 Inhibitors
The kinase activity of AS PKCδ was measured by fluorescence polarization using the Protein Kinase C Assay Kit with fluorescein-labeled substrate peptides (Invitrogen) (20). PKCδ (0.109 ng) or AS PKCδ (0.175 ng) was added to the PKC kinase buffer (20 mm HEPES, pH 7.4, 10 mm MgCl2, 0.02% Nonidet P-40, 0.03% Triton X-100, 0.1 μg of phosphatidylserine, and 0.05 mm sodium vanadate) with 250 nm substrate peptide (RFARKGSLRQKNV) modified from the PKCα pseudosubstrate domain (1, 2), 12.5 nm PMA, 1× fluorescein-labeled phosphopeptide, and 1× anti-phosphoserine antibody, according to the manufacturer's instructions, in a final volume of 90 μl. The kinase reaction was initiated by the addition of ATP (1–100 μm) or N6-(benzyl)-ATP (0.5–200 μm) in 10 μl. The plate was incubated at 22 °C, and the plane-polarized fluorescence was measured every 5 min using an Analyst HT plate reader (Molecular Devices) with excitation at 485 nm and emission at 530 nm. Polarization, Michaelis-Menten constant (Km), maximum velocity (Vmax), and catalytic constant (kcat) were calculated as described (20).
The effect of inhibitors was also measured using fluorescence polarization. Different concentrations of each inhibitor (0.0012–2.0 × 105 nm) were added to the reaction mixture in a final volume of 40 μl. The mixture was incubated at 22 °C for 5 min in the dark. The kinase reaction was initiated by the addition of ATP (final concentration 2.5 μm) and then allowed to proceed for 90 min in the dark at 22 °C. Next, 50 μl of solution containing 2× fluorescein-labeled phosphopeptide and 2× anti-phosphoserine antibody were added according to the manufacturer's instructions. After incubation for 30 min at 22 °C in the dark, plane-polarized fluorescence was measured as described above. The percentage of inhibition was calculated with the equation, (signal without inhibitor − signal with inhibitor)/(signal without inhibitor − signal in the presence of 20 μm staurosporine) × 100. Data from three triplicate experiments were used to calculate IC50 values (nm) by nonlinear regression analysis using Prism version 5.0c (GraphPad Software).
Generation of AS PKCδ Knock-in Mice
Knock-in mice were generated by Taconic Biosciences using homologous recombination with a targeting vector derived from the C57BL/6J mouse genomic BAC clone RP23-133F24. The targeting vector was composed of a 3.1-kb short arm carrying the M425A mutation, a Neo cassette flanked by two loxP sites, a 5.4-kb-long arm, and a diphtheria toxin A gene. NotI-linearized vector was electroporated into C57BL/6 ES cells and selected with 200 μg/ml G418. Surviving ES clones were screened by Southern blotting, and a PCR fragment encompassing the M425A mutation was generated and sequenced to confirm the mutation. The floxed-Neor cassette used for selection was deleted by electroporation of a Cre recombinase plasmid. Chimeric mice were generated following blastocyst injection of targeted ES cells. Heterozygous mutant mice were obtained by breeding chimeras with C57BL/6NTac mice. Heterozygous offspring were intercrossed to generate homozygous knock-in mutant mice. Mouse genotyping was performed by PCR using the primers G8-PCR-F (5′-GCTTTGGCTGAGTGTACTGGCAGAC) and G8–35-R (5′-GCCCACCAGTCCCATCGCC-3′). PKCδ and actin were detected in mouse tissues by Western blot analysis using a mouse monoclonal antibody against PKCδ (1:1000 dilution; BD Biosciences) and actin (1:2000 dilution; Sigma-Aldrich). All procedures were conducted in accordance with Institutional Animal Care and Use Committee policies.
Immunofluorescence Staining of Neutrophils
Neutrophils isolated by Percoll density gradient centrifugation (4, 19) were plated on glass coverslips coated with 20% fetal calf serum (FCS) for 10 min at 37 °C. The coverslips with attached neutrophils were treated with or without 200 nm PMA for 2 min at 37 °C and fixed in 2% paraformaldehyde in PBS for 10 min at room temperature (21). After permeabilization in 0.1% Triton X-100 in PBS for 5 min, neutrophils were blocked in 10% normal donkey serum, 0.2% BSA in PBS for 1 h and stained with mouse anti-PKCδ (1:200 dilution; BD Biosciences) antibody diluted in PBS containing 2% normal donkey serum and 0.2% BSA overnight at 4 °C. After three washes with PBS, neutrophils were incubated with the appropriate donkey fluorochrome-conjugated secondary antibodies (1:200 dilution; Jackson ImmunoResearch) and coverslipped in mounting medium containing DAPI (Vector Laboratories) to localize the nuclei. Images were captured using a Zeiss LSM 510 laser-scanning confocal microscope.
Neutrophil Superoxide Anion (O2⨪) Production
Neutrophils isolated by Percoll density gradient centrifugation (4, 19) were treated with 5 μm 1NA-PP1 for 15 min. O2⨪ production was then measured by a cytochrome c reduction assay using 100 nm PMA as a stimulus (19).
Statistical Analysis
Quantitative data were expressed as mean ± S.E. and analyzed using Prism version 5.0 (GraphPad). Analysis of variance with post hoc tests was used to determine statistical significance between means. A value of p less than 0.05 was considered to be statistically significant.
Homology Modeling
Human PKCδ homology models (amino acids 341–669) were generated using MOE version 2011.10 software and PKCι with ATP bound in the active kinase domain (Protein Data Bank (PDB) code 3A8W) (22) as the reference structure. During the homology modeling procedure in MOE, 10 models were developed, and the final model was used for the studies. The homology model developed by MOE did not have the ATP or bound water molecules for correct ATP positioning in the binding pocket. We therefore transferred the ATP and binding pocket water molecules from PKCι into the binding pocket of the PKCδ homology model. The complex of WT PKCδ and ATP/water molecules was then energy-minimized, and the pH of the system was set at pH 7.4, to correlate with experimental conditions. After the human PKCδ model was developed, we mutated the Met-427 to Ala using the side chain mutation function in MOE with energy minimized. AS PKA was generated by mutating Met-120 to Ala with energy minimized in the crystal structure of PKA (PDB code 1ATP) (23, 24). The interactions of ATP and inhibitors with the residues of the nucleotide-binding pocket were analyzed using the two-dimensional ligand-receptor interaction diagrams of Discovery Studio version 3.5.
Docking Studies
Docking of N6-(benzyl)-ADP (derived from PDB entry 1KSW) and 1NM-PP1 (PubChem code 5154691) was performed using the MOE software, which utilizes an induced fit model of the ligand and receptor for docking. Docking of 1NA-PP1 (PubChem code 4877) and 2MB-PP1 (modified from the co-crystal structure of Src-AS1/3MB-PP1 (PDB code 4LGG)) was performed using AutoDock Vina (25) integrated with Chimera version 1.7 (26). The pH of the induced fit docking procedure used in the MOE was set at 7.4. The top 10 poses returned from the docking studies were evaluated by visual inspection.
N6-(benzyl)-ATP (derived from PDB entry 1KSW) was docked in the soluble catalytic core of membrane-bound mammalian adenylyl cyclase (VC1:IIC2, PDB code 1TL7) (27) using AutoDock Vina (25) integrated with Chimera. The top 10 poses were evaluated by visual inspection. 2′(3′)-O-(N-Methylanthraniloyl)-guanosine 5′-triphosphate (MANT)-GTP, forskolin, and the chain C-containing GTP in the VC1:IIC2 crystal structure were deleted before docking. The adenine ring of 1NA-PP1 (PubChem code 4877) was aligned with the guanine ring of MANT-GTP in VC1:IIC2.
RESULTS
Generation of AS PKCδ
PKC isozymes belong to the AGC (PKA, PKG, and PKC) kinase group sharing a highly conserved catalytic domain. Based on the sequence homolog of amino acid residues in the catalytic domain of PKC isozymes and PKA (Fig. 1A), we identified Met-427 as the gatekeeper residue in human PKCδ and substituted it with alanine in AS PKCδ. Because mutations can destabilize proteins and reduce their abundance in cells, we expressed WT and AS PKCδ in two different cell lines, COS-7 (Fig. 1B) and Neuro2A (Fig. 1C). We found that both proteins expressed abundantly in these cell lines and that AS PKCδ showed levels of expression similar to WT PKCδ.
FIGURE 1.
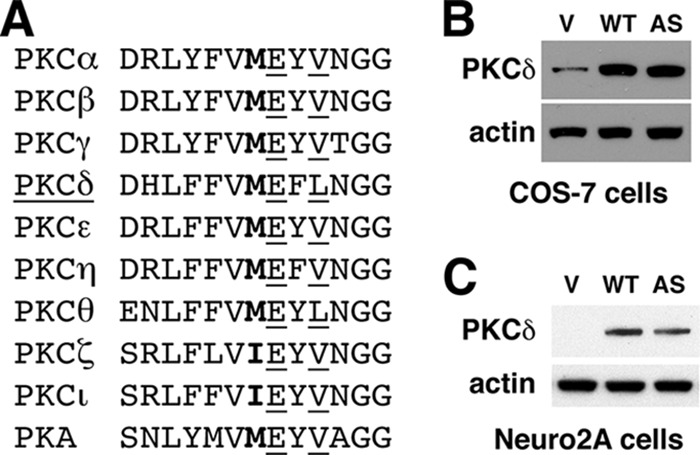
Design and expression of AS PKCδ. A, alignment of the gatekeeper (boldface type) and its surrounding residues within the nucleotide-binding domain of human PKC isozymes and PKA. Glu and Val/Leu residues are underlined. B and C, expression of WT and AS PKCδ in COS-7 (B) and Neuro2A cells (C) was detected by Western blot analysis using anti-PKCδ antibody. Transfection with the pcDNA3 vector (V) was used as a control.
Kinase Activity of AS PKCδ
To characterize AS PKCδ, we purified WT and AS PKCδ from COS-7 cells and found that both proteins utilized [γ-32P]ATP to phosphorylate a model substrate (histone 3) in mixed micelle PKC assays (Fig. 2, A and B). WT and AS PKCδ showed similar binding affinity (Km) and catalytic rate (kcat) for ATP in fluorescence polarization assays using a peptide substrate derived from the psuedosubstrate sequence of PKCα (Table 1). The similarities in Km and kcat values suggest that at the concentrations of ATP inside cells (2–10 mm), AS PKCδ can utilize ATP as efficiently as other WT enzymes.
FIGURE 2.

AS PKCδ, but not WT PKCδ, utilized N6-(benzyl)-ATP (BZ-ATP) to phosphorylate substrates. A, chemical structure of an ATP analog. R, site of N6 modification. B, phosphorylation of histone 3 by WT and AS PKCδ in the presence of [γ-32P]ATP or N6-(benzyl)-[γ-32P]ATP. Top, autoradiogram of phosphorylated histone 3; bottom, Coomassie-stained histone 3. C, mouse neutrophil lysates (100 μg) were labeled with AS PKCδ and N6-(benzyl)-ATPγS. After para-nitrobenzyl mesylate (PNBM) alkylation, the labeled substrates were detected by Western blot analysis using an antibody that detects thiophosphate esters. Immunoreactivity was increased in samples treated with the PKC activator PMA. D, the same amounts of neutrophil lysates as in D were loaded and visualized by silver staining.
TABLE 1.
Kinetic analysis of WT and AS PKCδ toward ATP and N6-(benzyl)-ATP (BZ-ATP)
| [Enyme] | ATP | Km | Vmax | kcat | kcat/Km | |
|---|---|---|---|---|---|---|
| nm | μm | nm min−1 | min−1 | m−1 min−1 | ||
| WT | 1.4 | ATP | 0.91 ± 0.15 | 1.61 ± 0.06 | 1.15 ± 0.10 | 1.26 × 106 |
| BZ-ATP | 500 ± 53.26 | 1.38 ± 0.07 | 0.98 ± 0.02 | 0.002 × 106 | ||
| AS | 2.24 | ATP | 1.31 ± 0.08 | 1.91 ± 0.03 | 0.85 ± 0.01 | 0.65 × 106 |
| BZ-ATP | 2.61 ± 0.18 | 1.70 ± 0.02 | 0.75 ± 0.01 | 0.29 × 106 |
In contrast, AS PKCδ was able to utilize an ATP analog with a bulky substitution (benzyl ring) at the N6 position (N6-(benzyl)-ATP) to phosphorylate histone 3 (Fig. 2, A and B). A similar result was observed in fluorescence polarization assays, which showed ∼200-fold lower Km and ∼150-fold higher efficiency (kcat/Km) for AS PKCδ than WT PKCδ when N6-(benzyl)-ATP was the phosphate donor (Table 1).
We previously found that AS PKCδ can utilize N6-(benzyl)-ATPγS to thiophosphorylate histone 3, which can be detected after alkylation with para-nitrobenzyl mesylate by immunoblot analysis using an antibody against thiophosphate esters (17). We decided to use this approach to determine whether we could use AS PKCδ and N6-(benzyl)-ATPγS to identify PKCδ substrates in cells. We were particularly interested in PKCδ substrates in neutrophils because of our work demonstrating an important role for neutrophils in cerebral ischemia and reperfusion injury (4). Because N6-(benzyl)-ATPγS does not cross cell membranes, we incubated neutrophil lysates in kinase buffer containing recombinant AS PKCδ (Fig. 2, C and D). Despite the presence of endogenous WT PKCδ in these lysates, we found several proteins that were specifically labeled, and the labeling intensity of some of these proteins was increased when lysates were incubated with the PKC activator PMA, suggesting that they are PKCδ substrates.
Sensitivity of AS PKCδ to Kinase Inhibitors
We next compared the ability of different kinase inhibitors to reduce phosphorylation of histone 3 or the PKCα pseudosubstrate peptide. WT PKCδ, like other wild type PKC isozymes we tested, was potently inhibited by the general kinase inhibitor staurosporine and by the more PKC-selective inhibitor bisindolylmaleimide I but not by PP1 analogs (Fig. 3 and Table 2). AS PKCδ was uniquely inhibited by 1NA-PP1, weakly inhibited by 1NM-PP1, and not at all inhibited by 2NM-PP1. Staurosporine and bisindolylmaleimide I inhibited AS PKCδ much less potently than WT PKCδ. Both WT and AS PKCδ were unable to phosphorylate histone 3 in the absence of phosphatidylserine and PMA (Fig. 3B). PMA mimics diacylglycerol, an endogenous activator of PKCδ (1, 2). Phosphatidylserine, an important phospholipid membrane component, is required for membrane binding and kinase activity of PKC. This result indicates that, like WT PKCδ, AS PKCδ requires known PKC lipid regulators for activation.
FIGURE 3.

The kinase activity of AS PKCδ is specifically inhibited by 1NA-PP1. A, structure of PP1 analogs. B, WT or AS PKCδ was incubated with [γ-32P]ATP, histone 3, and a 1 μm concentration of staurosporine (Stau), bisindolylmaleimide I (Bis), or PP1 analogs. Top, autoradiogram showing phosphorylated histone 3; bottom, histone 3 in a silver-stained gel. Inhibition of WT and AS PKCδ by staurosporine and bisindolylmaleimide I (C) and by PP1 analogs (D) was measured by fluorescence polarization assays (n = 3). Error bars, S.E.
TABLE 2.
IC50 values for the inhibition of PKC isozymes by inhibitors
| Staua | Bisb | 1NA-PP1 | 1NM-PP1 | 2NM-PP1 | |
|---|---|---|---|---|---|
| nm | nm | nm | nm | nm | |
| WT PKCδ | 41.2 ± 1.6 | 32.4 ± 3.0 | >100,000 | >100,000 | >1,000,000 |
| AS PKCδ | 1671.0 ± 45.5 | 221.1 ± 31.9 | 154.0 ± 14.3 | 2,598.7 ± 162.8 | >100,000 |
| WT PKCϵ | 1.5 ± 0.1 | 6.2 ± 1.1 | >100,000 | ||
| WT PKCγ | 18.1 ± 1.5 | 30.0 ± 2.2 | >100,000 |
a Stau, staurosporine.
b Bis, bisindolylmaleimide I.
Generation and Characterization of AS PKCδ Knock-in Mice
To investigate the functions of PKCδ in vivo, we used homologous recombination in embryonic stem cells to generate knock-in mice endogenously expressing AS PKCδ (Fig. 4, A and B). AS PKCδ mice were normal in appearance and behaved normally in a cage environment. Western blot analysis of neutrophil lysates from these knock-in mice demonstrated a 78-kDa PKCδ immunoreactive band of similar intensity in samples from WT mice (Fig. 4C). PKCδ immunoreactivity was present in the neutrophil cytoplasm of both genotypes and translocated to cell membranes after PMA activation (Fig. 5). We previously found that the superoxide anion (O2⨪) production from neutrophils is significantly reduced in PKCδ null mice (4). To confirm this phenotype and assess the effect of 1NA-PP1 in intact cells, we treated neutrophils from WT and AS PKCδ mice with 1NA-PP1 and found that it specifically reduced O2⨪ production from AS PKCδ but not from WT neutrophils (Fig. 6).
FIGURE 4.

Generation of AS PKCδ knock-in mice. A, organization of the mouse Prkcd gene, targeting constructs, and M425A mutant allele resulting from homologous recombination. Filled boxes, exons 7–18. Met-425 is in exon 13. Arrowheads, loxP sequences. The targeting vector was composed of a 3.1-kb short arm carrying the M425A mutation, a Neo expression cassette flanked by two loxP sites, a 5.4-kb-long arm, and a diphtheria toxin A (DTA) gene. B, verification of genotypes by PCR using the G8-PCR-F and G8–35-R primers. The wild type allele (+) generates a 0.47-kb product, and the M425A mutant allele (A) generates a 0.6-kb product. C, Western blot showing similar PKCδ immunoreactivity in neutrophils from WT and AS PKCδ knock-in (KI) mice.
FIGURE 5.
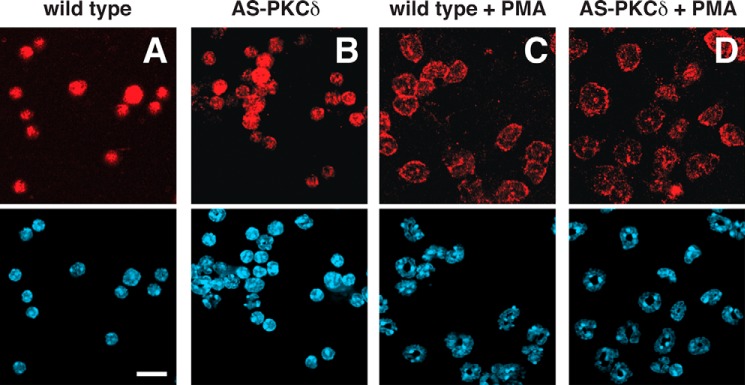
Pattern of PKCδ immunoreactivity in neutrophils of WT PKCδ (A and C) and AS PKCδ (B and D) mice. PKCδ (red) was detected diffusely in the cytoplasm at baseline (A and B) and at the plasma membrane after incubation with 200 nm PMA for 2 min (C and D). Nuclei (blue) were detected with DAPI in the bottom panels. Scale bar, 25 μm.
FIGURE 6.
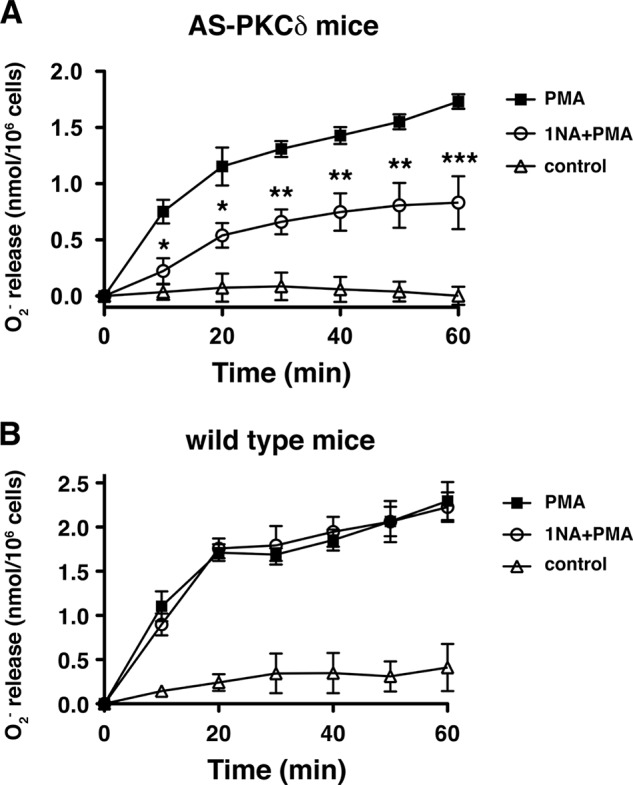
1NA-PP1 inhibits superoxide anion (O2⨪) production from PMA-stimulated neutrophils of AS PKCδ (A) but not WT (B) mice. A, PMA-stimulated (100 nm) O2⨪ production was significantly reduced by 5 μm 1NA-PP1 (Ftreatment (1,24) = 18.1, p = 0.0132; Ftime (6,24) = 57.5, p < 0.0001; Finteraction (6,24) = 5.7, p = 0.0009). *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with PMA-treated neutrophils (Bonferroni test). B, PMA-stimulated (100 nm) O2⨪ production from WT neutrophils was not reduced by 5 μm 1NA-PP1 (Ftreatment (1,24) = 0.00, p = 0.9822; Ftime (6,24) = 142.1, p < 0.0001; Finteraction (6,24) = 0.67, p = 0.6748). Error bars, S.E.
N6-(Benzyl)-ATP Fits in the Nucleotide-binding Pocket of AS PKCδ but Not WT PKCδ
To understand the molecular basis for differences in binding affinity and specificity of ATP and PP1 analogs in WT and AS PKCδ, we generated PKCδ models in silico. Because the x-ray crystal structure of PKCδ is not available, PKCδ homology models were created based on the crystal structure of human PKCι (PDB code 3A8W) (22). The root mean square deviation for the overlaid structure of WT and AS PKCδ is 0.279 Å (Fig. 7A), suggesting that the M427A mutation did not significantly change the three-dimensional structure of PKCδ (28). The position of ATP was nearly identical in the nucleotide-binding pockets of WT and AS PKCδ, with the adenine ring pointing toward the gatekeeper residues (Fig. 7B). In both WT and AS PKCδ, the adenine ring of ATP formed hydrogen bonds with Glu-428 and Leu-430 near the gatekeeper. The ATP phosphate groups were positioned by the charge interactions with the invariant Lys-378 and by hydrogen bonds with Ser-359, Phe-360, and Gly-361 in the glycine-rich loop (Fig. 7, C and D). These interactions are similar to those reported in previous studies of the crystal structure of ATP-bound PKA (PDB code 1ATP) (23, 24, 29, 30), PKCι (PDB code 3A8W) (22), and PKCβ (PDB code 3PFQ) (31), suggesting the accuracy of our PKCδ homology models. ATP interacted with the same residues in WT and AS PKCδ, but the electrostatic interaction with the gatekeeper residue (Met-427) in WT PKCδ was replaced by a weaker van der Waals interaction with Ala-427 in AS PKCδ. This finding suggests a molecular basis for the 2-fold lower specificity (kcat/Km) for ATP of AS PKCδ compared with WT PKCδ (Table 1).
FIGURE 7.

Comparison of ATP interactions in the nucleotide-binding pocket of WT and AS PKCδ. A, the homology model of WT PKCδ (gray) was superimposed with the AS PKCδ model (orange). The gatekeeper residue Met-427 in WT PKCδ is highlighted in green. B, ATP (yellow) docked in WT PKCδ was superimposed with ATP (colored by atom type: nitrogen (blue), carbon (black), oxygen (red), and phosphorus (orange)) docked in AS PKCδ. Shown are the ATP-interacting residues in the nucleotide-binding pocket of WT (C) and AS PKCδ (D). Residues involved in electrostatic (hydrogen bond, charge, or polar), van der Waals, covalent bond, water, and metal interactions with the ligand are shaded in pink, green, magenta, aquamarine, and dark gray, respectively. Hydrogen bond interactions with amino acid main chain and amino acid side chain residues are represented by green and blue dashed arrows directed toward the electron donor. Charged interaction is shown by a pink dashed arrow with heads on both sides. Pi interaction is represented by an orange line with, π indicating the interaction. In both WT and AS PKCδ, ATP forms hydrogen bonds with Glu-428 and Leu-430 (red number symbols) near the gatekeeper (red arrow) and Ser-359, Phe-360, and Gly-361 in the glycine-rich loop (red asterisks) and charged interactions with invariant Lys-378 (red arrowhead). The π-π interaction of ATP with Phe-633 is shown as an orange line in WT and AS PKCδ.
Because N6-(benzyl)-ADP has been crystalized with an analog-specific allele of c-Src (T338G) (32), we extracted N6-(benzyl)-ADP from the co-crystalized structure (PDB code 1KSW) and docked it in the PKCδ homology models. N6-(benzyl)-ADP and ATP docked at similar positions within the nucleotide-binding pocket of AS PKCδ (Fig. 8A). The benzyl ring of N6-(benzyl)-ADP was positioned deeper in the nucleotide-binding pocket and placed in a space enlarged by mutating Met-427 to Ala-427. This change allowed the phosphate groups of N6-(benzyl)-ADP to interact with the glycine-rich loop for potential phosphoryl transfer (Fig. 8C). In contrast, N6-(benzyl)-ADP was shifted outward in the nucleotide-binding pocket of WT PKCδ (Fig. 8B), preventing its phosphate groups from interacting with the glycine-rich loop (Fig. 8D). Moreover, the hydrogen bonds formed between ATP and Glu-428 and Leu-430 in AS PKCδ (Fig. 7D) were lost with N6-(benzyl)-ADP (Fig. 8C). This might explain why ATP showed 2 times higher affinity (Km) and specificity (kcat/Km) than N6-(benzyl)-ATP for AS PKCδ (Table 1).
FIGURE 8.

Comparison of N6-(benzyl)-ADP (BZ-ADP) interactions in the nucleotide-binding pocket of AS and WT PKCδ. Shown are the superimposed structures of BZ-ADP (colored by atom type) and ATP (yellow) in AS (A) and WT PKCδ (B). Ala-427 is highlighted in purple in AS PKCδ, and Met-427 is highlighted in green in WT PKCδ. Shown are the BZ-ADP-interacting residues in the nucleotide-binding pocket of AS (C) and WT PKCδ (D). ATP interacts with the residues in the glycine-rich loop (red asterisks) and the invariant Lys-378 (red arrowheads). The gatekeeper is indicated by a red arrow.
Mechanistic Studies of Inhibition of AS PKCδ by PP1 Analogs
To investigate the basis for specific 1NA-PP1 inhibition of AS PKCδ, we compared docking of 1NA-PP1 and ATP in AS PKCδ (Fig. 9A) and WT PKCδ (Fig. 9B). The adenine rings of 1NA-PP1 and ATP were aligned in AS PKCδ but not in WT PKCδ. Specifically, the adenine rings of 1NA-PP1 and ATP formed hydrogen bonds with the same residues (Glu-428 and Leu-430) in AS PKCδ (Figs. 7D and 9C). Moreover, the naphthyl ring of 1NA-PP1 fits in the space created by the M427A mutation, forming π-σ interactions with the invariant Lys-378 in AS PKCδ (Fig. 9C). Neither of these interactions was found with 1NA-PP1 docked in WT PKCδ (Fig. 9D) or with 1NM-PP1 or 2NM-PP1 docked in AS PKCδ (Fig. 10).
FIGURE 9.

Comparison of 1NA-PP1 interactions in the nucleotide-binding pocket of AS and WT PKCδ. A and B, superimposed structure of 1NA-PP1 (colored by atom type) and ATP (yellow) in AS (A) and WT PKCδ (B). Ala-427 is highlighted in purple in AS PKCδ, and Met-427 is highlighted in green in WT PKCδ. C and D, 1NA-PP1-interacting residues in the nucleotide-binding pocket of AS (C) and WT PKCδ (D). 1NA-PP1 forms hydrogen bonds with Glu-428 and Leu-430 (red number symbols), and π-σ interaction with Lys-378 (red arrowheads). The gatekeeper is indicated by a red arrow.
FIGURE 10.

Interactions of 1NM-PP1 and 2NM-PP1 with AS PKCδ. A, superimposed structures of 1NM-PP1 (colored by atom type) and ATP (yellow) in AS PKCδ. B, superimposed structures of 2NM-PP1 (colored by atom type) and ATP (yellow) in AS PKCδ. The mutated gatekeeper (Ala-427) in AS PKCδ is highlighted in purple. C and D, 1NM-PP1- (C) and 2NM-PP1-interacting (D) residues in the nucleotide-binding pocket of AS PKCδ. Glu-428 and Leu-430 are indicated by red number symbols.
PKA, another member of the AGC kinase family, has been engineered following the same approach (33). The AS PKA mutant (M120A) is sensitive to both 1NA-PP1 and 1NM-PP1 inhibitors. To investigate the structural basis for PP1 inhibition in AS PKA as well as to compare the mechanism with AS PKCδ, we docked 1NA-PP1, 1NM-PP1, and ATP in AS PKA. The adenine rings of 1NA-PP1 and 1NM-PP1 aligned with the adenine ring of ATP in AS PKA, with the naphthyl and naphthylmethyl rings situated in the engineered cavity (M120A) (Fig. 11, A and B). Interestingly, the adenine rings of 1NA-PP1 and 1NM-PP1 also formed hydrogen bonds with Glu-121 and Val-123 (corresponding to Glu-428 and Leu-430 in PKCδ) (Fig. 11, C and D).
FIGURE 11.

Interactions of 1NA-PP1 and 1NM-PP1 with AS PKA. A, superimposed structures of 1NA-PP1 (colored by atom type) and ATP (yellow) in AS PKA. B, superimposed structures of 1NM-PP1 (colored by atom type) and ATP (yellow) in AS PKA. The mutated gatekeeper (Ala-120) in AS PKA is highlighted in purple. C and D, 1NA-PP1- (C) and 1NM-PP1-interacting (D) residues in the nucleotide-binding pocket of AS PKA. The gatekeeper, invariant Lys, and residue forming hydrogen bonds with ligand are indicated by red arrows, arrowheads, and red number symbols, respectively.
To assess whether the in silico AS PKCδ model can be used to screen potential inhibitors, we docked novel PP1 analogs (6) and found that 2MB-PP1 (Fig. 12A) is better aligned with the adenine ring of ATP than 1NA-PP1 (Fig. 12B). 2MB-PP1 interacts with the same residues of AS PKCδ as 1NA-PP1, but the interaction is further strengthened by an additional π-π interaction with Phe-633 (Fig. 12C). This π-π stacking interaction has been implicated in stabilizing nucleic acid-protein complexes (34, 35). To test whether this enhanced π-π interaction improves the inhibitory effect of 2MB-PP1, we measured its IC50 using fluorescence polarization assays and found that the IC50 for 2MB-PP1 (40.6 nm) was almost 4 times lower than the IC50 for 1NA-PP1 (154.0 nm) (Fig. 12D). The results demonstrate that in silico modeling can predict the relative potency of inhibitors and support the use of this model to design potential modulators of PKCδ activity. Taken together, we conclude that PP1 analogs inhibit AS PKCδ and AS PKA by competing with ATP for interaction with Lys, Glu, Leu/Val, and Phe residues within the purine binding pockets of these mutant kinases.
FIGURE 12.

AS PKCδ is more sensitive to inhibition by 2MB-PP1 than 1NA-PP1. A, chemical structure of 2MB-PP1. B and C, interactions of 2MB-PP1 in the nucleotide-binding pocket of AS PKCδ. B, shown are the superimposed structure of 2MB-PP1 (colored by atom type) and ATP (yellow) in AS PKCδ. Ala-427 is highlighted in purple in AS PKCδ. C, 2MB-PP1-interacting residues in the nucleotide-binding pocket of AS PKCδ. 2MB-PP1 forms hydrogen bonds with Glu-428 and Leu-430 (red number symbols), π-σ interaction with Lys-378 (red arrowhead), and π-π interaction with Phe-633 (red dagger). The gatekeeper is indicated by a red arrow. D, inhibition of AS PKCδ, WT PKCδ, and a commercial mixture of PKC isozymes (PKC) (Calbiochem) by 2MB-PP1 and 1NA-PP1 was measured using fluorescence polarization assays (n = 3). Error bars, S.E.
DISCUSSION
In this report, we demonstrate that AS PKCδ can use N6-(benzyl)-ATP as a phosphate donor to phosphorylate immediate substrates of PKCδ. One of the limitations of this approach is that ATP analogs, such as N6-(benzyl)-ATP, are not cell-permeable (5, 6). One approach is to identify substrates in cell lysates, which may result in identifying proteins that do not have access to the kinase in intact cells. To compensate for this potential pitfall, ATP analogs have been successfully delivered to detergent-permeabilized cells to detect substrates within intact intracellular compartments (17, 36, 37).
We also found that AS PKCδ can be selectively inhibited by 1NA-PP1 and 2MB-PP1 in vitro and in intact neutrophils. Generation of O2⨪ by neutrophils is an important mechanism in innate immunity and contributes to the pathogenesis of many diseases (38). PKC isoforms have been implicated in O2⨪ production, because stimulation of neutrophils with PMA results in a robust oxidative burst (39). Using cytosol-depleted neutrophil cores, Brown et al. (40) identified the role of PKCδ in NADPH oxidase activation and O2⨪ generation. The marked reduction of O2⨪ production in PKCδ-deficient neutrophils supports PKCδ as the critical component for O2⨪ production (4). However, selective PKCδ peptide inhibitor did not show inhibitory effect on O2⨪ production (41). Although this peptide inhibitor was designed to disrupt protein-protein interactions with the regulatory domain of PKCδ, its design was not based on knowledge of PKCδ binding partners specific to neutrophils. Therefore, such a negative result with this peptide does not eliminate a role for PKCδ in neutrophil activation. Instead, our approach, which targets the catalytic activity of PKCδ, does not depend on prior knowledge about binding partners that may be cell type-specific. Indeed, 1NA-PP1 specifically reduced the O2⨪ production from the neutrophils of AS PKCδ knock-in mice (Fig. 6), suggesting that the kinase activity of PKCδ is indispensable in generating O2⨪ in neutrophils.
PP1 analogs are rapid, stable, reversible, cell-permeable inhibitors (5, 6), as shown for a single application of 100 nm 1NM-PP1 or 1NA-PP1, which gave sustained inhibition within 20 min and lasted for at least 3 days in cortical neurons from AS TrkB knock-in mice (44). Additionally, removal of 1NM-PP1 for 2 h restored the kinase activity of AS TrkB. Similarly, in mouse embryo fibroblasts from AS JNK2 knock-in mice, JNK2 kinase activity was completely inhibited by 10 μm 1NM-PP1 in 15 min and recovered 15 min following 1NM-PP1 removal (45).
Although N6-(benzyl)-ATP and PP1 analogs are highly selective for AS kinases, off-target effects have been described (5, 6). We investigated one possible off-target family of proteins, membrane-bound mammalian adenylyl cyclases (mAC), which possess large hydrophobic pockets in their catalytic domains that accommodate bulky substituents in nucleotides, such as MANT (27, 46). Our docking studies suggested that N6-ATP and PP1 analogs are unlikely to be specific ligands for mAC (Fig. 13). A recent paper examined off-target effects of PP1 analogs with several hundred WT kinases (6). Although some kinases are weakly inhibited by PP1 analogs in vitro, they are not likely to be bona fide targets in cells due to competition with the high concentration of ATP inside cells (2–10 mm).
FIGURE 13.

Interactions of N6-(benzyl)-ATP and 1NA-PP1 with mAC. A, superimposed structures of N6-(benzyl)-ATP (yellow) and MANT-GTP (colored by atom type) in the catalytic site of mAC. The residues (Thr-408, Ala-409, Gln-410, and Glu-411) in the hydrophobic pocket of mAC facing the MANT group are highlighted in green. The Trp-1020 residue close to the guanine ring of MANT-GTP is highlighted in magenta. B, superimposed structures of 1NA-PP1 (yellow) and MANT-GTP (colored by atom type) in the catalytic site of mAC. The naphthyl ring of 1NA-PP1 forms a steric clash with one of hydrophobic residues (Trp-1020) highlighted in magenta.
Our in silico docking studies support the chemical-genetics approach for AS kinases (5, 6). Placement of the benzyl ring of N6-(benzyl)-ATP in the cavity created by the M427A mutation allowed stable interaction with the glycine-rich loop for efficient phosphoryl transfer. The naphthyl and 2-methylbenzyl rings of 1NA-PP1 and 2MB-PP1 were similarly accepted in the cavity, permitting 1NA-PP1 and 2MB-PP1 to compete with ATP for stable interactions with Glu-428 and Leu-430 as well as the invariant Lys-378 and Phe-633 of AS PKCδ. In contrast, the insensitivity of WT PKCδ for PP1 and N6-ATP analogs could be explained by steric clash of the naphthyl ring of 1NA-PP1 and the benzyl ring of N6-(benzyl)-ATP with the large Met-427 gatekeeper.
The glycine-rich loop (G356KGSFGK), invariant Lys-378, gatekeeper Met-427, two residues near the gatekeeper (Glu-428 and Leu-430), and Phe-633 are associated with 1NA-PP1, 2MB-PP1, and N6-(benzyl)-ATP binding in AS PKCδ. These residues are in the amino-terminal lobe of the PKCδ catalytic domain. The catalytic domain for substrate phosphorylation is highly conserved in the AGC family (1, 29), and the phosphoryl transfer event has been well characterized in PKA (29, 30). The invariant Lys interacts with α- and β-phosphates, and the glycine-rich loop interacts with β- and γ-phosphates to orient ATP for catalysis. Thus, disrupting these interactions would be predicted to impede ATP binding and substrate phosphorylation.
The gatekeeper lies in the middle of the catalytic domain and is a key residue for determining the selectivity of inhibitors (1). This residue is conserved as a large hydrophobic amino acid. No kinases with a small Gly or Ala gatekeeper residue have been found in the human kinome (47). The side chain of the gatekeeper controls the relative accessibility of a hydrophobic pocket located adjacent to the N6 amine of ATP. Moreover, the gatekeeper residue may also regulate the autocatalytic activity of ERK2 (48) and the flexibility of PKA (49).
Staurosporine is a nonspecific kinase inhibitor, inhibiting more than 90% of kinases (50). Interestingly, we found that AS PKCδ is less sensitive to staurosporine inhibition (Fig. 3). Mutation of the gatekeeper residue usually causes minimal change in kinase activity (47) but might confer resistance to inhibitors. The size of the gatekeeper residue has been correlated with the binding affinity for staurosporine (51). The group of kinases with larger gatekeeper residues (e.g. Met or Phe) tends to have stronger binding affinity for staurosporine, whereas smaller gatekeepers (e.g. Thr or Leu) are associated with weaker binding affinities. Mutations at gatekeeper residues have been reported in BCR-ABL1 (breakpoint cluster region-Abelson murine leukemia viral oncogene homolog 1) (T315I), PDGFRα (platelet-derived growth factor receptor α) (T674I), EGF receptor (T790M), and KIT (T670I) (52). These mutations confer resistance to several kinase inhibitors (53).
PKC isozymes have unique and sometimes opposing roles in both normal cellular physiology and various disease states (1, 42). PKC transduces its signals by both protein-protein interactions and substrate phosphorylation. Developing specific PKC isozyme inhibitors has been challenging due to their closely related structures (43). Selective PKC isozyme translocation inhibitors have been developed to disrupt protein-protein interactions with receptors for activated C kinase (RACKs) (42, 43), but the respective RACK proteins have not been disclosed for every PKC isozyme (1). The chemical-genetics approach we used here provides not only a method to specifically interrogate the role of individual kinases in cell signaling but also a method to reveal kinase-specific sets of phosphorylated substrates. The AS PKCδ models herein will not only be useful in identifying PKCδ-specific substrates and designing modulators in silico to manipulate PKCδ kinase activity but will also provide a foundation for future application of this chemical-genetics approach to other protein kinases.
Acknowledgments
We thank Professor Shokat and members of the Shokat laboratory at the University of California, San Francisco, for providing the ATP and PP1 analogs.
This work was supported, in whole or in part, by National Institutes of Health Grants R21 NS057195 (to W. H. C.) and R01 AA018316 (to R. O. M.). This work was also supported by a grant from the Kent State University start-up fund (to W. H. C.).
- AS
- analog-specific
- PP1
- 4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- MANT
- 2′(3′)-O-(N-methylanthraniloyl)
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- PMA
- phorbol 12-myristate 13-acetate
- PDB
- Protein Data Bank
- mAC
- mammalian adenylyl cyclase(s)
- RACK
- receptor for activated C kinase
- 1NA-PP1
- 1-(tert-butyl)-3-(1-naphthyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine
- 2MB-PP1
- 1-(tert-butyl)-3-(2-methylbenzyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine
- 3MB-PP1
- 1-(tert-butyl)-3-(3-methylbenzyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine
- 1NM-PP1
- 1-tert-butyl-3-(1-naphthalenylmethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine
- 2NM-PP1
- 1-tert-butyl-3-(2-naphthalenylmethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine.
REFERENCES
- 1. Steinberg S. F. (2008) Structural basis of protein kinase C isoform function. Physiol. Rev. 88, 1341–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Newton A. C. (2010) Protein kinase C: poised to signal. Am. J. Physiol. Endocrinol. Metab. 298, E395–E402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Choi D. S., Wei W., Deitchman J. K., Kharazia V. N., Lesscher H. M., McMahon T., Wang D., Qi Z. H., Sieghart W., Zhang C., Shokat K. M., Mody I., Messing R. O. (2008) Protein kinase Cδ regulates ethanol intoxication and enhancement of GABA-stimulated tonic current. J. Neurosci. 28, 11890–11899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chou W. H., Choi D. S., Zhang H., Mu D., McMahon T., Kharazia V. N., Lowell C. A., Ferriero D. M., Messing R. O. (2004) Neutrophil protein kinase Cδ as a mediator of stroke-reperfusion injury. J. Clin. Invest. 114, 49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bishop A. C., Buzko O., Shokat K. M. (2001) Magic bullets for protein kinases. Trends Cell Biol. 11, 167–172 [DOI] [PubMed] [Google Scholar]
- 6. Zhang C., Lopez M. S., Dar A. C., Ladow E., Finkbeiner S., Yun C. H., Eck M. J., Shokat K. M. (2013) Structure-guided inhibitor design expands the scope of analog-sensitive kinase technology. ACS Chem. Biol. 8, 1931–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Allen J. J., Lazerwith S. E., Shokat K. M. (2005) Bio-orthogonal affinity purification of direct kinase substrates. J. Am. Chem. Soc. 127, 5288–5289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Habelhah H., Shah K., Huang L., Burlingame A. L., Shokat K. M., Ronai Z. (2001) Identification of new JNK substrate using ATP pocket mutant JNK and a corresponding ATP analogue. J. Biol. Chem. 276, 18090–18095 [DOI] [PubMed] [Google Scholar]
- 9. Shah K., Shokat K. M. (2002) A chemical genetic screen for direct v-Src substrates reveals ordered assembly of a retrograde signaling pathway. Chem. Biol. 9, 35–47 [DOI] [PubMed] [Google Scholar]
- 10. Eblen S. T., Kumar N. V., Shah K., Henderson M. J., Watts C. K., Shokat K. M., Weber M. J. (2003) Identification of novel ERK2 substrates through use of an engineered kinase and ATP analogs. J. Biol. Chem. 278, 14926–14935 [DOI] [PubMed] [Google Scholar]
- 11. Ubersax J. A., Woodbury E. L., Quang P. N., Paraz M., Blethrow J. D., Shah K., Shokat K. M., Morgan D. O. (2003) Targets of the cyclin-dependent kinase Cdk1. Nature 425, 859–864 [DOI] [PubMed] [Google Scholar]
- 12. Hindley A. D., Park S., Wang L., Shah K., Wang Y., Hu X., Shokat K. M., Kolch W., Sedivy J. M., Yeung K. C. (2004) Engineering the serine/threonine protein kinase Raf-1 to utilise an orthogonal analogue of ATP substituted at the N6 position. FEBS Lett. 556, 26–34 [DOI] [PubMed] [Google Scholar]
- 13. Larochelle S., Batliner J., Gamble M. J., Barboza N. M., Kraybill B. C., Blethrow J. D., Shokat K. M., Fisher R. P. (2006) Dichotomous but stringent substrate selection by the dual-function Cdk7 complex revealed by chemical genetics. Nat. Struct. Mol. Biol. 13, 55–62 [DOI] [PubMed] [Google Scholar]
- 14. Chou W. H., Wang D., McMahon T., Qi Z. H., Song M., Zhang C., Shokat K. M., Messing R. O. (2010) GABAA receptor trafficking is regulated by protein kinase Cϵ and the N-ethylmaleimide-sensitive factor. J. Neurosci. 30, 13955–13965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu D. F., Chandra D., McMahon T., Wang D., Dadgar J., Kharazia V. N., Liang Y. J., Waxman S. G., Dib-Hajj S. D., Messing R. O. (2012) PKCepsilon phosphorylation of the sodium channel NaV1.8 increases channel function and produces mechanical hyperalgesia in mice. J. Clin. Invest. 122, 1306–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hanke J. H., Gardner J. P., Dow R. L., Changelian P. S., Brissette W. H., Weringer E. J., Pollok B. A., Connelly P. A. (1996) Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J. Biol. Chem. 271, 695–701 [DOI] [PubMed] [Google Scholar]
- 17. Allen J. J., Li M., Brinkworth C. S., Paulson J. L., Wang D., Hübner A., Chou W. H., Davis R. J., Burlingame A. L., Messing R. O., Katayama C. D., Hedrick S. M., Shokat K. M. (2007) A semisynthetic epitope for kinase substrates. Nat. Methods 4, 511–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bell R. M. (1986) Protein kinase C activation by diacylglycerol second messengers. Cell 45, 631–632 [DOI] [PubMed] [Google Scholar]
- 19. Lowell C. A., Fumagalli L., Berton G. (1996) Deficiency of Src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J. Cell Biol. 133, 895–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qi Z. H., Song M., Wallace M. J., Wang D., Newton P. M., McMahon T., Chou W. H., Zhang C., Shokat K. M., Messing R. O. (2007) Protein kinase C epsilon regulates γ-aminobutyrate type A receptor sensitivity to ethanol and benzodiazepines through phosphorylation of γ2 subunits. J. Biol. Chem. 282, 33052–33063 [DOI] [PubMed] [Google Scholar]
- 21. Pongracz J., Webb P., Wang K., Deacon E., Lunn O. J., Lord J. M. (1999) Spontaneous neutrophil apoptosis involves caspase 3-mediated activation of protein kinase C-δ. J. Biol. Chem. 274, 37329–37334 [DOI] [PubMed] [Google Scholar]
- 22. Takimura T., Kamata K., Fukasawa K., Ohsawa H., Komatani H., Yoshizumi T., Takahashi I., Kotani H., Iwasawa Y. (2010) Structures of the PKC-ι kinase domain in its ATP-bound and apo forms reveal defined structures of residues 533–551 in the C-terminal tail and their roles in ATP binding. Acta Crystallogr. D Biol. Crystallogr. 66, 577–583 [DOI] [PubMed] [Google Scholar]
- 23. Zheng J., Knighton D. R., ten Eyck L. F., Karlsson R., Xuong N., Taylor S. S., Sowadski J. M. (1993) Crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MgATP and peptide inhibitor. Biochemistry 32, 2154–2161 [DOI] [PubMed] [Google Scholar]
- 24. Zheng J., Trafny E. A., Knighton D. R., Xuong N. H., Taylor S. S., Ten Eyck L. F., Sowadski J. M. (1993) 2.2 Å refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr. D Biol. Crystallogr. 49, 362–365 [DOI] [PubMed] [Google Scholar]
- 25. Trott O., Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 27. Mou T. C., Gille A., Fancy D. A., Seifert R., Sprang S. R. (2005) Structural basis for the inhibition of mammalian membrane adenylyl cyclase by 2′(3′)-O-(N-Methylanthraniloyl)-guanosine 5′-triphosphate. J. Biol. Chem. 280, 7253–7261 [DOI] [PubMed] [Google Scholar]
- 28. Carugo O., Pongor S. (2001) A normalized root-mean-square distance for comparing protein three-dimensional structures. Protein Sci. 10, 1470–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kornev A. P., Haste N. M., Taylor S. S., Eyck L. F. (2006) Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. U.S.A. 103, 17783–17788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Taylor S. S., Zhang P., Steichen J. M., Keshwani M. M., Kornev A. P. (2013) PKA: lessons learned after twenty years. Biochim. Biophys. Acta 1834, 1271–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leonard T. A., Różycki B., Saidi L. F., Hummer G., Hurley J. H. (2011) Crystal structure and allosteric activation of protein kinase C βII. Cell 144, 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Witucki L. A., Huang X., Shah K., Liu Y., Kyin S., Eck M. J., Shokat K. M. (2002) Mutant tyrosine kinases with unnatural nucleotide specificity retain the structure and phospho-acceptor specificity of the wild-type enzyme. Chem. Biol. 9, 25–33 [DOI] [PubMed] [Google Scholar]
- 33. Niswender C. M., Ishihara R. W., Judge L. M., Zhang C., Shokat K. M., McKnight G. S. (2002) Protein engineering of protein kinase A catalytic subunits results in the acquisition of novel inhibitor sensitivity. J. Biol. Chem. 277, 28916–28922 [DOI] [PubMed] [Google Scholar]
- 34. Burley S. K., Petsko G. A. (1985) Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science 229, 23–28 [DOI] [PubMed] [Google Scholar]
- 35. Boehr D. D., Farley A. R., Wright G. D., Cox J. R. (2002) Analysis of the π-π stacking interactions between the aminoglycoside antibiotic kinase APH(3′)-IIIa and its nucleotide ligands. Chem. Biol. 9, 1209–1217 [DOI] [PubMed] [Google Scholar]
- 36. Banko M. R., Allen J. J., Schaffer B. E., Wilker E. W., Tsou P., White J. L., Villén J., Wang B., Kim S. R., Sakamoto K., Gygi S. P., Cantley L. C., Yaffe M. B., Shokat K. M., Brunet A. (2011) Chemical genetic screen for AMPKα2 substrates uncovers a network of proteins involved in mitosis. Mol. Cell 44, 878–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moffat L. D., Brown S. B., Grassie M. E., Ulke-Lemée A., Williamson L. M., Walsh M. P., MacDonald J. A. (2011) Chemical genetics of zipper-interacting protein kinase reveal myosin light chain as a bona fide substrate in permeabilized arterial smooth muscle. J. Biol. Chem. 286, 36978–36991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mayadas T. N., Cullere X., Lowell C. A. (2014) The multifaceted functions of neutrophils. Annu. Rev. Pathol. 9, 181–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DeChatelet L. R., Shirley P. S., Johnston R. B., Jr. (1976) Effect of phorbol myristate acetate on the oxidative metabolism of human polymorphonuclear leukocytes. Blood 47, 545–554 [PubMed] [Google Scholar]
- 40. Brown G. E., Stewart M. Q., Liu H., Ha V. L., Yaffe M. B. (2003) A novel assay system implicates PtdIns(3,4)P(2), PtdIns(3)P, and PKCδ in intracellular production of reactive oxygen species by the NADPH oxidase. Mol. Cell 11, 35–47 [DOI] [PubMed] [Google Scholar]
- 41. Kilpatrick L. E., Sun S., Li H., Vary T. C., Korchak H. M. (2010) Regulation of TNF-induced oxygen radical production in human neutrophils: role of δ-PKC. J. Leukoc. Biol. 87, 153–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen L., Hahn H., Wu G., Chen C. H., Liron T., Schechtman D., Cavallaro G., Banci L., Guo Y., Bolli R., Dorn G. W., 2nd, Mochly-Rosen D. (2001) Opposing cardioprotective actions and parallel hypertrophic effects of δ PKC and ϵ PKC. Proc. Natl. Acad. Sci. U.S.A. 98, 11114–11119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mochly-Rosen D., Das K., Grimes K. V. (2012) Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 11, 937–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen X., Ye H., Kuruvilla R., Ramanan N., Scangos K. W., Zhang C., Johnson N. M., England P. M., Shokat K. M., Ginty D. D. (2005) A chemical-genetic approach to studying neurotrophin signaling. Neuron 46, 13–21 [DOI] [PubMed] [Google Scholar]
- 45. Jaeschke A., Karasarides M., Ventura J. J., Ehrhardt A., Zhang C., Flavell R. A., Shokat K. M., Davis R. J. (2006) JNK2 is a positive regulator of the cJun transcription factor. Mol. Cell 23, 899–911 [DOI] [PubMed] [Google Scholar]
- 46. Seifert R., Lushington G. H., Mou T. C., Gille A., Sprang S. R. (2012) Inhibitors of membranous adenylyl cyclases. Trends Pharmacol. Sci. 33, 64–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dar A. C., Shokat K. M. (2011) The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu. Rev. Biochem. 80, 769–795 [DOI] [PubMed] [Google Scholar]
- 48. Emrick M. A., Lee T., Starkey P. J., Mumby M. C., Resing K. A., Ahn N. G. (2006) The gatekeeper residue controls autoactivation of ERK2 via a pathway of intramolecular connectivity. Proc. Natl. Acad. Sci. U.S.A. 103, 18101–18106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schauble S., King C. C., Darshi M., Koller A., Shah K., Taylor S. S. (2007) Identification of ChChd3 as a novel substrate of the cAMP-dependent protein kinase (PKA) using an analog-sensitive catalytic subunit. J. Biol. Chem. 282, 14952–14959 [DOI] [PubMed] [Google Scholar]
- 50. Karaman M. W., Herrgard S., Treiber D. K., Gallant P., Atteridge C. E., Campbell B. T., Chan K. W., Ciceri P., Davis M. I., Edeen P. T., Faraoni R., Floyd M., Hunt J. P., Lockhart D. J., Milanov Z. V., Morrison M. J., Pallares G., Patel H. K., Pritchard S., Wodicka L. M., Zarrinkar P. P. (2008) A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132 [DOI] [PubMed] [Google Scholar]
- 51. Tanramluk D., Schreyer A., Pitt W. R., Blundell T. L. (2009) On the origins of enzyme inhibitor selectivity and promiscuity: a case study of protein kinase binding to staurosporine. Chem. Biol. Drug Des. 74, 16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Carter T. A., Wodicka L. M., Shah N. P., Velasco A. M., Fabian M. A., Treiber D. K., Milanov Z. V., Atteridge C. E., Biggs W. H., 3rd, Edeen P. T., Floyd M., Ford J. M., Grotzfeld R. M., Herrgard S., Insko D. E., Mehta S. A., Patel H. K., Pao W., Sawyers C. L., Varmus H., Zarrinkar P. P., Lockhart D. J. (2005) Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc. Natl. Acad. Sci. U.S.A. 102, 11011–11016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang J., Yang P. L., Gray N. S. (2009) Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39 [DOI] [PubMed] [Google Scholar]