

Background: cGKII acutely inhibits brush-border NHE3, but the mechanism is unknown.
Results: cGMP/cGKII phosphorylates NHE3 at three sites. All are necessary for NHE3 inhibition. One of these is also phosphorylated by SGK1 to stimulate NHE3.
Conclusion: cGKII inhibits NHE3 by phosphorylating and reducing NHE3 surface amount.
Significance: Phosphorylation of the same site in a protein can alter function differently based on phosphorylation of additional sites.
Keywords: Cyclic GMP (cGMP), Epithelial Cell, Intestine, Phosphorylation, Sodium-proton Exchange, Trafficking
Abstract
The epithelial brush-border Na+/H+ exchanger NHE3 is acutely inhibited by cGKII/cGMP, but how cGKII inhibits NHE3 is unknown. This study tested the hypothesis that cGMP inhibits NHE3 by phosphorylating it and altering its membrane trafficking. Studies were carried out in PS120/NHERF2 and in Caco-2/Bbe cells overexpressing HA-NHE3 and cGKII, and in mouse ileum. NHE3 activity was measured with 2′,7′-bis(carboxyethyl)-S-(and 6)carboxyfluorescein acetoxy methylester/fluorometry. Surface NHE3 was determined by cell surface biotinylation. Identification of NHE3 phosphorylation sites was by iTRAQ/LC-MS/MS with TiO2 enrichment and immunoblotting with specific anti-phospho-NHE3 antibodies. cGMP/cGKII rapidly inhibited NHE3, which was associated with reduced surface NHE3. cGMP/cGKII increased NHE3 phosphorylation at three sites (rabbit Ser554, Ser607, and Ser663, equivalent to mouse Ser552, Ser605, and Ser659), all of which had to be present at the same time for cGMP to inhibit NHE3. NHE3-Ser663 phosphorylation was not necessary for cAMP inhibition of NHE3. Dexamethasone (4 h) stimulated wild type NHE3 activity and increased surface expression but failed to stimulate NHE3 activity or increase surface expression when NHE3 was mutated to either S663A or S663D. We conclude that 1) cGMP inhibition of NHE3 is associated with phosphorylation of NHE3 at Ser554, Ser607, and Ser663, all of which are necessary for cGMP/cGKII to inhibit NHE3. 2) Dexamethasone stimulates NHE3 by phosphorylation of a single site, Ser663. The requirement for three phosphorylation sites in NHE3 for cGKII inhibition, and for phosphorylation of one of these sites for dexamethasone stimulation of NHE3, is a unique example of regulation by phosphorylation.
Introduction
The Na+/H+ exchanger NHE3 is expressed in the intestinal brush border. NHE3 is regulated by second messengers during digestion and inhibited in diarrheal diseases. cAMP and cGMP both inhibit NHE3 activity when added directly to intestinal cells and when intracellular cAMP and cGMP levels are increased by agents that produce diarrhea, for example, cholera toxin for cAMP and heat stable Escherichia coli enterotoxin for cGMP. cAMP acts by phosphorylating NHE3 primarily at two amino acids in the mouse intestine and kidney: Ser552 and Ser605 (our studies are with rabbit NHE3 and we refer to these amino acids as Ser554 and Ser607). Other cAMP consensus sequences in NHE3 have been reported, including Ser330, Ser514, Ser576, Ser662, Ser691, Ser692, and Ser805. However, none of these have been shown to affect NHE3 activity (1–3). cAMP also reduces the amount of plasma membrane NHE3, which appears to occur by an additional process that follows phosphorylation, and is likely to be stimulation by endocytosis (4). In contrast, how elevated cGMP, which is known to act via brush-border cGKII, inhibits NHE3 activity is unknown. Information lacking includes whether cGMP/cGKII directly phosphorylates NHE3 in vivo and what are the consequences of such phosphorylation on NHE3 trafficking.
In some cases, cGMP regulates intracellular events by mechanisms analogous to those demonstrated for cAMP. However, the effects of cGMP in the small intestine are not fully understood. The intrinsic ileal peptide guanylin and the E. coli heat-stable enterotoxin (STa) both bind to the same brush-border receptor, guanylate cyclase C, and subsequently increase intracellular cGMP content within minutes (5). STa, guanylin, and cGMP all rapidly inhibit small intestinal NaCl-linked absorption, principally at the level of NHE3, which is an essential component of this sodium-absorptive process (5). This effect on NHE3 is specific because other brush-border transporters, including NHE2 and SGLT1, are not acutely altered during this process. The downstream effect of cGMP on ion and fluid transport in the small intestinal enterocytes appears to occur entirely via activation of the type II isoform of cGMP-dependent protein kinase in the brush border (6, 7), which we showed previously was part of a NHE3 signaling complex (8). Moreover, previous studies of cGKII identified a number of its phosphorylated substrates, all of which were also phosphorylated by PKA (9).
NHE3 and cAMP-dependent protein kinase type II (PKAII) are part of the same signaling complex that is scaffolded by either NHERF1 or NHERF2, which are multi-PDZ domain scaffolding proteins (10, 11). Based on the cell type, cAMP inhibition of NHE3 requires NHERF1 or NHERF2 (12), both of which bind ezrin, which is currently thought to act as an A-kinase anchoring protein (AKAP) to position PKAII so it can phosphorylate NHE3 (11, 13, 14). Nonetheless, the role of ezrin in NHE3 phosphorylation has been questioned recently (15). We previously reported that cGMP inhibition of NHE3 requires NHERF2, an effect not duplicated by NHERF1, and that NHERF2 links cGKII into a NHE3 signaling complex (8).
Sites of NHE3 phosphorylation by cGMP/cGKII have not been identified. Therefore, the current study tested the hypothesis that cGMP regulates the brush-border Na+/H+ exchanger NHE3 by phosphorylating it at specific sites to reduce its plasma membrane expression.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Reagents and antibodies were from the following sources as indicated: 8-pCPT-cGMP3 (Life Science Institute); tetramethyl ammonium chloride and 8-Br-cAMP (Sigma); EZ-Link Sulfo-NHS-SS-biotin (Pierce Chemical, Rockford, IL); restriction endonucleases (New England Biolabs, Ipswich, MA); 2′7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester (BCECF-AM) (EMD Millipore, Billerica, MA); protein G-Sepharose (Amersham Biosciences); DNA primers (Operon Biotechnologies, Huntsville, AL); mouse monoclonal anti-hemagglutinin (HA) (Covance Research Products, Princeton, NJ); mouse monoclonal anti-phospho-Ser554 and -Ser607 antibodies and rabbit polyclonal anti-phospho-Ser554 and -Ser607 were from by Dr. Peter Aronson (Yale University, New Haven, CT) (numbers refer to rabbit NHE3).
PS120 Cell Mutagenesis and Transfection
PS120 fibroblasts, which lack all endogenous plasma membrane NHEs, were used for stable expression of rabbit NHE3-S554A, NHE3-S554D, NHE3-S607A, NHE3-S607D, NHE3-S663A, and NHE3-S663D, and NHE3-S554D,S607D,S663D, all with either a triple HA epitope tag at the N terminus (16) or a C-terminal vesicular stomatitis virus glycoprotein epitope tag (17). All mutations were made using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol. The template for mutagenesis was the pcDNA3.1/Neomycin+ vector (EMD Millipore) containing rabbit HA3-NHE3. PS120 cells stably transfected with human NHERF2 were transfected with each rabbit NHE3 plasmid construct using Lipofectamine 2000 (Invitrogen). Transfected cell lines resistant to G418 and hygromycin, where indicated, were additionally selected by exposing cells to repetitive cycles of acid loading, as described previously (18, 19). All PS120 cell lines were grown in DMEM supplemented with 25 mm NaHCO3, 10 mm HEPES, 50 units/ml of penicillin, 50 μg/ml of streptomycin, and 10% fetal bovine serum in a 5% CO2, 95% O2 incubator at 37 °C (also with 400 μg/ml of G418 (Neomycin), and 600 μg/ml of hygromycin).
Cell Culture
For transport assays, stably transfected PS120 cells were grown on glass coverslips to ∼70% confluence and studied after serum starvation for 3 h. For cGKII virus or empty vector infection, 50–60% confluent cells were incubated with cGKII viral particles or empty vector in serum-free medium at 37 °C for 6 h and then replaced with normal growth medium. OK proximal tubule cells, which lack endogenous NHERF2 and cGKII, were stably transfected with rabbit NHE3 with the HA3 epitope tag at the N terminus and human NHERF2. OK cells were maintained in high glucose Dulbecco's modified Eagle's medium supplemented with 25 mm NaHCO3, 10 mm HEPES, 1 mm sodium pyruvate, 10% fetal bovine serum, 100 units/ml of penicillin, and 50 μg/ml of streptomycin, 400 μg/ml of G418 (Neomycin) and/or 600 μg/ml of hygromycin in a 5% CO2, 95% O2 humidified incubator at 37 °C. For cGKII virus or empty vector infection, OK cells were treated with 6 mm EGTA in serum-free OK medium for 2 h at 37 °C before viral infection to allow virus exposure to the basolateral in addition to the apical surface. Appropriate amounts of cGKII viral particles (109-1010 particles/ml) were diluted in serum-free OK medium and added to EGTA-treated cells. Cells were infected by incubating at 37 °C for 6 h and then the medium was replaced with normal growth medium. For transport assays, OK cells were grown to confluence on glass coverslips and polarized cells were used ∼48 h after infection.
The Caco-2/Bbe cell line, originally derived from a human adenocarcinoma, was obtained from M. Mooseker (Yale University, New Haven, CT) and J. Turner (University of Chicago, Chicago, IL) (20). Cells were maintained in DMEM + 25 mm NaHCO3, 0.1 mm nonessential amino acids, 10% fetal bovine serum, glutamine (4 mm), 50 units/ml of penicillin, and 50 μg/ml of streptomycin, pH 7.4, at 37 °C in 5% CO2. To achieve polarity, cells were cultured on 6-well collagen-coated dishes (Transwell filters, 0.02 μm, Corning) for 12 days after becoming confluent. For virus infection, cells were first exposed to 6 mm EGTA in serum-free medium for 2 h at 37 °C to increase virus access to the basolateral surface; then 109–1010 viral particles/ml in serum-free medium were added to both Transwell chambers. After incubating for more than 6 h, cells were rinsed and incubated further in normal growth medium.
Mass Spectrometry Analysis and Database Searching
Total membrane proteins were extracted from mouse ileum pretreated with an equal concentration of DMSO as a control or 100 μm 8-pCPT-cGMP. Each protein extract was digested with trypsin and the peptides were labeled with iTRAQ reagent according to the manufacturer's instructions. iTRAQ-labeled peptides from different samples (up to 8) were mixed and separated into 24 fractions by strong cation exchange chromatography. Ten percent of the peptides in each strong cation exchange fraction were analyzed by reverse phase chromatography coupled to a tandem mass spectrometer (LC-MS/MS) using a Waters Nanoacquity UPLC interfaced with a Thermo Scientific LTQ Orbitrap Velos with 0.03 Da mass accuracy on fragment ions. The remaining 90% of the strong cation exchange fractions were enriched for phosphopeptides using TiO2 before LC-MS/MS analysis. The MS/MS spectra were extracted and searched for phosphorylation on Ser, Thr, and Tyr against the Refseq database using Mascot (Matrix Science) through Proteome Discoverer software (version 1.2, Thermo Scientific) with extract, specifying species, enzyme allowing one missed cleavage, fixed cysteine methylthiolation and variable methionine oxidation, and 8-plex-iTRAQ labeling of N-terminal Lys and Tyr. Peptide identifications from Mascot searches were processed within Proteome Discoverer to identify peptides with a confidence threshold 1% false discovery rate, based on a concatenated decoy database search to calculate the median protein and peptide ratios. Technical variation was <10% as determined from four iTRAQ 8-plex experiments using 32 technical replicates of immuno-depleted human plasma samples.
Normalization of Mass Spectrometry Data
Quantitative mass spectrometry data from total membrane proteins from the ileum with and without exposure to 8-pCPT-cGMP underwent normalization to quantitate differential phosphorylation at three sites in NHE3. Peptides from tryptic digests of protein extracted from control or cGMP-treated ileum were labeled with unique iTRAQ isobaric mass tags and identified using mass spectrometry before and after TiO2 enrichment for phosphopeptides. Fragmentation spectra revealed the peptide sequences and reporter ions corresponding to each iTRAQ tag. The relative intensities of the reported ions reflect the relative amounts of the peptide present in each sample. To normalize for variations in sample preparation, the median reporter ion signal per sample was first determined for all peptide sequences in a sample. All sample values were then normalized to minimize technical variations between samples, such as amount of loading. The median value for each sample was divided by the normalized median of the total sample to determine their relative concentrations, and thus the normalization factor per sample. Because both samples from control and the 8-pCPT-cGMP samples were mixed prior to MS analysis and TiO2 enrichment of phosphopeptides, the same sample-specific normalization factor was applied to each phosphopeptide identified after TiO2 enrichment. This yielded the relative fold-changes in the three NHE3 phosphopeptides as shown in Fig. 3, A–C.
FIGURE 3.


Quantitative mass spectrometry and truncation studies identified a novel NHE3 phosphorylation site, Ser663, which is necessary for cGMP inhibition of NHE3. A–C, identification and quantification of phosphosites in iTRAQ-labeled phosphopeptides: RLESFK (A), VQIPNSPSNFR (B), SVDSFLQADGHEEQLQPAAPESTHM (C). Fragmentation spectrum (MS2) of ion 383.2 m/z identifying RLESFK phosphorylation at Ser4 (mouse Ser659 (rabbit Ser663)), VQIPNSPSNFR phosphorylation at Ser6 (Ser789 (rabbit Ser797)), and SVDSFLQADGHEEQLQPAAPESTHM phosphorylation at Ser4 (Ser808 (rabbit Ser816)). Results in the box show the normalized quantitative mass spectrometry amount of phosphopeptide in the presence/absence of 8-pCPT-cGMP with all data determined using the reporter ions and normalization performed as described under “Experimental Procedures.” Reporter ions are on the left with low mass range. i = iTRAQ labeled, p = phosphorylated, o = oxidized. In mass spectroscopy studies of Ser or Thr phosphopeptides, fragmentation removes the phosphate group plus a water thus reducing the peptide mass by 98 Da (mass of H3PO4) from the calculated phosphopeptide mass and produces a neutral loss ion. This neutral loss ion from the phosphorylated Ser/Thr phosphopeptide is typically the most intense ion in the fragmentation spectrum and is diagnostic of a phosphopeptide. Moreover, if the charge is on the N terminus of the neutral loss peptide, when the peptide bond breaks, the convention is to label the fragment ion a b ion. If the charge is on the C terminus, the observed fragment ion is called a y ion. The phosphorylation site in a phosphopeptide can be determined when losses of H3PO4 start to occur in the b or y ion series. Thus in the example of Ser659, y1 and y2 show no loss of water-phosphate but starting at y3 there is loss of 98 Da (H3PO4) from the calculated y3 mass. D, in PS120/NHERF2/HA-NHE3/cGKII cells, 8-pCPT-cGMP had no effect on NHE3 activity when NHE3 was truncated to amino acid 660 (NHE3/Δ660); however, 8-pCPT-cGMP inhibited NHE3 activity when truncated to amino acid 690 (NHE3/Δ690) (p < 0.05), similarly to that in wild type cells (mean ± S.E., n = 3).
Measurement of Na+/H+ Exchange Activity
Na+/H+ exchange activity in cells expressing HA-NHE3 was determined fluorometrically using the intracellular pH-sensitive dye BCECF-AM (10 μm). Polarized OK cells on glass coverslips were studied 3 days after confluence. PS120 cells were studied when ∼70% confluent as described previously (21). Cells were loaded for 30 min at 37 °C with 10 μm BCECF-AM in Na+/NH4Cl solution (98 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgSO4, 1 mm NaH2PO4, 25 mm glucose, 20 mm HEPES, and 40 mm NH4Cl, pH 7.4). During dye loading and NH4Cl prepulse, cells were treated with 100 μm 8-pCPT-cGMP, 25 μm forskolin (FSK), or a DMSO concentration control. 2 μm Dexamethasone was preincubated for 4 h. The slides were placed in a fluorometer (Photon Technology International, Lawrenceville, NJ), cells were initially perfused with TMA+ solution (130 mm tetramethylammonium chloride (TMF), 5 mm KCl, 2 mm CaCl2, 1 mm MgSO4, 1 mm NaH2PO4, 25 mm glucose, 20 mm HEPES, pH 7.4), which was then switched to Na+ solution (130 mm NaCl instead of TMA-Cl) for the Na+-dependent pHi recovery. At the end of each experiment, the fluorescence ratio was calibrated to pHi using the high potassium/nigericin method (17, 19). For PS120 cells, Na+/H+ exchange activity data were calculated as the product of the change in pHi (ΔpHi/dt) × the intracellular buffering capacity at each pHi at multiple pHi values using at least three coverslips per condition in a single experiment. Kinetics of Na+/H+ exchange were analyzed by a Hill plot using Origin (Microcal Software) to estimate Vmax and K′(H+)i in an individual experiment (19). Mean ± S.E. were determined from at least three experiments. For OK epithelial cells, initial rates of Na+-dependent intracellular alkalinization were calculated starting at pHi 6.3 over the initial 1-min Na+ exposure (linear phase of pHi change) and expressed as ΔpH/ΔT in an individual experiment. Mean ± S.E. were determined from at least three experiments.
Measurement of Buffering Capacity
PS120/NHERF2 or PS120/NHERF2/cGKII cells were incubated with/without 8-pCPT-cGMP (100 μm) during the 10 μm BCECF-AM dye loading in Na+ solution (130 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgSO4, 1 mm Na-PO4, 25 mm glucose, and 20 mm HEPES, pH 7.4) for 30 min at 37 °C. The cells were then initially washed with Na+-free TMA+ solution (see above) to remove excess dye. Cells were then perfused with TMA+ solution and exposed to stepwise changes in extracellular concentrations of Na+ free-NH4Cl (15, 10, 8, 6, 4, 2, and 0 mm) from high to low concentration followed by exposure to three K-Clamps (pH 6.0, 6.6, and 7.3) with the K+/nigericin method. The stepwise Na+-free NH4Cl solutions were prepared by dilution of 40 mm Na+-free NH4Cl solution (40 mm NH4Cl, 90 mm TMA, 5 mm KCl, 2 mm CaCl2, 1 mm MgSO4, 1 mm TMA-PO4, 25 mm glucose, and 20 mm HEPES, pH 7.4) with the TMA+ solution. pH was adjusted with TMA-OH.
Buffering Capacity (mm/pH) was calculated for each pH interval according to the following equation (22),
 |
where the intracellular NH4+ concentration was calculated according to the Henderson-Hasselbach equation,
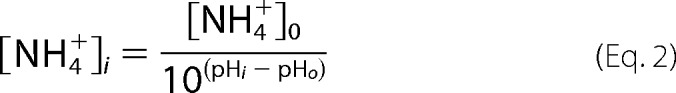 |
[NH3]i was ignored in the physiological pH range (pH < 7.5) and the pH for each buffering capacity interval was determined as the average value over that pH interval.
Quantitation of Surface NHE3 by Biotinylation
NHS-SS-biotin was used to determine the percentage of total NHE3 on the surface of PS120 cells, as described previously (23). PS120 cells grown on 10-cm dishes were infected with cGKII virus or empty vector and studied 48 h later. PS120 cells were serum-starved for 3 h, rinsed three times in cold PBS (150 mm NaCl, 20 mm Na2HPO4, pH 7.4), and once in borate buffer (154 mm NaCl, 1.0 mm boric acid, 7.2 mm KCl, and 1.8 mm CaCl2, pH 8.0), then 1 mg/ml of NHS-SS-biotin in borate buffer was added at 0 and 30 min. Cells were then washed three times with quenching buffer (120 mm NaCl, 20 mm Tris, pH 7.4) to remove unreacted biotin, washed three times in PBS and solubilized with 1 ml of N+ buffer (60 mm HEPES, pH 7.4,150 mm NaCl, 3 mm KCl, 5 mm Na3EDTA, 3 mm EGTA, and 1% Triton X-100). The cell lysate was centrifuged at 5000 rpm for 10 min and the postnuclear supernatant was incubated with streptavidin-agarose beads from 3 h to overnight at 4 °C. After precipitation, the supernatant was retained as the intracellular fraction and the avidin-agarose beads were washed five times in N− buffer (60 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm KCl, 5 mm Na3EDTA, and 3 mm EGTA) with 0.1% Triton X-100 to remove nonspecifically bound proteins. The avidin-agarose bead-bound proteins, which represent plasma membrane NHE3, were solubilized in loading buffer (5 mm Tris-HCl, pH 6.8, 1% SDS, 10% glycerol, and 1% 2-mercaptoethanol), boiled for 10 min, size fractionated by SDS-PAGE (10% gel), and then electrophoretically transferred to nitrocellulose. After blocking with 5% nonfat milk in PBS, the blots were probed with monoclonal anti-HA antibody. Western analysis and quantification of the surface fractions were performed using the Odyssey system and software (LI-COR). Multiple volumes of the total and surface samples were used, and the signal intensity was derived by linear regression to obtain a single value for each sample. The percentage of surface NHE3 was calculated from a ratio ((surface NHE3 signal/total NHE3 signal) × dilution factor of surface and total NHE3 samples) and expressed as percentage of total NHE3 with normalization to GAPDH.
Isolation of Ileal Total Membranes from Mice and Lysate from Cells and Immunoblotting
Ileum was rinsed with ice-cold 0.9% saline and opened along the mesenteric border. Ileum harvested as above was preincubated for 15 min at 37 °C in Ringer's HCO3, 25 mm glucose, gassed at 95% O2, 5% CO2, and then exposed at 37 °C to 20 μm forskolin for 15 min or 100 μm 8-pCPT-cGMP or an equal concentration of DMSO (as control) for 30 min. Then ileal villus cells were scraped and placed into homogenization buffer A (in mm: 60 mannitol, 2.4 Tris, pH 7.1, 1 EGTA, 1 β-glycerol-PO4, 2 Na3VO4, and 1 phenylalanine). Protease inhibitor mixture (1:100; Sigma) and phosphorhamidon (1:1,000) were added to the buffer. Cells were then homogenized at 4 °C with a Polytron (10 times for 10 s at speed 5 with a 20-s interval between each burst) followed by homogenization of samples in a glass-Teflon homogenizer. The homogenates were centrifuged at 5,000 × g for 10 min at 4 °C to remove cell debris and nuclei and designated as total lysates. Lysates were then centrifuged at 40,000 × g for 60 min, and total membrane (TM) pellets were collected. The resulting TMs were resuspended in buffer B (in mm: 300 mannitol, 20 HEPES, pH 7.4, 5 Mg-gluconate, 1 Na3VO4, 1 β-glycerol-PO4, 1 phenylalanine, and protease inhibitors added as in buffer A). The protein concentrations of the TM samples were measured with Bradford protein assay (Sigma) and subsequently analyzed by SDS-PAGE and Western blotting using primary antibodies for NHE3, phospho-NHE3 (Ser554 and Ser607), and actin with fluorescently labeled secondary goat anti-mouse or anti-rabbit IRDye TM 700 or TM 800 antibodies (Rockland). The fluorescence intensity of detected protein bands was visualized by the Odyssey system (LI-COR).
For isolating cell lysates from PS120/NHERF2/NHE3 or Caco-2/Bbe cells, cells were incubated with 100 μm 8-pCPT-cGMP or 100 μm 8-Br-cAMP or an equal concentration of DMSO for 30 min or 20 μm FSK for 15 min, and then lysed in lysis buffer (50 mm Tris-Cl, pH 7.4, 150 mm NaCl, 0.1 mm phenylmethylsulfonyl fluoride, 5 μg/ml of aprotinin, 1 μm pepstatin, 1 mm iodoacetamide, 5 μg/ml of leupeptin, and 1% Triton X-100), followed by centrifugation at 5,000 × g at 4 °C for 10 min. The supernatant were resolved by SDS-PAGE and Western blotting with anti-HA and anti-phospho-NHE3 (Ser554 and Ser607) as the primary antibody.
Statistics
Results were expressed as mean ± S.E. Statistical evaluation was by analysis of variance or Student's t test.
RESULTS
cGMP/cGKII Inhibits NHE3 in PS120 and Polarized OK Cells in a Time-dependent Manner and Reduces NHE3 Surface Expression
Our previous studies done in PS120, OK, or Caco-2/Bbe cells, and mouse ileum showed that cGMP inhibits NHE3 activity using a NHERF2 and cGKII-dependent process (8, 12, 24). To identify the mechanism for this cGMP regulation, we first defined the time course of cGMP inhibition of NHE3. Studies were performed in PS120/NHERF2/NHE3 and OK/NHERF2/NHE3 cells, both transiently infected with replication-deficient adenovirus containing the coding sequence of rat cGKII (25). OK/NHERF2/NHE3 cells were exposed to 100 μm 8-pCPT-cGMP for 15, 30, 45, and 60 min and then Na+/H+ exchange activity was measured. cGMP inhibition of NHE3 activity occurred rapidly, was significant at 15 min after addition of 8-pCPT-cGMP, maximum at 45 min, and lasted for at least 60 min (Fig. 1, A and B). cGMP inhibition of NHE3 also occurred rapidly in PS120/NHERF2/NHE3 cells, being significant by 15 min and maximum by 60 min (Fig. 1, C and D). Buffering capacity was not changed by cGMP, cGKII, or both (Fig. 1E). As a negative control, the inhibition of NHE3 by 8-pCPT-cGMP did not occur in cells infected with empty vector and cells without NHE3 (Fig. 1F).
FIGURE 1.

cGMP inhibits NHE3 and decreases cell surface but not total NHE3. A and B, in OK/NHERF2/HA-NHE3 cells infected with adenovirus-cGKII, 8-pCPT-cGMP inhibition of NHE3 activity occurred rapidly. NHE3 activity was assessed as the initial rates of Na+-dependent intracellular alkalinization (ΔpH/min). A is a single experiment and B is mean ± S.E. of three experiments. C and D, transport activity was measured kinetically in adenovirus-cGKII infected PS120/NHERF2/HA-NHE3 cells. Results were similar to that in OK cells, with rapid inhibition of the NHE3 Vmax (μm/s) (C is a single experiment, and D is mean ± S.E. of three experiments). E, pHi dependence of intrinsic intracellular buffering capacity. Buffering capacity corresponding to each pHi was calculated by measuring ΔpHi in response to different concentrations of NH4Cl (see “Experimental Procedures” for details). PS120/NHERF2 and PS120/NHERF2/cGKII cells were treated with/without 8-pCPT-cGMP (100 μm, 30 min). Curve fitting was generated using a first order exponential decay equation. F, as a negative control, NHE3 transport activity was measured in PS120/NHERF2/HA-NHE3 and PS120/NHERF2 cells with adenovirus-cGKII or empty vector infection. Without cGKII, there was no inhibition of NHE3 activity by 8-pCPT-cGMP. Results are mean ± S.E. of three experiments. G and H, surface NHE3 was decreased in PS120/NHERF2/HA-NHE3 cells by 8-pCPT-cGMP. PS120 cells were grown in 10-cm dishes, and cells were infected with adenovirus-cGKII at 50% confluence. 48 h after infection, cells were serum starved for 3 h and then treated with 100 μm 8-pCPT-cGMP for 45 min at 37 °C before cell surface biotinylation. Detection and quantitation of total and surface amounts of NHE3 are shown in F (single experiments) and G (mean ± S.E., n = 3). Surface expression of NHE3 was determined as a percent of total NHE3 and normalized to GAPDH. 8-pCPT-cGMP-treated cells had ∼35% less surface NHE3 expression than non-treated cells (p < 0.05).
Rapid regulation of NHE3 activity in epithelial cells usually involves, at least in part, changes in the amount of NHE3 at the plasma membrane by changes in regulated endocytosis and/or exocytosis (26, 27). We therefore tested the hypothesis that cGMP inhibition of NHE3 activity might occur by a reduction in the amount of NHE3 on the cell surface. The surface NHE3 amount was determined by cell surface biotinylation in PS120 cells after exposure to 100 μm 8-pCPT-cGMP for 45 min. As shown in Fig. 1, G and H, cell surface NHE3 was reduced ∼35% by 8-pCPT-cGMP exposure (p < 0.05) without a change in total NHE3 expression. Thus, cGMP inhibition of NHE3 activity includes a decrease in NHE3 surface amount, which is consistent with a role for trafficking in this effect. In these studies, normalization of total NHE3 was to GAPDH with different conclusions if actin were used for normalization. We speculate that this might be due to changes in cytoskeleton caused by cGMP/cGKII.
cGMP/cGKII Increases NHE3 Phosphorylation at Ser554 and Ser607, Which Does Not Require PKA
Multiple studies have implicated that two sites of NHE3 phosphorylation are necessary for inhibition of NHE3 in response to stimuli, which act by elevating cAMP/PKA, including dopamine (28–30). In many cases, cGMP regulates intracellular events by mechanisms analogous to those demonstrated for cAMP. Therefore, we tested the hypothesis that cGMP/cGKII phosphorylates NHE3 at the same sites as cAMP. Studies were undertaken by determining NHE3 phosphorylation using well characterized phospho-specific antibodies, anti-Ser(P)554 and anti-Ser(P)607, in PS120 cells (3). As shown in Fig. 2, A-C, both Ser554 and Ser607 were significantly phosphorylated by cGMP/cGKII (p < 0.05) in the same samples in which cAMP/PKA phosphorylated these amino acids. The cGMP/cGKII-, plus cAMP-induced phosphorylations were slightly but not significantly greater than either alone.
FIGURE 2.

Specific phosphoantibody assay identified that 8-pCPT-cGMP/cGKII increased NHE3 phosphorylation at Ser554 and Ser607, with this phosphorylation of Ser554 not requiring PKA. A–C, specific phosphoantibody immunoblotting identified that 8-pCPT-cGMP/cGKII increased NHE3 phosphorylation at two serines: Ser554 and Ser607. A is a single experiment and B (Ser554) and C (Ser607) are densitometric analysis of the phosphorylation levels from multiple experiments (mean ± S.E., n = 3). Both 8-Br-cAMP and 8-pCPT-cGMP increased the phosphorylation levels of Ser554 and Ser607 (p < 0.05); cAMP and 8-pCPT-cGMP do not have statistically significant additive effects on phosphorylation. D and E, in PS120 cells and Caco-2 cells, H89 pretreatment prevented 8-Br-cAMP or forskolin-induced phosphorylation of NHE3 at Ser554 (P-NHE3-Ser554), but it did not prevent the phosphorylation by 8-pCPT-cGMP/cGKII (p < 0.05). Results are the quantitation of phosphorylation levels (mean ± S.E., n = 3), with untreated control set to 100% for each experiment.
In some cases cGMP acts via PKA (31, 32). Thus, we determined whether the phosphorylation of NHE3 by cGMP required PKA. H89 at low concentrations is a relatively specific inhibitor of PKA (30, 33–35). PS120 and Caco-2/Bbe cells were preincubated with 10 μm H89 for 30 min, 20 μm forskolin for 15 min, or 100 μm 8-Br-cAMP or 100 μm 8-pCPT-cGMP for 30 min at 37 °C. Immunoblotting was done with anti-HA and anti-Ser(P)554 antibodies. As shown in Fig. 2, D and E, H89 significantly prevented the phosphorylation of NHE3 at Ser554 by 8-Br-cAMP and forskolin in PS120 (Fig. 2D) and Caco-2/Bbe cells (Fig. 2E), respectively (p < 0.05). However, H89 did not prevent the phosphorylation of NHE3 at Ser554 by 8-pCPT-cGMP/cGKII (Fig. 2, D and E). These results support that phosphorylation of NHE3 at Ser554 by cGMP/cGKII does not involve PKA and that both cGKII and PKA stimulate the phosphorylation of NHE3-Ser554.
Quantitative Mass Spectrometry and Truncation Studies Identify a Novel cGMP/cGKII-induced NHE3 Phosphorylation Site, Ser663
Previous studies of cGKII have identified a number of its phosphorylated substrates, all of which have also been phosphorylated by PKA (9, 37, 38). To identify cGMP-specific phosphorylation sites in NHE3, if they exist, total membrane proteins were extracted from mouse ileum by ultracentrifugation and digested by trypsin. The generated tryptic peptides were then analyzed by quantitative mass spectrometry. As shown in Fig. 3, A–C, three of the identified phosphopeptides from mouse ileum containing Ser659, Ser789, and Ser808, had the amount of phosphorylation increased by cGMP. These are equivalent to Ser663, Ser797, and Ser816 in rabbit NHE3.
Truncation mutations were performed to determine whether any of these sites was functionally important and required for cGMP inhibition of NHE3 activity. As shown in Fig. 3D, unlike cGMP inhibition of wild type NHE3 activity, cGMP had no effect on NHE3 truncated to amino acid 660 (NHE3/Δ660). However, cGMP inhibited the activity of NHE3 truncated to amino acid 690 (NHE3/Δ690V) to a similar extent as that in wild type cells. These results are consistent with NHE3 phosphorylation at Ser663 but not Ser797 and Ser816 being necessary for cGMP/cGKII inhibition of NHE3 activity.
Ser554, Ser607, and Ser663 Must All Be Present for cGMP/cGKII to Inhibit NHE3
Having shown that cGMP/cGKII stimulates the phosphorylation of NHE3 at three sites, Ser554, Ser607, and Ser663, we next studied how these three sites affect the cGMP regulation of NHE3 activity. All three sites were singly mutated from Ser to Ala and Ser to Asp, and triple mutated from Ser to Asp. The engineered constructs were transfected into PS120/NHERF2 cells to make stable cell lines: NHE3-S554A and -S554D, NHE3-S607A and -S607D, NHE3-S663A and -S663D, and NHE3-S554D,S607D,S663D. As shown in Fig. 4, A and B, unlike wild type NHE3 containing cells, which had significant inhibition of NHE3 activity after being exposed to 8-pCPT-cGMP (p < 0.05), single mutant S554A, S607A, and S663A cells completely lost the cGMP/cGKII inhibition of NHE3 activity. Three phosphomimetic mutations, NHE3-S554D, NHE3-S607D, NHE3-S663D, however, were inhibited by cGMP similarly to wild type NHE3 (p < 0.05). These results indicate that the inhibition of NHE3 activity by cGMP requires phosphorylation of all three sites, Ser554, Ser607, and Ser663. To confirm these results, NHE3 activity was measured in NHE3-S554D,S607D,S663D triple mutant cells infected with cGKII. As shown in Fig. 4C, Ser to Asp triple mutant cells lost the inhibition of NHE3 by 8-pCPT-cGMP/cGKII. This result confirmed that changes in phosphorylation in all three sites are required for 8-pCPT-cGMP/cGKII inhibition of NHE3 activity.
FIGURE 4.

The activity of NHE3 in S554A, S607A, and S663A single mutants was not inhibited by 8-pCPT-cGMP/cGKII. A, NHE3 activity (Vmax, μm/s) was determined in adenovirus cGKII-infected PS120/NHERF2/HA-NHE3 WT, and NHE3-S554A, -S607A, -S663A, and -S663D single mutant cells. NHE3 activity was decreased in wild type and S663D cells pretreated with 8-pCPT-cGMP for 45 min (p < 0.05), but not changed significantly by cGMP in all Ser to Ala single mutant cell lines (mean ± S.E., n = 3 except S663D in which n = 5). Also noted is that basal NHE3 activity was significantly increased in S663A mutant cells and significantly reduced in S554A and S607A mutant cell lines (mean ± S.E., n = 3) (p < 0.05). B, NHE3 basal activity was decreased in S554D and S607D mutant cell lines compared with WT cells, and NHE3 activity was significantly further decreased in both cell lines by 8-pCPT-cGMP. C, NHE3 basal activity was decreased in the S554D,S607D,S663D triple mutant cell lines compared with WT cells, and triple mutant cells lost inhibition of NHE3 activity by 8-pCPT-cGMP. D–I, total and cell surface NHE3 expression in WT and mutant cell lines. There were no changes of total NHE3 in NHE3-S554A or -S554D, NHE3-S607A or -S607D, and NHE3-S663A or -S663D mutants compared with wild type cells. Surface NHE3 significantly decreased in NHE3-S554A and -S554D and NHE3-S607A and -S607D cell lines was significantly increased in S663A cells (mean ± S.E., n = 3) (p < 0.05), and not significantly changed in S663D cells. Surface NHE3 in NHE3-S554D and NHE3-S607D cells was significantly reduced by 8-pCPT-cGMP, but not in NHE3-S554A, NHE3-S607A, and NHE3-S663A and -S663D cells. In each experiment, surface expression of WT NHE3 was set to 100%. J, NHE3 activity (Vmax, μm/s) was inhibited by FSK in PS120/NHERF2/HA-NHE3-S663A single mutant cells similarly to WT cells (mean ± S.E., n = 3) (p < 0.05).
These mutants also had changes in basal NHE3 activity, which was dramatically increased in the single mutant NHE3-S663A cell line (p < 0.05) and significantly decreased in single mutant NHE3-S554A, -S554D, -S607A, -S607D and triple mutant NHE3-S554D,S607D,S663D cell lines. To evaluate the explanation for the increase in basal activity of S663A and decrease in basal activity of S554A, S554D, S607A, and S607D, total NHE3 and the percentage of the surface amounts were determined. As shown in Fig. 4, D–I, there was no significant change in total NHE3 expression in NHE3-S554A, -S554D, -S607A, -S607D, -S663A, and -S663D mutant cells compared with wild type cells. However, the amount of surface NHE3 expressed as a percent of total NHE3 was significantly decreased in NHE3-S554A, -S554D, -S607A, and -S607D (p < 0.05) and significantly increased in S663A cells (p < 0.05), but there was no significant change in S663D cells (Fig. 4I). Surface NHE3 was significantly decreased by 8-pCPT-cGMP in NHE3-S554D and -S607D cell lines, but not in NHE3-S554A, -S607A, and -S663A cell lines, which is consistent with changes in NHE3 activity by 8-pCPT-cGMP. The surface amount of NHE3 was not reduced by 8-pCPT-cGMP (Fig. 4, H and I) in S663D mutant cells, which is different from the inhibition of activity of this mutant by 8-pCPT-cGMP. These data further support our hypothesis that cGMP regulates the brush-border Na+/H+ exchanger NHE3 by stimulating the phosphorylation of NHE3 at specific sites to reduce its plasma membrane expression.
To determine whether Ser663 is a specific site for cGMP/cGKII inhibition of NHE3 activity and not for PKA, PS120/NHERF2/NHE3-S663A cells were preincubated with FSK, and NHE3 activity was determined. FSK significantly inhibited NHE3 activity to a similar extent in both wild type cells and S663A-mutated cells (p < 0.05) (Fig. 4J). These data showed that Ser554, Ser607, and Ser663 all are required for cGMP/cGKII to inhibit NHE3, but only Ser663 is a specific site for cGKII inhibition of NHE3.
Dexamethasone Fails to Stimulate NHE3 Activity or Increase Surface Expression When NHE3 Is Mutated to S663A or S663D
NHE3-Ser663 has been reported to be a specific phosphorylation site for SGK1 and to be necessary for dexamethasone to stimulate NHE3 over short times, not involving transcriptional regulation (39, 40). Because our studies showed that cGMP/cGKII phosphorylation of NHE3 at this site inhibited NHE3 activity, we initially confirmed that dexamethasone stimulates NHE3 activity under basal conditions through phosphorylation of Ser663 and increases NHE3 surface expression (Fig. 5, A and B). Further evidence of the role of NHE3-Ser663 phosphorylation in dexamethasone stimulation of NHE3 was provided by studies of S663A and S663D mutants. Dexamethasone did not induce a change of activity in NHE3-S663A or NHE3-S663D (Fig. 5A) or increase the surface expression of these mutants (Fig. 5B).
FIGURE 5.
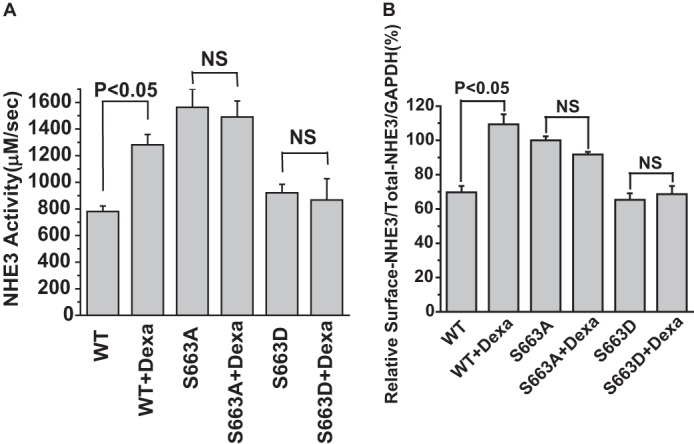
Dexamethasone failed to stimulate NHE3 activity or increase the surface NHE3 amount when NHE3 was mutated to S663A or S663D. A, transport activity (initial rates) was measured in adenovirus-cGKII-infected PS120/NHERF2/HA-NHE3 WT and S663A and S663D single mutant cells. Dexamethasone increased the activity of NHE3 in control cells (p < 0.05), but failed to increase NHE3 activity in either S663A or S663D cells (mean ± S.E., n = 3). B, the surface NHE3 amount was not changed by dexamethasone treatment for 4 h in S663A and S663D mutant cells (mean ± S.E., n = 3). PS120 cells were grown in 10-cm dishes, and cells were infected with adeno-cGKII virus. 48 h after infection, cells were serum starved for 3 h and then treated with 2 μm dexamethasone for 4 h at 37 °C before biotinylation. Surface expression of NHE3 was determined as a percent of total NHE3.
The NHE3 Inhibition by cGMP/cGKII Involves Phosphorylation of NHE3 at Ser554 and Ser607 in Mouse Ileum
To confirm that inhibition of NHE3 activity by cGMP/cGKII involves phosphorylating NHE3 at Ser554, and Ser607, we used polyclonal specific anti-P-Ser554 and anti-P-Ser607 to examine the phosphorylation level of NHE3 in mouse ileum. Western blots were prepared from mouse ileum total membrane. In mouse ileum, NHE3 was phosphorylated at Ser554 and Ser607 by both FSK and cGMP (Fig. 6).
FIGURE 6.
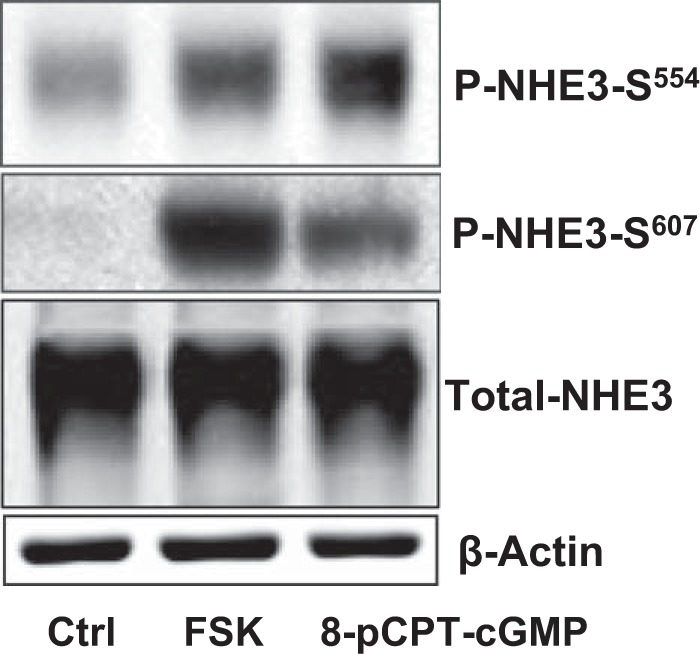
NHE3 phosphorylation by cGMP at Ser554 and Ser607 was verified by specific phosphoantibodies in mouse ileum. Total membrane protein was isolated from mouse ileum after exposure to 100 μm 8-pCPT-cGMP for 30 min or 20 μm FSK for 15 min. Detection of total NHE3 and phospho-NHE3 is shown in a single experiment, which was repeated 4 times with similar results. In mouse ileum, both FSK and cGMP increased the phosphorylation levels of Ser554 and Ser607.
DISCUSSION
This study identified three amino acids of NHE3, which were phosphorylated by cGMP/cGKII, an effect that inhibited NHE3 activity by decreasing the surface expression but not total expression over the short time (up to 1 h) of these studies. These results are consistent with an effect via trafficking, and established the requirement for all three phosphorylation sites at the same time in NHE3 for inhibition by cGKII. These results provide evidence of a unique form of regulation by phosphorylation of NHE3, because we identified a single multifunctional site that, when phosphorylated, causes both stimulation and inhibition of NHE3. In addition, the difference in magnitude of the cGMP/cGKII effect on NHE3 activity, which exceeds the effect on surface expression, suggests an effect on the turnover number as well as an effect on trafficking. These mechanisms of the cGKII/cGMP effects are very similar to the effects of cAMP elevation on NHE3 regulation, as reported in the past (1).
Previous studies of cGKII have identified a number of its phosphorylated substrates, all of which have also been phosphorylated by PKA (9, 37, 38). Serulle et al. (9) showed that cGKII phosphorylates GluR1 at Ser845 in the brain, a site also phosphorylated by PKA. By quantitative mass spectrometry and further truncation studies, we identified Ser663 as a novel cGKII phosphorylation site on NHE3 and showed by functional studies that it is not involved in NHE3 regulation by PKA. By using specific anti-phosphoantibodies, we showed that cGKII also increased the NHE3 phosphorylation at Ser554 and Ser607. The inhibition of NHE3 activity by cGKII was prevented by the single mutation of each of these three sites from Ser to Ala and expression in a model in which WT NHE3 was inhibited by cGMP/cGKII. However, the Ser to Asp single mutation of Ser663, Ser554, and Ser607 still preserved the cGKII inhibition of NHE3, which we suggest is due to the other two sites still being able to be phosphorylated by cGMP/cGKII, such that all three sites act as if they are phosphorylated. Triple mutation studies (Ser to Asp) confirmed that all three sites must be able to be phosphorylated at the same time (or show increases in phosphorylation) for NHE3 to be inhibited by cGMP/cGKII. These results indicate that all three sites, Ser554, Ser607, and Ser663, are phosphorylated by cGKII and must be phosphorylated for cGKII to inhibit NHE3 activity.
The requirement for multiple sites of NHE3 or its homologues to be phosphorylated at the same time has been demonstrated for two other kinases/kinase cascades: Mos-MEK-MAPK-p90RSK and CaMKII. In starfish NHE3, the Mos-MEK-MAPK-p90RSK pathway stimulates NHE3 via Rsk phosphorylation of NHE3 at three distal C-terminal sites, all of which are necessary for stimulation of NHE3 activity (41). Also in mammalian NHE3, CaMKII inhibits basal NHE3 activity via phosphorylation of three sites, all of which must be present for this effect of CaMKII (42). In these examples, the phosphorylated sites are clustered relatively close to each other based on linear modeling of the NHE3 C terminus (within 84, 110, and 118 amino acids) (Fig. 7, A–C). We suggest that the total negative charges might alter the environment of the intracellular domain of NHE3, including by affecting protein-protein interactions that have been shown to form on the NHE3 C terminus and to be involved in NHE3 regulation (43). We speculate that this might indicate a more complicated regulation of NHE3 by kinases that involves changes in signaling complexes affected by NHE3 phosphorylation and/or interaction with components of the trafficking machinery. In this regard, CaMKII inhibition of NHE3 does not appear to involve changes in NHE3 surface expression (42), favoring a function of the multiple phosphorylations via NHE3 complex formation. Other recent studies have identified clustering of sites phosphorylated by single kinases and provided evidence that clustered phosphorylation sites tend to be activated by the same kinase and may act to increase the robustness of the phosphorylation-dependent response (36).
FIGURE 7.
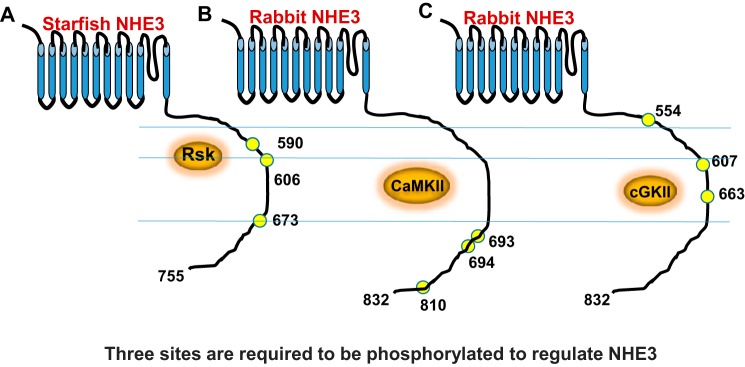
A schematic demonstrates the relative locations of the multiple phosphorylation sites on the NHE3 C terminus needed to be present at the same time for regulation by Rsk (41) (A), CaMKII (42) (B), and cGKII (C).
Ser663 was previously reported to be phosphorylated by SGK1 in response to the short exposure to dexamethasone (∼4 h) to stimulate NHE3 by an effect that did not involve changes in transcriptional stimulation of NHE3 (20). Our studies confirmed the role of Ser663 in dexamethasone stimulation of NHE3 activity over short intervals, but also showed that Ser663 was phosphorylated by cGKII to inhibit NHE3. These results suggest that phosphorylation of Ser663 involves a unique bifunctional regulation in which separate pools are affected, brush-border NHE3 is inhibited by endocytosis, and intracellular NHE3 is stimulated by exocytosis. The concept that a single site when phosphorylated alone stimulates and when phosphorylated along with several additional sites inhibits transport function has not been described previously. We suggest that there are separate changes in the C terminus of NHE3 when this site is phosphorylated alone versus with other amino acids, which accounts for the divergent regulation, although how that occurs is unknown.
The studies correlating the effects of NHE3 Ser to Ala and Ser to Asp mutants (NHE3-S554D,S607D,S663D) on NHE3 activity with surface expression under basal and after cGMP/cGKII exposure are summarized in Table 1. Mutating any of the three amino acids Ser to Asp eliminated cGMP/cGKII inhibition of NHE3 activity and the reduction of surface NHE3. The Ser to Asp mutations reconstituted cGMP inhibition of NHE3 activity. Of great interest was that only the S663D mutation failed to duplicate the reduction in surface NHE3 by cGMP/cGKII. We suggest that this finding indicates that multiple phosphorylation sites interact in a complex manner, perhaps involving differences in timing of the effect or with different kinetics.
TABLE 1.
Summary of changes of NHE3 activity and surface expression by cGMP in different cell lines
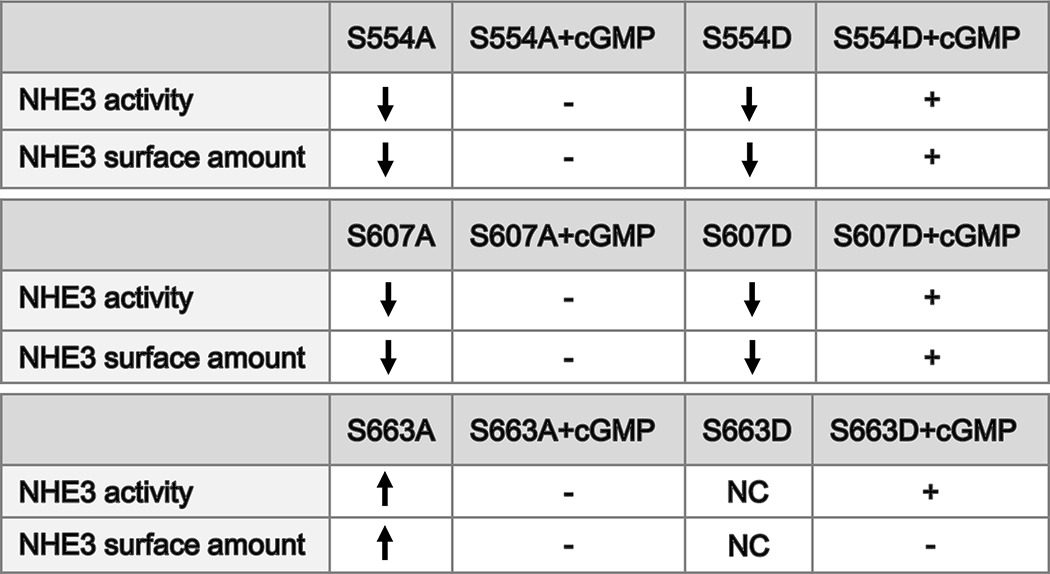
−, no inhibition; +, inhibited by cGMP; NC, no change.
In addition to the changes in cGMP and dexamethasone regulation of NHE3 related to phosphorylation of Ser663, mutation of this amino acid to Ala stimulated basal NHE3 activity without changing total NHE3 expression. The effect of the S663A mutation to increase both NHE3 activity and surface expression is consistent with removing the inhibitory effect of a kinase, which acts to inhibit NHE3 under basal conditions. Please note that our studies did not emphasize identification of the kinase(s), which regulates basal NHE3 activity and we did not further explore the mechanisms of these changes in basal NHE3 activity.
Thus we have described a new phenomenon in which phosphorylation of the same amino acid in a single protein can stimulate or inhibit function of the protein based on whether there is simultaneous phosphorylation of additional sites. Understanding in more detail how this occurs is likely to increase understanding of NHE3 regulation by signaling complexes, which form on its C terminus to dynamically regulate its activity (43).
This work was supported, in whole or in part, by National Institutes of Health NIDDK Grants R01-DK26523, R01-DK61765, P01-DK072084, and P30-DK89502, The Hopkins Digestive Diseases Basic and Translational Research Core Center, and the Hopkins Center for Epithelial Disorders.
- 8-pCPT-cGMP
- 8-(4-chlorophenylthio)-cGMP
- TMA
- tetramethylammonium
- TM
- total membrane
- DMSO
- dimethyl sulfoxide
- FSK
- forskolin
- BCECF-AM
- 2′,7′-bis(carboxyethyl)-S-(and 6)carboxyfluorescein acetoxy methylester
- 8-Br-cAMP
- 8-bromo-cAMP
- CaM
- calmodulin.
REFERENCES
- 1. Moe O. W. (1999) Acute regulation of proximal tubule apical membrane Na+/H+ exchanger NHE-3: role of phosphorylation, protein trafficking, regulatory factors. J. Am. Soc. Nephrol. 10, 2412–2425 [DOI] [PubMed] [Google Scholar]
- 2. Yip J. W., Ko W. H., Viberti G., Huganir R. L., Donowitz M., Tse C. M. (1997) Regulation of the epithelial brush border Na+/H+ exchanger isoform 3 stably expressed in fibroblasts by fibroblast growth factor and phorbol esters is not through changes in phosphorylation of the exchanger. J. Biol. Chem. 272, 18473–18480 [DOI] [PubMed] [Google Scholar]
- 3. Kocinsky H. S., Girardi A. C., Biemesderfer D., Nguyen T., Mentone S., Orlowski J., Aronson P. S. (2005) Use of phospho-specific antibodies to determine the phosphorylation of endogenous Na+/H+ exchanger NHE3 at PKA consensus sites. Am. J. Physiol. Renal Physiol. 289, F249–F258 [DOI] [PubMed] [Google Scholar]
- 4. Kocinsky H. S., Dynia D. W., Wang T., Aronson P. S. (2007) NHE3 phosphorylation at serines 552 and 605 does not directly affect NHE3 activity. Am. J. Physiol. Renal Physiol. 293, F212–F218 [DOI] [PubMed] [Google Scholar]
- 5. Vaandrager A. B. (2002) Structure and function of the heat-stable enterotoxin receptor/guanylyl cyclase C. Mol. Cell. Biochem. 230, 73–83 [PubMed] [Google Scholar]
- 6. Vaandrager A. B., Bot A. G., Ruth P., Pfeifer A., Hofmann F., De Jonge H. R. (2000) Differential role of cyclic GMP-dependent protein kinase II in ion transport in murine small intestine and colon. Gastroenterology 118, 108–114 [DOI] [PubMed] [Google Scholar]
- 7. Vaandrager A. B., Bot A. G., De Jonge H. R. (1997) Guanosine 3′,5′-cyclic monophosphate-dependent protein kinase II mediates heat-stable enterotoxin-provoked chloride secretion in rat intestine. Gastroenterology 112, 437–443 [DOI] [PubMed] [Google Scholar]
- 8. Cha B., Kim J. H., Hut H., Hogema B. M., Nadarja J., Zizak M., Cavet M., Lee-Kwon W., Lohmann S. M., Smolenski A., Tse C. M., Yun C., de Jonge H. R., Donowitz M. (2005) cGMP inhibition of Na+/H+ antiporter 3 (NHE3) requires PDZ domain adapter NHERF2, a broad specificity protein kinase G-anchoring protein. J. Biol. Chem. 280, 16642–16650 [DOI] [PubMed] [Google Scholar]
- 9. Serulle Y., Zhang S., Ninan I., Puzzo D., McCarthy M., Khatri L., Arancio O., Ziff E. B. (2007) A novel GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron 56, 670–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lamprecht G., Weinman E. J., Yun C. H. (1998) The role of NHERF and E3KARP in the cAMP-mediated inhibition of NHE3. J. Biol. Chem. 273, 29972–29978 [DOI] [PubMed] [Google Scholar]
- 11. Yun C. H., Lamprecht G., Forster D. V., Sidor A. (1998) NHE3 kinase A regulatory protein E3KARP binds the epithelial brush border Na+/H+ exchanger NHE3 and the cytoskeletal protein ezrin. J. Biol. Chem. 273, 25856–25863 [DOI] [PubMed] [Google Scholar]
- 12. Sarker R., Valkhoff V. E., Zachos N. C., Lin R., Cha B., Chen T. E., Guggino S., Zizak M., de Jonge H., Hogema B., Donowitz M. (2011) NHERF1 and NHERF2 are necessary for multiple but usually separate aspects of basal and acute regulation of NHE3 activity. Am. J. Physiol. Cell Physiol. 300, C771–C782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zizak M., Lamprecht G., Steplock D., Tariq N., Shenolikar S., Donowitz M., Yun C. H., Weinman E. J. (1999) cAMP-induced phosphorylation and inhibition of Na+/H+ exchanger 3 (NHE3) are dependent on the presence but not the phosphorylation of NHE regulatory factor. J. Biol. Chem. 274, 24753–24758 [DOI] [PubMed] [Google Scholar]
- 14. Dransfield D. T., Bradford A. J., Smith J., Martin M., Roy C., Mangeat P. H., Goldenring J. R. (1997) Ezrin is a cyclic AMP-dependent protein kinase anchoring protein. EMBO J. 16, 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hayashi H., Tamura A., Krishnan D., Tsukita S., Suzuki Y., Kocinsky H. S., Aronson P. S., Orlowski J., Grinstein S., Alexander R. T. (2013) Ezrin is required for the functional regulation of the epithelial sodium proton exchanger, NHE3. PLoS One 8, e55623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Murtazina R., Kovbasnjuk O., Donowitz M., Li X. (2006) Na+/H+ exchanger NHE3 activity and trafficking are lipid Raft-dependent. J. Biol. Chem. 281, 17845–17855 [DOI] [PubMed] [Google Scholar]
- 17. Levine S. A., Nath S. K., Yun C. H., Yip J. W., Montrose M., Donowitz M., Tse C. M. (1995) Separate C-terminal domains of the epithelial specific brush border Na+/H+ exchanger isoform NHE3 are involved in stimulation and inhibition by protein kinases/growth factors. J. Biol. Chem. 270, 13716–13725 [DOI] [PubMed] [Google Scholar]
- 18. Levine S. A., Montrose M. H., Tse C. M., Donowitz M. (1993) Kinetics and regulation of three cloned mammalian Na+/H+ exchangers stably expressed in a fibroblast cell line. J. Biol. Chem. 268, 25527–25535 [PubMed] [Google Scholar]
- 19. Tse C. M., Levine S. A., Yun C. H., Brant S. R., Pouyssegur J., Montrose M. H., Donowitz M. (1993) Functional characteristics of a cloned epithelial Na+/H+ exchanger (NHE3): resistance to amiloride and inhibition by protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 90, 9110–9114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anderson J. M., Van Itallie C. M., Peterson M. D., Stevenson B. R., Carew E. A., Mooseker M. S. (1989) ZO-1 mRNA and protein expression during tight junction assembly in Caco-2 cells. J. Cell Biol. 109, 1047–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sarker R., Grønborg M., Cha B., Mohan S., Chen Y., Pandey A., Litchfield D., Donowitz M., Li X. (2008) Casein kinase 2 binds to the C terminus of Na+/H+ exchanger 3 (NHE3) and stimulates NHE3 basal activity by phosphorylating a separate site in NHE3. Mol. Biol. Cell 19, 3859–3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boyarsky G., Ganz M. B., Sterzel R. B., Boron W. F. (1988) pH regulation in single glomerular mesangial cells I. Acid extrusion in absence and presence of HCO3−. Am. J. Physiol. 255, C844–C856 [DOI] [PubMed] [Google Scholar]
- 23. Cha B., Tse M., Yun C., Kovbasnjuk O., Mohan S., Hubbard A., Arpin M., Donowitz M. (2006) The NHE3 juxtamembrane cytoplasmic domain directly binds ezrin: dual role in NHE3 trafficking and mobility in the brush border. Mol. Biol. Cell 17, 2661–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murtazina R., Kovbasnjuk O., Chen T. E., Zachos N. C., Chen Y., Kocinsky H. S., Hogema B. M., Seidler U., de Jonge H. R., Donowitz M. (2011) NHERF2 is necessary for basal activity, second messenger inhibition, and LPA stimulation of NHE3 in mouse distal ileum. Am. J. Physiol. Cell Physiol. 301, C126–C136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vaandrager A. B., Tilly B. C., Smolenski A., Schneider-Rasp S., Bot A. G., Edixhoven M., Scholte B. J., Jarchau T., Walter U., Lohmann S. M., Poller W. C., de Jonge H. R. (1997) cGMP stimulation of cystic fibrosis transmembrane conductance regulator Cl− channels co-expressed with cGMP-dependent protein kinase type II but not type Iβ. J. Biol. Chem. 272, 4195–4200 [DOI] [PubMed] [Google Scholar]
- 26. Li X., Zhang H., Cheong A., Leu S., Chen Y., Elowsky C. G., Donowitz M. (2004) Carbachol regulation of rabbit ileal brush border Na+/H+ exchanger 3 (NHE3) occurs through changes in NHE3 trafficking and complex formation and is Src dependent. J. Physiol. 556, 791–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Donowitz M., Cha B., Zachos N. C., Brett C. L., Sharma A., Tse C. M., Li X. (2005) NHERF family and NHE3 regulation. J. Physiol. 567, 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bacic D., Kaissling B., McLeroy P., Zou L., Baum M., Moe O. W. (2003) Dopamine acutely decreases apical membrane Na+/H+ exchanger NHE3 protein in mouse renal proximal tubule. Kidney Int. 64, 2133–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hu M. C., Fan L., Crowder L. A., Karim-Jimenez Z., Murer H., Moe O. W. (2001) Dopamine acutely stimulates Na+/H+ exchanger (NHE3) endocytosis via clathrin-coated vesicles: dependence on protein kinase A-mediated NHE3 phosphorylation. J. Biol. Chem. 276, 26906–26915 [DOI] [PubMed] [Google Scholar]
- 30. Wieprecht M., Wieder T., Geilen C. C. (1994) N-[2-Bromocinnamyl(amino)ethyl]-5-isoquinolinesulphonamide (H-89) inhibits incorporation of choline into phosphatidylcholine via inhibition of choline kinase and has no effect on the phosphorylation of CTP:phosphocholine cytidylyltransferase. Biochem. J. 297, 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Müller U., Hildebrandt H. (2002) Nitric oxide/cGMP-mediated protein kinase A activation in the antennal lobes plays an important role in appetitive reflex habituation in the honeybee. J. Neurosci. 22, 8739–8747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zaccolo M., Movsesian M. A. (2007) cAMP and cGMP signaling cross-talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ. Res. 100, 1569–1578 [DOI] [PubMed] [Google Scholar]
- 33. Xu R., Salpeter M. M. (1995) Protein kinase A regulates the degradation rate of Rs acetylcholine receptors. J. Cell. Physiol. 165, 30–39 [DOI] [PubMed] [Google Scholar]
- 34. Azarani A., Orlowski J., Goltzman D. (1995) Parathyroid hormone and parathyroid hormone-related peptide activate the Na+/H+ exchanger NHE-1 isoform in osteoblastic cells (UMR-106) via a cAMP-dependent pathway. J. Biol. Chem. 270, 23166–23172 [DOI] [PubMed] [Google Scholar]
- 35. Visconti P. E., Moore G. D., Bailey J. L., Leclerc P., Connors S. A., Pan D., Olds-Clarke P., Kopf G. S. (1995) Capacitation of mouse spermatozoa: II. protein tyrosine phosphorylation and capacitation are regulated by a cAMP-dependent pathway. Development 121, 1139–1150 [DOI] [PubMed] [Google Scholar]
- 36. Schweiger R., Linial M. (2010) Cooperativity within proximal phosphorylation sites is revealed from large-scale proteomics data. Biol. Direct. 26, 5–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roche K. W., O'Brien R. J., Mammen A. L., Bernhardt J., Huganir R. L. (1996) Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 16, 1179–1188 [DOI] [PubMed] [Google Scholar]
- 38. Chikuda H., Kugimiya F., Hoshi K., Ikeda T., Ogasawara T., Shimoaka T., Kawano H., Kamekura S., Tsuchida A., Yokoi N., Nakamura K., Komeda K., Chung U. I., Kawaguchi H. (2004) Cyclic GMP-dependent protein kinase II is a molecular switch from proliferation to hypertrophic differentiation of chondrocytes. Genes Dev. 18, 2418–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. He P., Lee S. J., Lin S., Seidler U., Lang F., Fejes-Toth G., Naray-Fejes-Toth A., Yun C. C. (2011) Serum- and glucocorticoid-induced kinase 3 in recycling endosomes mediates acute activation of Na+/H+ exchanger NHE3 by glucocorticoids. Mol. Biol. Cell 22, 3812–3825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang D., Zhang H., Lang F., Yun C. C. (2007) Acute activation of NHE3 by dexamethasone correlates with activation of SGK1 and requires a functional glucocorticoid receptor. Am. J. Physiol. Cell Physiol. 292, C396–C404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harada K., Fukuda E., Hirohashi N., Chiba K. (2010) Regulation of intracellular pH by p90Rsk-dependent activation of an Na+/H+ exchanger in starfish oocytes. J. Biol. Chem. 285, 24044–24054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zizak M., Chen T., Bartonicek D., Sarker R., Zachos N. C., Cha B., Kovbasnjuk O., Korac J., Mohan S., Cole R., Chen Y., Tse C. M., Donowitz M. (2012) Calmodulin kinase II constitutively binds, phosphorylates, and inhibits brush border Na+/H+ exchanger 3 (NHE3) by a NHERF2 protein-dependent process. J. Biol. Chem. 287, 13442–13456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Donowitz M., Li X. (2007) Regulatory binding partners and complexes of NHE3. Physiol. Rev. 87, 825–872 [DOI] [PubMed] [Google Scholar]