

Background: Both inhibition of Plk1 and usage of metformin are reported to achieve strong antineoplastic functions in many cancers, including prostate cancer (PCa).
Results: Plk1 inhibitor BI2536 synergizes with metformin in controlling PCa cell growth.
Conclusion: Plk1 inhibition improves the antineoplastic function of metformin in PCa through both signaling and metabolic pathways.
Significance: The proposed combination therapy shows great potential for clinical trials.
Keywords: Cell Cycle, Mitosis, p53, Prostate Cancer, Serine/Threonine Protein Kinase, Polo-like Kinase 1, Metformin
Abstract
The widely used anti-diabetic drug metformin has been shown to exert strong antineoplastic actions in numerous tumor types, including prostate cancer (PCa). In this study, we show that BI2536, a specific Plk1 inhibitor, acted synergistically with metformin in inhibiting PCa cell proliferation. Furthermore, we also provide evidence that Plk1 inhibition makes PCa cells carrying WT p53 much more sensitive to low-dose metformin treatment. Mechanistically, we found that co-treatment with BI2536 and metformin induced p53-dependent apoptosis and further activated the p53/Redd-1 pathway. Moreover, we also show that BI2536 treatment inhibited metformin-induced glycolysis and glutamine anaplerosis, both of which are survival responses of cells against mitochondrial poisons. Finally, we confirmed the cell-based observations using both cultured cell-derived and patient-derived xenograft studies. Collectively, our findings support another promising therapeutic strategy by combining two well tolerated drugs against PCa proliferation and the progression of androgen-dependent PCa to the castration-resistant stage.
Introduction
Prostate cancer (PCa)2 is the most common form of malignancy in men and is the second leading cause of cancer-related death in males in the United States (1). Because PCa cells require androgen for proliferation and development, androgen deprivation (castration) is an effective treatment for patients with late-stage PCa. However, although nearly 80% of patients initially respond well to castration therapy, castration-resistant prostate cancer (CRPC) eventually occurs in most of these patients after several years and then progresses to metastatic diseases (2–4). With very limited methods to treat advanced PCa, novel drugs with new cellular targets are urgently needed.
Metformin is an anti-diabetic drug used to treat type 2 diabetes in almost 120 million people. It alleviates hyperglycemia by lowering hepatic glucose production and increasing glucose uptake by peripheral tissues (5). Increasing evidence suggests that metformin also decreases viability of various cancer cells and inhibits xenograft tumor growth in nude mice (6–10). Of note, metformin use was also shown to reduce the development of CRPC in one clinical trial, in which 3000 CRPC patients were analyzed (11). More importantly, the drug is inexpensive and well tolerated and actively participates in metabolism, which make it quite attractive in cancer therapeutics (12, 13). However, there are still many issues that need to be addressed before metformin can be widely used in cancer treatments. First, the detailed antineoplastic mechanisms behind this drug remain to be elucidated. Previously, the fundamental effect of metformin in cancer treatment was believed to be due to its activation of AMP-activated protein kinase (AMPK) and the subsequent inhibition of both cell cycle progression and mTORC1 (mammalian target of rapamycin complex 1), a critical regulator of protein synthesis and cell proliferation (14–17). However, metformin was also shown to achieve the same effect via the p53/Redd-1 pathway independent of AMPK in PCa cells (6, 18). Furthermore, metformin inhibits mTORC1 in a Rag GTPase-dependent manner (19). More recently, another group reported that the inhibition of mTOR1 by metformin was due to the enhanced binding between PRAS40 and Raptor, both of which are components of mTORC1, thus independent of AMPK (20). The second concern is that intake of metformin into the cell requires the expression of OCT1 (organic cation transporter 1) (21, 22), and the concentrations of metformin used in current in vitro or preclinical antiproliferative studies are much higher than the recommended therapeutic dose in humans (23). In other words, the positive results in animal studies cannot indicate success in clinical trials if we do not use a similar dose of metformin. Thus, how to increase the efficacy of this drug to avoid the high dose-induced side effects and how to efficiently deliver it to the desired organs should be the major tasks to make metformin a real candidate for cancer therapy.
Plk1 (Polo-like kinase 1) is an essential serine/threonine kinase involved in many mitotic events, such as mitotic entry, bipolar spindle formation, and sister chromatid segregation (24). Plk1 is overexpressed in many types of human cancers, and Plk1 inhibitors have been preclinically evaluated as potential drugs for cancer treatment (25). BI2536, the first Plk1 inhibitor to enter clinical trials, has already been studied in phases I and II, and it is well tolerated in humans regardless of the limited therapeutic effects in some types of tumors (26–28). Of note, it was recently reported that Plk1 is up-regulated in androgen-insensitive PCa cells and that its inhibition leads to necroptosis (29). In addition, Plk1 not only promotes androgen receptor signaling (30), but also acts as a negative regulator of tumor suppressor p53 (31, 32), which is crucial for mediating metformin treatment of PCa (18).
In this study, we investigated a new therapeutic strategy against PCa using a combination treatment of metformin and Plk1 inhibitor BI2536. In addition to both in vitro and in vivo observations, we also provide possible mechanisms for the synergy in signaling and metabolic pathways.
EXPERIMENTAL PROCEDURES
Chemicals
BI2536 was purchased from Symansis Ltd. (Timaru, New Zealand) and dissolved in dimethyl sulfoxide as a working solution. Metformin was purchased from Sigma (D150959) and dissolved in distilled water.
Cell Culture, Viral Infection, and RNAi
LNCaP (WT p53), C4-2, DU145 (mutant p53), PC3 (p53-null), HEK293A, and RWPE-1 (non-transformed prostate epithelial) cells were purchased from American Type Culture Collection and cultured at 37 °C in 5% CO2. LNCaP (androgen-dependent) and C4-2 (derived from LNCaP cells but androgen-independent) cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 100 units/ml penicillin, and 100 units/ml streptomycin. DU145, PC3 and HEK293A cells were cultured in Dulbecco's modified Eagle's medium. RWPE-1 cells were cultured in keratinocyte serum-free medium (Invitrogen). Lentivirus constructs were generated, and viral infections were performed as described previously (33). Adenovirus was generated using the pAdEasy XL adenoviral vector system (Stratagene, La Jolla, CA) following the manufacturer's instructions. The virus was then amplified as follows: HEK293A cells were infected with adenovirus for 3 days, harvested, resuspended in 1 ml of sterile PBS, and lysed by four rounds of freeze/thawing using a dry ice/methanol bath and a 37 °C water bath. The supernatant was collected after centrifugation at 12,000 × g for 10 min as viral stocks and stored at −80 °C. The p53 shRNA construct was transfected into the cells with Lipofectamine 2000 reagent (Invitrogen). Puromycin (Clontech) was used to select single positive clones after transfection using the method described previously (33). After a 2-month selection, monoclones were picked up, and p53-deleted stable cell lines were generated.
Western Blotting
Cells were lysed in 20 mm Tris (pH 8.0), 150 mm NaCl, 1.5 mm EDTA, 5 mm EGTA, 0.5% Nonidet P-40, and 0.5 mm Na3VO4 supplemented with protease inhibitors (Sigma). Western blotting was then performed with antibodies against cleaved poly(ADP-ribose) polymerase (PARP; EMD Millipore AB3565), uncleaved PARP (Cell Signaling 9542P), phospho-AKT (Cell Signaling 4060), AKT (Cell Signaling 9272), phospho-S6 (Cell Signaling 4858), S6 (Cell Signaling 2217), Plk1 (Santa Cruz Biotechnology sc-17783), β-actin (Sigma A5441), α-tubulin (Sigma T6199), Erk2 (Santa Cruz Biotechnology sc-154), Redd-1 (Proteintech 10638-1-AP), p53 (Santa Cruz Biotechnology sc-126), and PKM2 (pyruvate kinase M2; Cell Signaling 3198).
Cell Viability Assay
Cells were grown in 96-well plates, and viable cell numbers were determined by assaying conversion of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide to formazan. The IC50 values were obtained from the average viability curves generated by four independent measurements of each condition. The combination index of BI2536 and metformin was measured using the following equation: combination index = (Am)50/(As)50 + (Bm)50/(Bs)50, where (Am)50 is the IC50 of metformin in the combination with half of the concentration of the BI2536 IC50, (As)50 is the concentration of metformin that will produce the identical level of effect alone, (Bm)50 is the IC50 of BI2536 in the combination with half of the concentration of the metformin IC50, and (Bs)50 is the IC50 of BI2536 after single administration. Combination indices of >1, 1, and <1 indicate antagonism, an additive effect, and synergy, respectively.
Glucose/Lactate Assays
Glucose consumption and lactate production were calculated by measuring the remaining glucose and lactate in the cell culture medium using glucose and l-lactate assay kits (Eton Bioscience) following the procedures recommended by the manufacturer.
Intracellular ATP Measurement
Control and treated cells were counted and then harvested to test intracellular ATP levels following the procedure described by Roche Applied Science.
α-Ketoglutarate Assay
α-Ketoglutarate levels in cultured cells were measured using an α-ketoglutarate colorimetric assay kit (BioVision) following the procedure recommended by the manufacturer.
Colony Formation Assay
Cells (500–1000) were seeded in 6-well plates and cultured in medium alone or containing different drugs for 20 days, with medium refreshment every 2 days. After culturing, cells were fixed in 10% formalin and stained with 0.5% crystal violet, and colony numbers were counted.
FACS Analysis
Cells were harvested after trypsin digestion, fixed in 75% ethanol, stained with propidium iodide solution at a final concentration of 50 μg/ml, and then subjected to FACS analysis.
Mouse Xenograft Model
LNCaP cells (5 × 106 cells/mouse) were mixed with an equal volume of Matrigel (Collaborative Biomedical Products) and inoculated into the right flanks of 24 athymic nude mice (Harlan Laboratories). One week later, the mice were randomly separated into four groups (six mice/group). At day 17 after implantation, castration was performed in all 24 mice. Metformin was dissolved in distilled water and given to the mice by gavage. BI2536 was dissolved in 0.1 n HCl, diluted in 0.9% NaCl, and injected into the tail veins. Both drugs were injected twice per week for 10 weeks, during which body weights and tumor volumes were measured. Tumor volumes were estimated using the following formula: V (mm3) = L (mm) × W2 (mm)/2. The length and width were measured using digital calipers twice per week.
Patient-derived Xenograft Model
Mice carrying LuCaP35CR tumors were obtained from Dr. Robert Vessella at the University of Washington (34). Tumors were amplified by cutting the original tumors into ∼20–30-mm3 pieces, followed by implantation into precastrated nude mice. After amplifying enough tumors, tumors were harvested and cut into ∼20–30-mm3 pieces before implantation into 16 precastrated nude mice. When tumors reached 250–300 mm3, mice were randomly separated into four groups (four mice/group) for different treatments.
Serum Prostate-specific Antigen (PSA) Measurement
Blood was collected from mice by retro-orbital bleeding once per week to determine serum PSA levels. PSA levels were measured using a PSA (human) ELISA kit (Abnova KA0208) following the procedure recommended by the manufacturer.
Tumor Western Blotting
Tumors were harvested and frozen at −80 °C before conducting further analysis. For Western blotting, tumors were melted on ice and homogenized using radioimmune precipitation assay buffer (150 mm NaCl, 1 mm PMSF, 1 mm EDTA, 1% Triton X-100, and 0.1% SDS) supplemented with protease inhibitors. Proteins were detected using specific antibodies following the procedure described above.
Statistical Analysis
Standard two-tailed Student's t tests were performed to determine the significance of differences between experimental conditions and treatments.
RESULTS
Plk1 Inhibition and Metformin Synergistically Decrease Viability and Colony Formation of PCa Cells
To test whether inhibition of Plk1 synergizes with metformin in inhibiting PCa cell proliferation, we conducted cell viability assays to calculate the combination indices of metformin and BI2536, a well recognized Plk1 inhibitor, in two PCa cell lines: androgen-dependent LNCaP and LNCaP-derived but androgen-independent C4-2. The combination indices of the two drugs in both cell lines are ∼0.4, suggesting that they act in a synergistic manner in both cell lines (Tables 1 and 2). To further confirm these initial observations, we performed extensive Western blot analysis to follow the protein level of cleaved PARP, a marker for apoptosis. Consistent with the cell viability assays, we found that there was a significantly increased level of cleaved PARP in both LNCaP and C4-2 cells after they were treated with both metformin and BI2536 in comparison with monotherapy (Fig. 1, A–C). However, we did not observe a similar result in PC3 cells, a p53-null PCa cell line (Fig. 1D). More importantly, we also found that these two drugs, either working alone or together, failed to induce detectable apoptosis in the non-transformed prostate epithelial cell line RWPE-1 (Fig. 1E). Finally, we also found that BI2536 and metformin acted synergistically to inhibit colony formation of LNCaP (Fig. 1F) and C4-2 (Fig. 1G) cells.
TABLE 1.
IC50 values of BI2536 and metformin in LNCaP cells
Combination index = 0.384.
| Drug | IC50 |
|---|---|
| BI2536 | 91.3 nm |
| Metformin | 5 mm |
| BI2536 + 2.5 mm metformin | 35 nm |
| Metformin + 50 nm BI2536 | 5.5 μm |
TABLE 2.
IC50 values of BI2536 and metformin in C4-2 cells
Combination index = 0.374.
| Drug | IC50 |
|---|---|
| BI2536 | 8 nm |
| Metformin | 5 mm |
| BI2536 + 2.5 mm metformin | 2.2 nm |
| Metformin + 4 nm BI2536 | 495 μm |
FIGURE 1.

BI2536 and metformin inhibit PCa cell proliferation in a synergistic manner. A–E, inhibition of Plk1 potentiates the lethality of metformin in WT p53 PCa cells, but not in PC3 and non-transformed prostate cells. LNCaP (A and B), C4-2 (C), PC3 (D), or RWPE-1 (E) cells were treated with metformin (met), BI2536 (BI), or both at the indicated concentrations for the indicated times and harvested for immunoblotting with antibodies against cleaved PARP (c-PARP), a marker for apoptosis. In E, 50 μm sodium arsenite (As) was used as a positive control for cell death. CTRL, control. F, BI2536 and metformin inhibit colony formation synergistically in LNCaP cells. LNCaP cells (1 × 103) were seeded on soft agar in 6-well plates; treated with 5 μm metformin, 10 nm BI2536, or both for 3 weeks; and stained with 0.005% crystal violet (upper panel). Quantification results are shown (lower panel). *, p < 0.05; **, p < 0.01. G, BI2536 and metformin inhibit colony formation synergistically in C4-2 cells. C4-2 cells (1 × 103) were seeded on soft agar in 6-well plates; treated with 0.5 mm metformin, 1 nm BI2536, or both for 3 weeks; and stained with 0.005% crystal violet (upper panel). Quantification results are shown (lower panel). *, p < 0.05; **, p < 0.01.
Plk1 Status Affects Cellular Response to Metformin
Because inhibition of Plk1 potentiated metformin-mediated inhibition of proliferation of LNCaP and C4-2 cells, two PCa cell lines with elevated levels of Plk1, we then asked whether overexpression of Plk1 in RWPE-1 cells, which have an almost undetectable level of Plk1, affects their response to metformin. Accordingly, we overexpressed Plk1 in RWPE-1 cells by infection with adenovirus. One immediate outcome of Plk1 overexpression in RWPE-1 cells was activation of the PI3K/AKT/mTOR cell survival pathway, as indicated by the increased phosphorylation levels of AKT and S6, two major downstream effectors of PI3K (Fig. 2A). Plk1 elevation-mediated activation of the PI3K/AKT/mTOR pathway has been addressed in detail in a separate study (35). Interestingly, we observed that Plk1 overexpression causes RWPE-1 cells, which normally do not respond to even very high concentrations of metformin, to be much more sensitive to this drug (Fig. 2, B and C). This was not due to the cell cycle effect because Plk1 overexpression did not change the cell cycle of RWPE-1 cells (Fig. 2D). In contrast, we found an opposite trend in the prostate cancer cell line LNCaP, i.e. Plk1-depleted LNCaP cells (Fig. 2E) were much more sensitive to metformin than control cells with a normal Plk1 level, as indicated by both cleaved PARP Western blotting (Fig. 2F) and cell viability assays (Fig. 2G). Importantly, the increased cell death was not due to the Plk1 inhibition itself, as cells infected with the Plk1 depletion virus did not have a significantly higher apoptosis level compared with cells infected with the control virus (Fig. 2H). In addition, the protein level of Redd-1, which is induced after treatment with high concentrations of metformin (18), was much lower in Plk1-overexpressing cells than in Plk1-depleted cells (Fig. 2E). Furthermore, Plk1 depletion also potentiated metformin-associated cell death in C4-2 cells (Fig. 2I). Consistent with the observation in Fig. 2E, we found that co-treatment with BI2536 further enhanced the level of Redd-1 induced by metformin in LNCaP cells, but not in PC3 and DU145 cells (Fig. 2J), both of which do not have WT p53. Taken together, these data suggest that the roles of Plk1 in cellular responses to metformin are different between PCa cells and non-transformed prostate epithelial cells. In addition, p53 status may play an important role in the cellular response to the combination treatment with Plk1 inhibition and metformin.
FIGURE 2.

Plk1 status affects the cellular response to metformin. A, RWPE-1 cells were infected with adenovirus expressing GFP or GFP-Plk1 for 2 days and harvested for immunoblotting with the indicated antibodies. CTRL, control. B, RWPE-1 cells expressing GFP or GFP-Plk1 were treated with 0.5 mm metformin for 24 h (upper panel) or directly harvested for immunoblotting (lower panel). C, RWPE-1 cells were infected with lentivirus (empty vector (lv-ctrl) or Plk1-overexpressing (lv-Plk1+)) for 1 day, harvested, and reseeded in 96-well plates at different concentrations of metformin for 3 days, followed by cell viability measurement. *, p < 0.05. D, RWPE-1 cells were infected with lentivirus for 3 days and harvested for FACS analysis. E, LNCaP cells were infected with lentivirus (empty vector, Plk1-overexpressing, or Plk1-depleted) for 3 days, treated with 5 mm metformin for 4 h, and harvested. F, LNCaP cells were infected with lentivirus for 2 days and treated with 0.5 mm metformin for 24 h. G, LNCaP cells were infected with lentivirus for 1 day, harvested, and reseeded in 96-well plates at different concentrations of metformin for 3 days, followed by cell viability assay. *, p < 0.05. H, 5 × 105 LNCaP cells were seeded in 6-well plates and infected with lentivirus for 3 days, and cell viability was measured by counting the number of viable cells. I, C4-2 cells were infected with lentivirus for 2 days and treated with 0.5 mm metformin (Met) for 24 h. J, LNCaP, DU145 and PC3 cells were treated with the indicated drugs for 4 h. BI, BI2536.
Metformin and BI2536 Induce p53-dependent Apoptosis
Because p53 is likely involved in mediating the synergic effect of BI2536 and metformin and because p53-dependent synergy between metformin and 2-deoxyglucose was reported previously (36), we asked whether the p53 level was changed upon BI2536 and metformin treatment. Although we did not detect any obvious change in the p53 level in LNCaP cells using lower drug concentrations (Fig. 3A), a significant increase with combination treatment compared with single-drug treatments was observed when we increased the concentrations of both metformin and BI2536 to about their IC50 values (Fig. 3B). Furthermore, the combination treatment led to a significant increase in the p53 level in C4-2 cells even at lower concentrations (Fig. 3C). Of note, we repeatedly noticed that C4-2 cells were much more sensitive to BI2536 compared with LNCaP cells during the experimentation, suggesting that Plk1 is much more critical for CRPC cell survival. Next, to further determine whether p53 is important for the combination effect, we generated several p53-depleted LNCaP cell lines (Fig. 3D) and tested their sensitivities to different drug treatments. Compared with WT LNCaP cells, p53-depleted LNCaP cells showed similar sensitivities to either metformin or BI2536 alone, but were much less sensitive to the combination treatment, as indicated by cell viability after drug treatments (Fig. 3E). Consistent with this observation, we found that there was a significant decrease in the level of cleaved PARP in p53-depleted LNCaP cells compared with control cells with the same combination treatment (Fig. 3F). Furthermore, we conducted a colony formation assay to confirm the results (Fig. 3, G and H).
FIGURE 3.
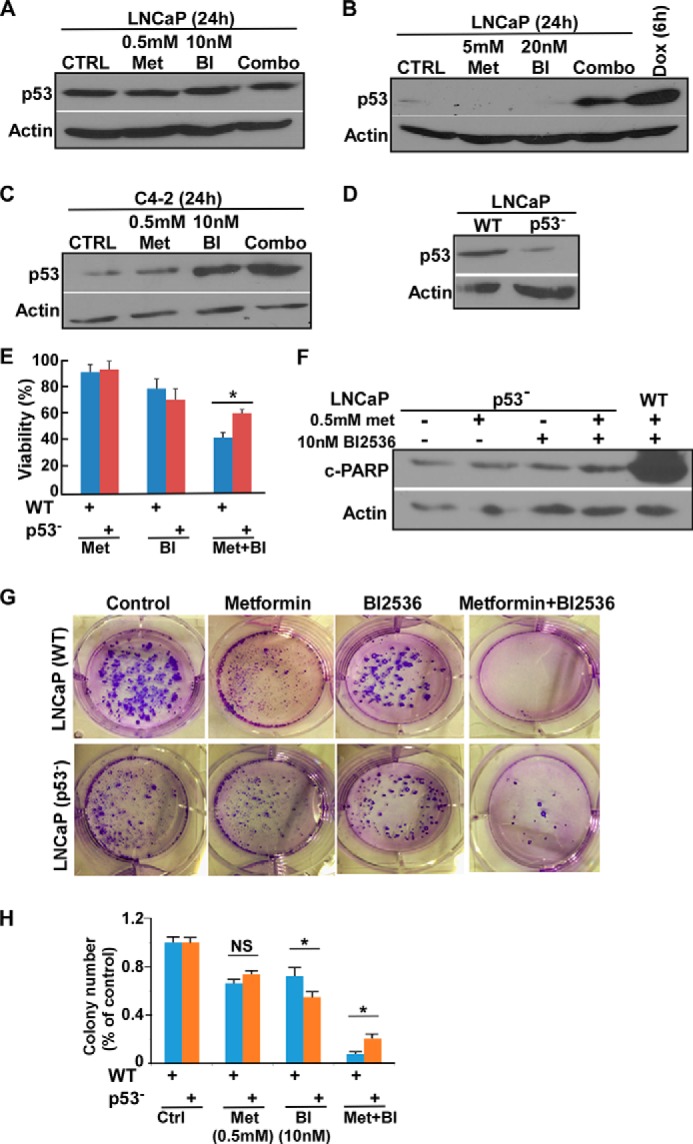
Inhibition of Plk1 and metformin induce p53-dependent apoptosis. A and B, LNCaP cells were treated with lower (A) or higher (B) concentrations of drugs for 24 h and harvested for immunoblotting. Doxorubicin (Dox) was used as a positive control to induce elevation of p53 levels in B. CTRL, control; Met, metformin; BI, BI2536. C, C4-2 cells were treated with indicated drugs. D, immunoblot of WT and p53-depleted LNCaP cell clones. E, 5 × 105 LNCaP cells (WT or p53-depleted) were seeded in 6-well plates; treated with 0.5 mm metformin, 10 nm BI2536 or both for 24 h; and harvested for viability assay. *, p < 0.05. F, LNCaP cells (WT or p53-depleted) were treated with the indicated drugs and subjected to immunoblotting. G, 1 × 103 LNCaP cells (WT and p53-depleted) were seeded on soft agar in 6-well plates; treated with 0.5 mm metformin, 10 nm BI2536; or both for 3 weeks; and stained with 0.005% crystal violet. H, quantification of colony numbers. The results were normalized to the control group of each cell line. *, p < 0.05; NS, not significant.
BI2536 Hinders Glycolysis and Glutamine Anaplerosis Induced by Metformin
As metformin actively participates in many important metabolic pathways, we investigated the changes in cell metabolism after addition of the Plk1 inhibitor with metformin. Similar to previous reports on the effect of metformin on glycolysis, we found that metformin treatment significantly up-regulated glucose consumption and lactate production in LNCaP cells (Fig. 4A), whereas addition of BI2536 down-regulated metformin-induced glycolysis, as indicated by the decrease in glucose consumption and lactate production (Fig. 4B). Mechanistically, metformin treatment increased the level of PKM2, a rate-limiting factor of glycolysis, in LNCaP cells, but addition of BI2536 antagonized such an effect (Fig. 4C). Glutamine anaplerosis was recently found to be another important cell survival pathway after metformin treatment (37). Thus, we asked whether Plk1 inhibition has any effects on glutamine metabolism as well. At a high concentration of metformin (2.5 mm), the majority of cellular α-ketoglutarate is generated from glutamine (but not glucose) metabolism in PCa cell lines (37). We investigated whether BI2536 inhibits glutamine anaplerosis by measuring cellular α-ketoglutarate levels under this condition. As expected, treatment with 2.5 mm metformin up-regulated glutamine anaplerosis in LNCaP cells, but addition together with BI2536 significantly antagonized this effect (Fig. 4D). Taken together, these data suggest that inhibition of Plk1 negatively affects the two crucial cellular anti-stress metabolic responses induced by metformin treatment, consequently resulting in rapid cell death due to energy crisis.
FIGURE 4.
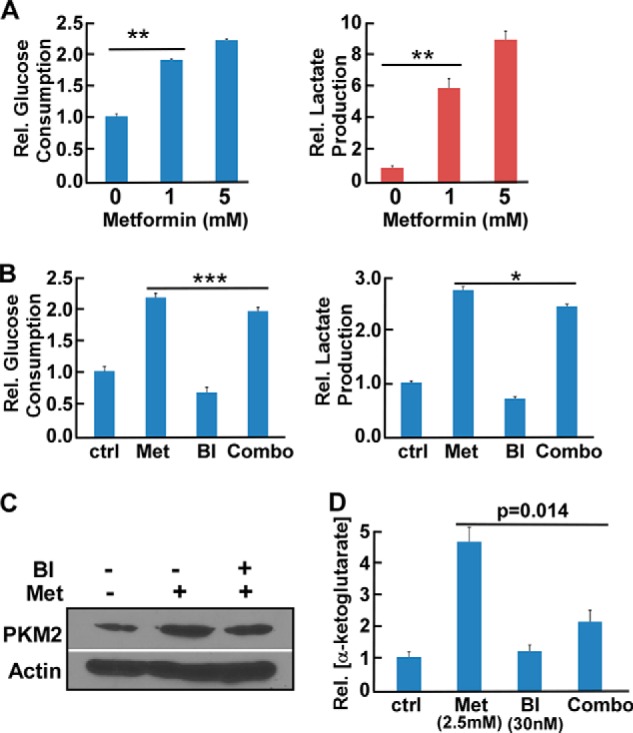
Metformin and BI2536 alter glucose and glutamine metabolism. A, LNCaP cells were treated with metformin for 24 h. After cell numbers were counted, the media were collected to measure glucose consumption and lactate production upon normalizing the cell numbers. **, p < 0.01. B, LNCaP cells were treated with 0.5 mm metformin (Met), 10 nm BI2536 (BI), or both for 24 h. After cell numbers were counted, the media were collected to measure glucose consumption and lactate production. *, p < 0.05; ***, p < 0.001. ctrl, control. C, LNCaP cells were treated with 0.5 mm metformin alone or with 10 nm BI2536 for 24 h. D, LNCaP cells were treated with the indicated drugs for 24 h and harvested to measure α-ketoglutarate levels.
BI2536 and Metformin Act Synergistically in an LNCaP Xenograft Model
To assess the in vivo effect of metformin and BI2536 treatment alone or in combination, we conducted an LNCaP xenograft model study. To mimic the progression of tumors to the androgen-independent stage, we castrated the mice after detectable tumors were formed. As indicated in Fig. 5 (A, B, and D), neither metformin nor BI2536 treatment alone had an obvious effect in preventing tumor growth, whereas a strong inhibition of tumor growth was detected in the combination group, supporting a synergistic effect of metformin and BI2536 in vivo. Moreover, the trend was similar to tumor volume measurements when the serum PSA levels in these mice were followed (Fig. 5C).
FIGURE 5.
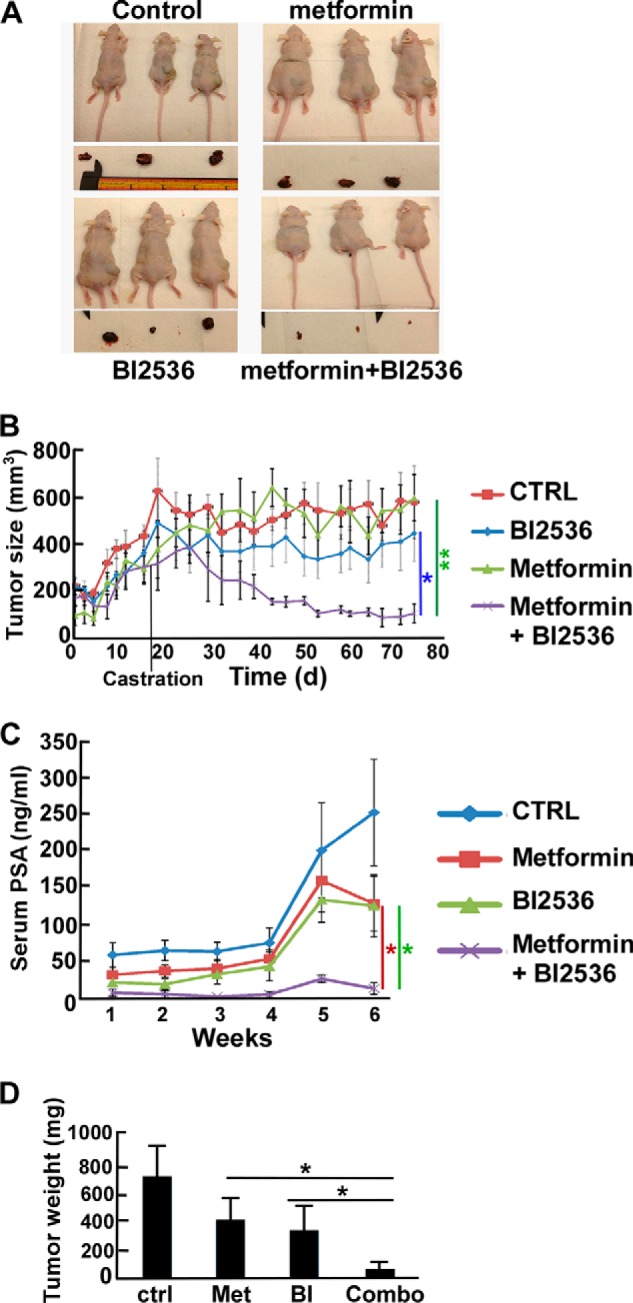
BI2536 and metformin synergistically inhibit growth of LNCaP-derived tumors. LNCaP cells (5 × 106) were subcutaneously inoculated into the flanks of Nu/Nu nude mice to form tumors. Mice were castrated at day 17, intravenously injected twice per week with BI2536 (BI; 5 mg/kg), given metformin (Met; 5 mg/kg) by gavage, or both and followed for 75 days. A, images of the mice at the end of the study. B, tumor growth curves of the study. CTRL, control. C, inhibition of PSA levels by BI2536 and metformin. Blood was collected beginning day 30 (once per week), and serum PSA levels were measured using the PSA ELISA kit for 6 weeks. D, tumor weight measurement upon sacrifice. *, p < 0.05; **, p < 0.01.
BI2536 and Metformin Act Synergistically in a Patient-derived Xenograft Model
To better mimic the tumor growth conditions and follow the situation at the relatively late CRPC stage, we conducted a patient-derived xenograft study using the LuCaP35CR model, which was originally generated from a 66-year-old patient (34). Of note, this model was not indicated to harbor any p53 mutation in a whole-exome sequencing study of the LuCaP series (38). After ∼40 days of drug treatment, we found that metformin treatment alone had nearly no effect and that BI2536 alone showed a very limited effect in inhibiting tumor growth. In contrast, we observed a significantly lower growth rate of tumors treated with both metformin and BI2536 in comparison with monotherapy (Fig. 6, A, B, and D). Importantly, this result was also consistent with the serum PSA levels and Western blot analysis of the harvested tumors (Fig. 6, C and E). Thus, these experiments support the notion that BI2536 and metformin also have a strong synergy in a patient-derived xenograft model.
FIGURE 6.
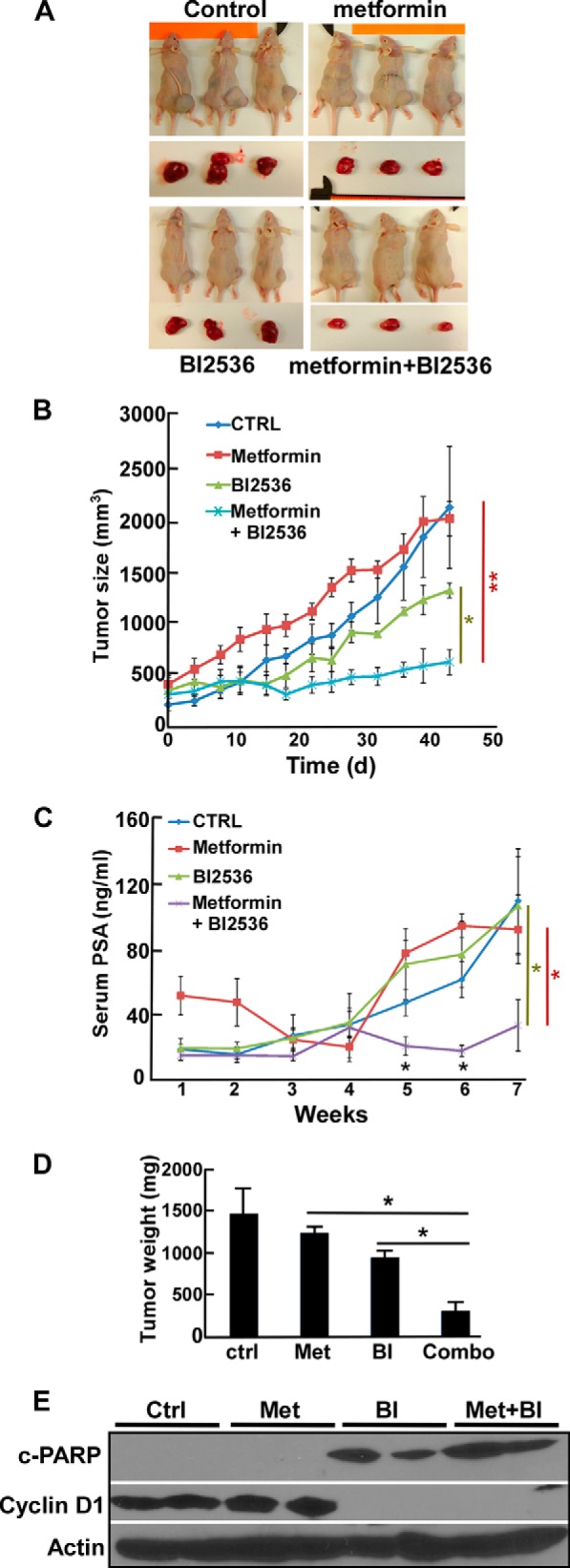
BI2536 and metformin synergistically inhibit LuCaP35CR xenografts. LuCaP35CR tumors were inoculated into nude mice that had been castrated 2 weeks prior. After tumors reached sizes of 250–300 mm3, mice were intravenously injected twice per week with BI2536 (BI; 30 mg/kg), given metformin (Met; 15 mg/kg) by gavage, or both and followed for an additional 43 days. A, images of the mice at the end of the study. B, tumor growth curves of the study. CTRL, control. C, serum PSA levels of the mice. D, tumor weight measurement upon sacrifice. E, immunoblot of the harvested tumor samples. c-PARP, cleaved PARP. *, p < 0.05; **, p < 0.01.
DISCUSSION
Because of its high safety levels and remarkable functions in inhibiting growth of multiple tumor cell lines, physicians are considering the potential use of metformin in the treatment of cancer. However, in addition to the undefined molecular action, there are many other concerns about the dose and cellular uptake of the drug when considering the clinical applications. Thus, how to achieve the antineoplastic function with recommended therapeutic doses for diabetic patients or even lower doses for non-diabetic patients is a challenging task for physicians. Herein, we presented a novel therapeutic strategy that can dramatically increase the efficiency of metformin in inhibiting the growth of PCa cells by combining with Plk1 inhibition.
Beyond the well identified cell cycle-related functions, Plk1 also plays important roles in many pivotal signaling pathways of cancer cells. In addition to the previous finding that Plk1 acts as a negative regulator of p53, we also showed that Plk1 contributes to activation of the PI3K/AKT/mTOR pathway (Fig. 2A), one of the most frequently activated cell survival pathways in numerous cancer cells (39–41). Thus, we proposed to challenge the hypothesis that Plk1 inhibition could potentially be one novel avenue to increase metformin efficiency against PCa.
First, we found that the Plk1 inhibitor BI2536 and metformin showed a strong synergy in LNCaP cells and its androgen-independent derivative, C4-2 cells (42), as we noticed that BI2536 strikingly increased metformin-associated lethality in these two cell lines. In LNCaP cells, we could use ∼5 μm metformin to induce apoptosis and prevent colony formation when combined with a low concentration of BI2536 (Fig. 1, B and F). Another interesting observation is that C4-2 cells were much more sensitive to Plk1 inhibition (IC50 = 8 nm) compared with LNCaP cells (IC50 = 90 nm) even though they share the same genetic background (Table 2 and Fig. 1, C and G), suggesting that Plk1 plays additional important roles as PCa cells progress to the castration-resistant stage. In contrast, normal prostate cells were quite resistant to these two drugs (Figs. 1E and 2C), thus offering selectivity for this proposed therapeutic strategy against cancer cells. Consistent with these observations, Plk1 depletion sensitized LNCaP cells to low concentrations of metformin (Fig. 2, F and G). Furthermore, we observed that overexpression of Plk1 in RWPE-1 cells somehow made these non-transformed cells more sensitive to metformin (Fig. 2, B and C). Consistent with our previous findings that Plk1 negatively regulates p53, we found that p53 was indeed down-regulated when we overexpressed Plk1 (Fig. 2B, lower panel). However, a significant induction of p53 was observed in Plk1-overexpressing RWPE-1 cells upon metformin treatment (Fig. 2B, upper panel). One possible explanation is that Plk1 overexpression changes the cell signaling network (such as the PI3K/AKT/mTOR pathway) in normal prostate epithelial cells, somehow making them behave like cancer cells, which are more sensitive to metformin, and that p53 induction correlates with the onset of apoptosis.
Although mitochondrial inhibition as the premise for metformin downstream functions is well accepted, the notion that the drug exerts its antitumor functions through the AMPK pathway is plausible, as alternative pathways are supported by increasing evidence. Particularly in PCa, activation of the p53/Redd-1 axis was found to be crucial in inhibition of both cell cycle progression and the mTOR pathway (18). Even though the exact role of p53 in metformin treatment is still under debate, as opposite results were observed in colon cancer (43), we do not believe that these two observations actually counteract each other. Because colon cancer cells without p53 cannot efficiently make metabolic alterations in response to the mitochondrial stress induced by metformin, they are more sensitive to metformin than colon cancer cells with WT p53 (43). However, for the p53/Redd-1 axis in PCa cells carrying WT p53, positive regulation of p53 will lead to enhanced repression of cell proliferation. Indeed, after LNCaP and C4-2 cells were co-treated with BI2536 and metformin, the p53 and Redd-1 levels were both up-regulated (Figs. 2J and 3, B and C). Because Redd-1 was found to be a potent tumor suppressor downstream of p53 (44–46), we propose that the up-regulated p53/Redd-1 pathway could be one of the explanations for the observed synergy in PCa. Consistently, we found that p53 knockdown made LNCaP cells significantly less sensitive to the combination treatment of BI2536 and metformin (Fig. 3, E–G), although we have not discovered the exact molecular mechanism. Interestingly, we also found that BI2536 treatment significantly inhibited metformin-induced glycolysis and glutamine anaplerosis (Fig. 4, B–D), two important cell survival responses to mitochondrial stress (37, 47). Similar to our findings, an inhibitor of glycolysis strongly synergizes with metformin in PCa cells (36), but the difference is that in comparison with metformin treatment alone, we did not observe a sharp decrease in ATP levels when combining metformin and BI2536 (data not shown). In short, we propose that inhibition of Plk1 promotes the cytotoxicity of metformin to PCa cells through both signaling and metabolic pathways.
Previously, the lowest dose of metformin used in mouse xenograft studies was 40 mg/kg/day (23), and there was a detectable reduction of tumor growth in the LNCaP xenograft model using 1 mg/day metformin (6). To determine whether we could significantly reduce the dose of metformin when combined with BI2536 treatment, we injected 5 mg/kg metformin only two times per week, which equals ∼0.03 mg/day. Of note, the dose of BI2536 was also significantly lower than the previously reported dose in a PCa xenograft study (48). Upon castration, all the tumors in the LNCaP xenograft study grew much slower, and neither of these two single treatments had an obvious effect on tumor repression compared with the control group. In contrast, the tumors treated with both drugs nearly stopped growing upon castration (Fig. 5B), strongly suggesting that these two drugs act together in preventing progression from the androgen-sensitive to androgen-independent stage. LuCaP35CR is the androgen-independent form of LuCaP35, a new patient-derived xenograft tumor model used to study progression to androgen independence (34). For our purpose, LuCaP35CR could be an excellent model to mimic the relatively late stage of PCa. Because these tumors grow much faster than tumors generated from LNCaP cells, we increased the doses of metformin and BI2536 to 30 and 15 mg/kg (two injections/week), respectively. Similar to the LNCaP xenograft study, we did not detect any obvious change with single-drug treatment, as both of the doses were still significantly lower than what has been generally used in mouse studies. In striking contrast, the mice treated with both drugs not only had smaller tumors and lower serum PSA levels (Fig. 6, B and C), but also showed reduced angiogenesis potentials as the tumors became transparent (Fig. 6A).
In summary, our in vitro and in vivo data support a strong synergy of Plk1 inhibition and metformin in inhibiting PCa cell proliferation. In addition, we are the first to provide evidence that metformin can still achieve its antineoplastic function at doses significantly lower than those used in other studies when combined with BI2536. Thus, the combination strategy can be considered for clinical trials to increase the efficiency of metformin in PCa.
This work was supported, in whole or in part, by National Institutes of Health Grants R01CA157429 (to X. L.) and R01AR059130 (to N. A.). This work was also supported by National Science Foundation Grant MCB-1049693 and American Cancer Society Grant RSG-13–073 (to X. L.) and by the China Scholarship Council (to C. S.). Xenograft data were acquired by a Purdue Center for Cancer Research facility supported by National Institutes of Health Grant P30 CA023168.
- PCa
- prostate cancer
- CRPC
- castration-resistant prostate cancer
- AMPK
- AMP-activated protein kinase
- PARP
- poly(ADP-ribose) polymerase
- PSA
- prostate-specific antigen.
REFERENCES
- 1. Jemal A., Siegel R., Ward E., Hao Y., Xu J., Murray T., Thun M. J. (2008) Cancer statistics, 2008. CA Cancer J. Clin. 58, 71–96 [DOI] [PubMed] [Google Scholar]
- 2. Goldenberg S. L., Gleave M. E., Taylor D., Bruchovsky N. (1999) Clinical experience with intermittent androgen suppression in prostate cancer: minimum of 3 years' follow-up. Mol. Urol. 3, 287–292 [PubMed] [Google Scholar]
- 3. Bruchovsky N., Klotz L. H., Sadar M., Crook J. M., Hoffart D., Godwin L., Warkentin M., Gleave M. E., Goldenberg S. L. (2000) Intermittent androgen suppression for prostate cancer: Canadian Prospective Trial and related observations. Mol. Urol. 4, 191–199; discussion 201 [PubMed] [Google Scholar]
- 4. Gleave M., Goldenberg S. L., Bruchovsky N., Rennie P. (1998) Intermittent androgen suppression for prostate cancer: rationale and clinical experience. Prostate Cancer Prostatic Dis. 1, 289–296 [DOI] [PubMed] [Google Scholar]
- 5. Kirpichnikov D., McFarlane S. I., Sowers J. R. (2002) Metformin: an update. Ann. Int. Med. 137, 25–33 [DOI] [PubMed] [Google Scholar]
- 6. Ben Sahra I., Laurent K., Loubat A., Giorgetti-Peraldi S., Colosetti P., Auberger P., Tanti J. F., Le Marchand-Brustel Y., Bost F. (2008) The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 27, 3576–3586 [DOI] [PubMed] [Google Scholar]
- 7. Dowling R. J., Zakikhani M., Fantus I. G., Pollak M., Sonenberg N. (2007) Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 67, 10804–10812 [DOI] [PubMed] [Google Scholar]
- 8. Zakikhani M., Dowling R., Fantus I. G., Sonenberg N., Pollak M. (2006) Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 66, 10269–10273 [DOI] [PubMed] [Google Scholar]
- 9. Karnevi E., Said K., Andersson R., Rosendahl A. H. (2013) Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer 13, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sikka A., Kaur M., Agarwal C., Deep G., Agarwal R. (2012) Metformin suppresses growth of human head and neck squamous cell carcinoma via global inhibition of protein translation. Cell Cycle 11, 1374–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spratt D. E., Zhang C., Zumsteg Z. S., Pei X., Zhang Z., Zelefsky M. J. (2013) Metformin and prostate cancer: reduced development of castration-resistant disease and prostate cancer mortality. Eur. Urol. 63, 709–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cairns R. A., Harris I. S., Mak T. W. (2011) Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95 [DOI] [PubMed] [Google Scholar]
- 13. Vander Heiden M. G. (2011) Targeting cancer metabolism: a therapeutic window opens. Nat. Rev. Drug Discov. 10, 671–684 [DOI] [PubMed] [Google Scholar]
- 14. Jones R. G., Plas D. R., Kubek S., Buzzai M., Mu J., Xu Y., Birnbaum M. J., Thompson C. B. (2005) AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293 [DOI] [PubMed] [Google Scholar]
- 15. Bolster D. R., Crozier S. J., Kimball S. R., Jefferson L. S. (2002) AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 277, 23977–23980 [DOI] [PubMed] [Google Scholar]
- 16. Turner N., Li J. Y., Gosby A., To S. W., Cheng Z., Miyoshi H., Taketo M. M., Cooney G. J., Kraegen E. W., James D. E., Hu L. H., Li J., Ye J. M. (2008) Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes 57, 1414–1418 [DOI] [PubMed] [Google Scholar]
- 17. Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N., Musi N., Hirshman M. F., Goodyear L. J., Moller D. E. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ben Sahra I., Regazzetti C., Robert G., Laurent K., Le Marchand-Brustel Y., Auberger P., Tanti J. F., Giorgetti-Peraldi S., Bost F. (2011) Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 71, 4366–4372 [DOI] [PubMed] [Google Scholar]
- 19. Kalender A., Selvaraj A., Kim S. Y., Gulati P., Brûlé S., Viollet B., Kemp B. E., Bardeesy N., Dennis P., Schlager J. J., Marette A., Kozma S. C., Thomas G. (2010) Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 11, 390–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu X., Chhipa R. R., Pooya S., Wortman M., Yachyshin S., Chow L. M., Kumar A., Zhou X., Sun Y., Quinn B., McPherson C., Warnick R. E., Kendler A., Giri S., Poels J., Norga K., Viollet B., Grabowski G. A., Dasgupta B. (2014) Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc. Natl. Acad. Sci. U.S.A. 111, E435–E444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sogame Y., Kitamura A., Yabuki M., Komuro S. (2009) A comparison of uptake of metformin and phenformin mediated by hOCT1 in human hepatocytes. Biopharm. Drug Dispos. 30, 476–484 [DOI] [PubMed] [Google Scholar]
- 22. Segal E. D., Yasmeen A., Beauchamp M. C., Rosenblatt J., Pollak M., Gotlieb W. H. (2011) Relevance of the OCT1 transporter to the antineoplastic effect of biguanides. Biochem. Biophys. Res. Commun. 414, 694–699 [DOI] [PubMed] [Google Scholar]
- 23. Ben Sahra I., Le Marchand-Brustel Y., Tanti J. F., Bost F. (2010) Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Mol. Cancer Ther. 9, 1092–1099 [DOI] [PubMed] [Google Scholar]
- 24. Lapenna S., Giordano A. (2009) Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 8, 547–566 [DOI] [PubMed] [Google Scholar]
- 25. Strebhardt K. (2010) Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. 9, 643–660 [DOI] [PubMed] [Google Scholar]
- 26. Hofheinz R. D., Al-Batran S. E., Hochhaus A., Jäger E., Reichardt V. L., Fritsch H., Trommeshauser D., Munzert G. (2010) An open-label, phase I study of the polo-like kinase-1 inhibitor, BI 2536, in patients with advanced solid tumors. Clin. Cancer Res. 16, 4666–4674 [DOI] [PubMed] [Google Scholar]
- 27. McInnes C., Wyatt M. D. (2011) PLK1 as an oncology target: current status and future potential. Drug Discov. Today 16, 619–625 [DOI] [PubMed] [Google Scholar]
- 28. Schöffski P., Blay J. Y., De Greve J., Brain E., Machiels J. P., Soria J. C., Sleijfer S., Wolter P., Ray-Coquard I., Fontaine C., Munzert G., Fritsch H., Hanft G., Aerts C., Rapion J., Allgeier A., Bogaerts J., Lacombe D. (2010) Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network Of Core Institutes (NOCI). Eur. J. Cancer 46, 2206–2215 [DOI] [PubMed] [Google Scholar]
- 29. Deeraksa A., Pan J., Sha Y., Liu X. D., Eissa N. T., Lin S. H., Yu-Lee L. Y. (2013) Plk1 is upregulated in androgen-insensitive prostate cancer cells and its inhibition leads to necroptosis. Oncogene 32, 2973–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hou X., Li Z., Huang W., Li J., Staiger C., Kuang S., Ratliff T., Liu X. (2013) Plk1-dependent microtubule dynamics promotes androgen receptor signaling in prostate cancer. Prostate 73, 1352–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang X., Li H., Zhou Z., Wang W. H., Deng A., Andrisani O., Liu X. (2009) Plk1-mediated phosphorylation of Topors regulates p53 stability. J. Biol. Chem. 284, 18588–18592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ando K., Ozaki T., Yamamoto H., Furuya K., Hosoda M., Hayashi S., Fukuzawa M., Nakagawara A. (2004) Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J. Biol. Chem. 279, 25549–25561 [DOI] [PubMed] [Google Scholar]
- 33. Liu X., Lei M., Erikson R. L. (2006) Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol. Cell. Biol. 26, 2093–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Corey E., Quinn J. E., Buhler K. R., Nelson P. S., Macoska J. A., True L. D., Vessella R. L. (2003) LuCaP 35: a new model of prostate cancer progression to androgen independence. Prostate 55, 239–246 [DOI] [PubMed] [Google Scholar]
- 35. Li Z., Li J., Bi P., Lu Y., Burcham G., Elzey B. D., Ratliff T., Konieczny S. F., Ahmad N., Kuang S., Liu X. (2014) Plk1 phosphorylation of PTEN causes a tumor-promoting metabolic state. Mol. Cell. Biol. 34, 3642–3661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ben Sahra I., Laurent K., Giuliano S., Larbret F., Ponzio G., Gounon P., Le Marchand-Brustel Y., Giorgetti-Peraldi S., Cormont M., Bertolotto C., Deckert M., Auberger P., Tanti J. F., Bost F. (2010) Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 70, 2465–2475 [DOI] [PubMed] [Google Scholar]
- 37. Fendt S. M., Bell E. L., Keibler M. A., Davidson S. M., Wirth G. J., Fiske B., Mayers J. R., Schwab M., Bellinger G., Csibi A., Patnaik A., Blouin M. J., Cantley L. C., Guarente L., Blenis J., Pollak M. N., Olumi A. F., Vander Heiden M. G., Stephanopoulos G. (2013) Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 73, 4429–4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kumar A., White T. A., MacKenzie A. P., Clegg N., Lee C., Dumpit R. F., Coleman I., Ng S. B., Salipante S. J., Rieder M. J., Nickerson D. A., Corey E., Lange P. H., Morrissey C., Vessella R. L., Nelson P. S., Shendure J. (2011) Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc. Natl. Acad. Sci. U.S.A. 108, 17087–17092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Engelman J. A. (2009) Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat. Rev. Cancer 9, 550–562 [DOI] [PubMed] [Google Scholar]
- 40. Liu P., Cheng H., Roberts T. M., Zhao J. J. (2009) Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 8, 627–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bitting R. L., Armstrong A. J. (2013) Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr. Relat. Cancer 20, R83–R99 [DOI] [PubMed] [Google Scholar]
- 42. Liu A. Y., Brubaker K. D., Goo Y. A., Quinn J. E., Kral S., Sorensen C. M., Vessella R. L., Belldegrun A. S., Hood L. E. (2004) Lineage relationship between LNCaP and LNCaP-derived prostate cancer cell lines. Prostate 60, 98–108 [DOI] [PubMed] [Google Scholar]
- 43. Buzzai M., Jones R. G., Amaravadi R. K., Lum J. J., DeBerardinis R. J., Zhao F., Viollet B., Thompson C. B. (2007) Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 67, 6745–6752 [DOI] [PubMed] [Google Scholar]
- 44. DeYoung M. P., Horak P., Sofer A., Sgroi D., Ellisen L. W. (2008) Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 22, 239–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ellisen L. W., Ramsayer K. D., Johannessen C. M., Yang A., Beppu H., Minda K., Oliner J. D., McKeon F., Haber D. A. (2002) REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol. Cell 10, 995–1005 [DOI] [PubMed] [Google Scholar]
- 46. Shoshani T., Faerman A., Mett I., Zelin E., Tenne T., Gorodin S., Moshel Y., Elbaz S., Budanov A., Chajut A., Kalinski H., Kamer I., Rozen A., Mor O., Keshet E., Leshkowitz D., Einat P., Skaliter R., Feinstein E. (2002) Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol. Cell. Biol. 22, 2283–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Choi Y. W., Lim I. K. (2014) Sensitization of metformin-cytotoxicity by dichloroacetate via reprogramming glucose metabolism in cancer cells. Cancer Lett. 346, 300–308 [DOI] [PubMed] [Google Scholar]
- 48. Liu X. S., Song B., Elzey B. D., Ratliff T. L., Konieczny S. F., Cheng L., Ahmad N., Liu X. (2011) Polo-like kinase 1 facilitates loss of Pten tumor suppressor-induced prostate cancer formation. J. Biol. Chem. 286, 35795–35800 [DOI] [PMC free article] [PubMed] [Google Scholar]