

Background: Molecular mechanisms of RANKL/RANK-mediated formation of multinucleated osteoclasts are not fully understood.
Results: PIRO (progranulin (PGRN)-induced receptor-like gene during osteoclastogenesis) is a direct target for the formation of multinucleated osteoclasts by PGRN upon RANK activation.
Conclusion: Progranulin/PIRO axis is a new regulatory axis in osteoclastogenesis.
Significance: This noble regulatory axis offers new avenues for the development of effective therapeutic strategies for osteoporosis.
Keywords: Bone, Cytokine, Genomics, Osteoclast, Osteoporosis, Receptor, Cell Fusion
Abstract
Homeostatic bone remodeling is vital to maintain healthy bone tissue. Although the receptor activator of nuclear factor κB ligand (RANKL)/RANK axis is considered the master regulator of osteoclastogenesis, the underlying mechanisms including cell fusion remain incompletely defined. Here, we introduce a new axis in the formation of multinucleated cells via RANK signaling: the progranulin (PGRN)/PIRO (PGRN-induced receptor-like gene during osteoclastogenesis) axis. When mouse bone marrow-derived macrophages were stimulated with PGRN in the presence of RANKL, explosive OC formation was observed. PGRN knockdown experiments suggested that endogenous PGRN is an essential component of the RANKL/RANK axis. Our efforts for identifying genes that are induced by PGRN unveiled a remarkably induced (20-fold) gene named PIRO. Substantial PGRN and PIRO expression was detected after 2 and 3 days, respectively, suggesting that their sequential induction. PIRO was predicted to be a five transmembrane domain-containing receptor-like molecule. The tissue distribution of PGRN and PIRO mRNA expression suggested that bone marrow cells are the most suitable niche. Mouse and human PIRO are part of a multigene family. Knockdown experiments suggested that PIRO is a direct target for the formation of multinucleated cells by PGRN. PGRN levels were also substantially higher in ovariectomized mice than in sham control mice. These observations suggest that PGRN and PIRO form a new regulatory axis in osteoclastogenesis that is included in RANK signaling in cell fusion and OC resorption of osteoclastogenesis, which may offer a novel therapeutic modality for osteoporosis and other bone-associated diseases.
Introduction
Bone remodeling is vital for the maintenance of healthy bone tissue in adult humans. Imbalances in this process can cause various pathological conditions, including osteoporosis. Although the receptor activator of nuclear factor κB ligand (RANKL)/RANK4 axis is believed to be a master regulator of osteoclastogenesis (1, 2), and several other cell communication factors have been described over the last decade, and the details of the underlying mechanisms, such as the cell fusion or resorption of osteoclasts (OCs) remains largely undefined. At least one in three women and one in five men are expected to suffer from osteoporotic fractures during their lifetime (3). However, only a limited number of therapeutic modalities are currently available. Although anti-RANKL has been used and anti-sclerostin human antibodies are likely to soon be available, and their long-term side effects remain to be determined because these proteins also play many other important roles in the differentiation of various cell types, such as dendritic and neuronal cells (4–8). Therefore, there is an urgent need for new therapeutic strategies to effectively treat osteoporosis and other bone diseases. The identification of the RANKL, its cognate receptor RANK, and its decoy receptor osteoprotegerin has brought a new molecular perspective to OC biology and bone homeostasis (9). The interaction between RANKL and RANK is required for osteoclast differentiation. Osteoprotegerin is a decoy ligand produced by both osteoblasts (OBs) and osteocytes that acts as a soluble receptor antagonist of RANKL, which prevents it from binding to and activating RANK. Because RANKL and osteoprotegerin are produced by different cell types, the expression of these ligands must be tightly regulated. Dysregulation of their expression leads to pathological conditions such as bone loss, bone tumor-associated osteolysis, and autoimmune diseases.
GRN encodes progranulin (PGRN), which was originally identified as a wound healing factor (6). PGRN was first purified as a growth factor from conditioned tissue culture media (10, 11) and is known to play a critical role in multiple physiological and pathological conditions, including cell growth, wound healing, tumorigenesis, and neurodegenerative diseases, such as frontotemporal dementia (12). Recently, it has been demonstrated that PGRN binds directly to tumor necrosis factor receptor (TNFR) and disturbs TNF-α-TNFR interaction, suggesting a role as a physiological antagonist of TNF-α signaling (13). However, Matsubara et al. (14) identified PGRN as a novel proinflammatory adipokine by differential proteome analysis of cellular models of insulin resistance. They showed that PGRN expression was induced by TNF-α or dexamethasone and decreased during adipocyte differentiation. Several subsequent studies have failed to show PGRN binding to TNFR (15, 16). Hence, the importance of PGRN in inflammation remains quite controversial and may need to be clarified in other inflammatory diseases involving osteoporosis. More recently, it has been shown that OBs produce a great deal of PGRN, which affects chondrogenesis (17). Therefore, we are interested in the roles of PGRN in bone biology. We report here a new RANK-dependent axis of potent osteoclastogenic factors, PGRN and PGRN-induced receptor-like gene during osteoclastogenesis (PIRO), whose primary functions are centered on the formation of multinucleated OCs, which are largely responsible for bone resorption.
EXPERIMENTAL PROCEDURES
Mice and Reagents
Ten-week-old C57BL/6J female mice were purchased from Damul Science (Daejeon, Korea). The mice were maintained at 22–24 °C and 55–60% humidity in a controlled environment under a 12-h light/dark cycle. All experiments were performed in accordance with the guidelines for animal experimentation from the Institute Committee of Wonkwang University. Control mice were injected with PBS (n = 9). Mice were sacrificed after 8 days, and blood samples were collected. Ovariectomized model mice (OVX, n = 9) and sham-operated mice (n = 9) were operated on at 9 weeks and sacrificed at 14 weeks, at which time blood samples were collected. Mouse progranulin (mPGRN) and human PGRN (hPGRN) were obtained from AdipoGen International (San Diego, CA). Soluble, recombinant human M-CSF and human RANKL were purchased from PeproTech EC, Ltd. (London, UK). FBS, α-minimum essential medium, and penicillin/streptomycin were purchased from Invitrogen. All other chemicals were of analytical or cell culture grade. Experiments were performed in accordance with the animal experiment guidelines of the Institutional Commmittee of Wonkwang University (Approval WKU14-17). All human subjects were reviewed and approved by the Wonkwang University institutional review board (Approval WKUH-HRBR-032).
Human and Mouse Bone Marrow Macrophage Preparation
Human bone marrow cells (HBMCs) and peripheral blood mononuclear cells (PBMCs) were obtained from healthy volunteers and were separated by density gradient centrifugation using Ficoll-Histopaque (Sigma-Aldrich). These cells were cultured for 7 days in the presence of M-CSF (100 ng/ml). Mouse bone marrow cells were obtained from 10-week-old C57BL/6J female mice by flushing the femurs and tibias and were seeded on culture dishes in α-minimum essential medium supplemented with 10% FBS and M-CSF (10 ng/ml). Nonadherent cells were transferred to 10-cm Petri dishes and cultured in the presence of M-CSF (30 ng/ml) for an additional 3 days.
In Vitro Osteoclastogenesis Assay
To examine the effect of PGRN on osteoclast differentiation, HBMCs and PBMCs were cultured for 8 and 15 days, respectively, in the presence of M-CSF (100 ng/ml) and RANKL (100 ng/ml), with or without PGRN (500 ng/ml). To examine the effect of PGRN on OC differentiation from MBMMs, these cells were cultured for 4 days with PGRN, M-CSF (30 ng/ml), and RANKL (100 ng/ml). The cells were fixed in 3.7% formalin for 10 min, permeabilized with 0.1% Triton X-100, and then stained with TRAP (Sigma). TRAP+ multinucleated cells containing more than five nuclei were counted as OCs.
Cytotoxicity Assay
Mouse bone marrow cells (1 × 104 cells/well) were cultured in 96-well plates with various concentrations of PGRN and incubated for 3 days in the presence of M-CSF (30 ng/ml). Then XTT solution (50 μl) was added to each well, and cultures were incubated for 4 h. The plate was read at 450 nm using an ELISA reader (Molecular Devices, Sunnyvale, CA).
Actin Ring Formation Assay
Mouse bone marrow cells were incubated with M-CSF (30 ng/ml) and RANKL (100 ng/ml) in the presence of various concentrations of PGRN for 4 days. Cells were fixed by incubation with 3.7% formaldehyde for 20 min, permeabilized with 0.1% Triton X-100 for 10 min, and stained with rhodamine-conjugated phalloidin for 10 min to visualize F-actin. Fluorescence images were acquired using a Leica fluorescent microscope (Wetzlar, Germany).
Pit Formation Assay
HBMCs (1 × 107 cells) and primary OBs (1 × 106 cells) were seeded on collagen gel-coated culture dishes and cultured for 7 days in the presence of 10−8 m 1,25-dihydroxyvitamin D3 (Sigma) and 10−6 m prostaglandin E2 (Sigma). The co-cultured cells were detached by treatment with 0.1% collagenase at 37 °C for 10 min and were then replated onto dentin slices or on hydroxyapatite-coated plates (Corning Inc., Corning, NY). The cells were incubated in the presence or absence of PGRN. After 24 or 48 h, the cells were removed, and the total resorption pits were photographed and analyzed using Image-Pro Plus software version 4.0 (Media Cybernetics, Silver Spring, MD).
RNA-Seq, Bioinformatics, and Identification of the PIRO Supergene Family
Expression profiling of human PGRN was extensively established by using GeneCards, BioGPS, and GEO. One of total RNA from differentiating OCs incubated in the presence or absence of PGRN was applied to an oligo(dT) bead column (Qiagen). A library was constructed with the TruSeq RNA sample preparation kit (Illumina, San Diego, CA), and deep sequencing was conducted with using a HiSeq 2500 instrument (TheraGen Bio, Suwon, Korea). Approximately 30,000 expressed mRNAs were sequenced to 100-fold coverage, and a comprehensive UniGene was established. Using the GM10800 full-length cDNA sequence, multiple BLASTN searches of the ENSEMBL Genome Database were conducted, which identified 26 PIRO family members in the mouse genome. The 26 PIRO cDNA sequences were aligned, and their sequence homology was calculated using DNASTAR software (Madison, WI). To isolate the human PIRO homologs, multiple translated sequences of mPIRO-1 were subjected to a protein sequence BLAST (BLASTP) (National Center for Biotechnology Information), which identified a human PIRO partial cDNA. Two human PIRO homologs located on chromosome 7 and 18 were identified by multiple homology searches of the human High Throughput Genomic Sequences database. These full-length cDNAs were assembled by expression sequence data (EST) in the National Center for Biotechnology Information or direct sequence analysis of the RT-PCR products by Sanger sequencing. A full-length human PIRO cDNA sequence was deposited into the DNA Data Bank of Japan (DDBJ accession number AB968079).
Quantitative Real Time RT-PCR
Total RNA was isolated using QIAzol reagent (Qiagen) according to the manufacturer's instructions. Equal amounts of total RNA were reverse-transcribed into cDNA using reverse transcript IITM (Promega, Madison, WI). PCR was performed using an ExicyclerTM 96 real time quantitative thermal block (Bioneer, Seoul, Korea). The relative expression level of each gene was calculated from the cycle threshold (Ct) value using the Ct method. The sequences of the primers used for real time PCR are listed in Table 1.
TABLE 1.
Sequences of primers, siRNA sequences, and shRNA sequences used in real time PCR experiments
| Name | Sequence |
|---|---|
| Primers | |
| mPGRN | Forward 5′-TTC ACA CAC GAT GCG TTT CA-3′ |
| Reverse 5′-AGG GCA CAC AGA AAA AG-3′ | |
| mPIRO | Forward 5′-CCT TTT TCA GTT TTC CTC GCC-3′ |
| Reverse 5′-TGC ACA CTG AAG GAC CTG GAA-3′ | |
| mc-Fos | Forward 5′-GGT GAA GAC CGT GTC AGG AG-3′ |
| Reverse 5′-TAT TCC GTT CCC TTC GGA TT-3′ | |
| mNFATc1 | Forward 5′-GAG TAC ACC TTC CAG CAC CTT-3′ |
| Reverse 5′-TAT GAT GTC GGG GAA AGA GA-3′ | |
| mTRAP | Forward 5′-TCA TGG GTG GTG CTG CT-3′ |
| Reverse 5′-GCC CAC AGC CAC AAA TCT-3′ | |
| mOSCAR | Forward 5′-GGA ATG GTC CTC ATC TGC TT-3′ |
| Reverse 5′-GGA ATG GTC CTC ATC TGC TT-3′ | |
| mGAPDH | Forward 5′-TCA AGA AGG TGG TGA AGC AG-3′ |
| Reverse 5′-AGT GGG AGT TGC TGT TGA AGT-3′ | |
| hPGRN | Forward 5′-TCC CCC TAA CCA AAT TCT CC-3′ |
| Reverse 5′-GGG ATG GCA GCT TGT AAT GT-3′ | |
| hPIRO | Forward 5′-CCT TTT TCA GTT TTC CTC GCC-3′ |
| Reverse 5′-TGC ACA CTG AAG GAC CTG GAA-3′ | |
| hGAPDH | Forward 5′-TCA AGA AGG TGG TGA AGC AG-3′ |
| Reverse 5′-GGT GGA GGA GTG GGT GTC-3′ | |
| siRNA sequences (duplex) | |
| mPIRO-siRNA 1 | Sense 5′-CUC CAU AUU CCA GGU CCU U(dTdT)-3′ |
| Antisense 5′-AAG GAC CUG GAA UAU GGA G(dTdT)-3′ | |
| mPIRO-siRNA 2 | Sense siRNA 2 5′-CGU CCU AAA GUG UGU AUU U(dTdT)-3′ |
| Antisense siRNA 2 5′-AAA UAC ACA CUU UAG GAC G(dTdT)-3′ | |
| mPIRO-siRNA 3 | Sense siRNA 3 5′-CGU CCU AAA GUG UGU AUU U(dTdT)-3′ |
| Antisense siRNA 3 5′-AAA UAC ACA CUU UAG GAC G(dTdT)-3′ | |
| hPIRO-siRNA1 | Sense siRNA 3 5′-CAC UUA UGA GUG AGA ACA U(dTdT)-3′ |
| Antisense siRNA 3 5′-AUG UUC UCA CUC AUA AGU G(dTdT)-3′ | |
| hPIRO-siRNA2 | Sense siRNA 3 5′-CUC CAU UUU UCA AGU CUC A(dTdT)-3′ |
| Antisense siRNA 3 5′-UGA GAC UUG AAA AAU GAC G(dTdT)-3′ | |
| hPIRO-siRNA3 | Sense siRNA 3 5′-CUG UCU UCU UGC CAU AUU U(dTdT)-3′ |
| Antisense siRNA 3 5′-AAA UAU GGC AAG ACA G(dTdT)-3′ | |
| shRNA sequences (vector: pMLP-GFP) | |
| mPGRN-shRNA 1 | 5′-AAAGGAGGTGAAGTGCGACATATAGTGAAGCCACAGATGTATATGTCGCACTTCACCTCCTTC-3′ |
| mPGRN-shRNA 2 | 5′-ATGACCTGATCCAGAGTAAGTATAGTGAAGCCACAGATGTATACTTACTCTGGATCAGGTCAC-3′ |
| mPGRN-shRNA 3 | 5′-AGAGAAGGGCATTTCTGCCATATAGTGAAGCCACAGATGTATATGGCAGAAATGCCCTTCTCC-3′ |
| hPGRN-shRNA 1 | 5′-ATGCCCTGATGGTTCTACCTGATAGTGAAGCCACAGATGTATCAGGTAGAACCATCAGGGCAC-3′ |
| hPGRN-shRNA 2 | 5′-AAAGGACACTTCTGCCATGATATAGTGAAGCCACAGATGTATATCATGGCAGAAGTGTCCTTC-3′ |
| hPGRN-shRNA 3 | 5′-ACAACGTGAAGGCTCGATCCTATAGTGAAGCCACAGATGTATAGGATCGAGCCTTCACGTTGC-3′ |
| hPGRN-shRNA 4 | 5′-ACCCAGGGCTACACGTGTGTAATAGTGAAGCCACAGATGTATTACACACGTGTAGCCCTGGGG-3′ |
siRNA and shRNA Experiments
Mouse and human PIRO siRNAs were designed and synthesized by Bioneer (shown in Table 2). Differentiating mouse or human OCs were transfected with 100 pmol of siRNA oligonucleotides using Lipofectamine 3000 (Invitrogen) according to the manufacturer's instructions. Mouse PGRN and human PGRN retroviral shRNAs were purchased from TransOMIC Technologies (Huntsville, AL). Retroviral particles were generated by transfecting the plasmids into Plat-E cells using X-tremeGENE 9 (Roche) according to the manufacturer's instructions. Bone marrow cells were incubated with the viral supernatant in the presence of Polybrene (8 g/ml), and then the cells were cultured for an additional 2 days.
ELISAs
We used mouse kits (Adipogen International) to determine PGRN levels in culture supernatant and mouse serum. Supernatant from cultures of differentiating osteoclasts was collected on specified days and was centrifuged at 3,000 rpm for 10 min to remove cell debris. To collect mouse sera, mice were decapitated under CO2 anesthesia, and blood was drained from the trunk into 1.5-ml polypropylene tubes and incubated at room temperature for 30 min. After centrifuging the blood at 3,000 rpm for 15 min at 4 °C, mouse serum samples were stored at −80 °C until use. The culture media and mouse serum samples were diluted at 1:5, 1:1,000, and 1:200, respectively, in 1× diluent solution. Assays were read at 450 nm.
OB Culture and Assays
To culture OBs, the calvariae of neonatal mice were digested with 0.1% collagenase and 0.2% dispase five times, and the cells isolated from the last three digestions were pooled and cultured in α-minimum essential medium containing 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. To measure alkaline phosphatase activity, primary OBs were inoculated at a density of 2 × 104 cells/well and cultured in the absence or presence of 50 μg/ml ascorbic acid and 10 mm β-glycerol phosphate. On day 7 of differentiation, the cells were harvested and sonicated in 50 mm Tris-HCl buffer (pH 7.4) containing 1% Triton X-100, 150 mm NaCl, and 1 mm EDTA. Then 100 ml of substrate (p-nitrophenylphosphate; Sigma) was added to the cells, and the plate was incubated at 37 °C for 30 min. The amount of p-nitrophenol released was determined by measuring the absorbance at 405 nm using a microplate reader. For alkaline phosphatase staining, cells were fixed in 70% ethanol and stained for 10–20 min with a solution containing 0.01% naphthol, AS-MX phosphate, 1% N,N-dimethyl formamide, and 0.06% fast blue BB (Sigma). For alizarin red S staining, on day 21 of differentiation, the cells were fixed using 3.7% formalin and stained for 10 min with 2% alizarin red S, pH 4.2 (Sigma). The bound ARS was dissolved in a 10% cetylpyridinium chloride monohydrate solution (pH 7.0), and the absorbance of this solution was measured at 540 nm using a microplate reader.
DC Generation
Human PBMCs were isolated from heparinized peripheral blood obtained from healthy adult volunteers by Ficoll-Hypaque (GE Healthcare) density gradient centrifugation. The study protocol was approved by the Institutional Review Board of the International St. Mary's Hospital (Incheon, Korea) and met the standards for blood donation. Immature dendritic cells were generated from the monocytes. Briefly, monocytes were isolated from PBMCs using the CD14+ monocyte isolation kit (Miltenyi Biotec, Auburn, CA). The CD14+ monocytes (5 × 105 cells/ml) were then cultured in the presence of 50 ng/ml human recombinant GM-CSF (Peprotech) and 20 ng/ml human recombinant IL-4 (Peprotech) in RPMI medium. After 5 days of incubation, the nonadherent immature dendritic cells were harvested and cultured in the presence of GM-CSF, IL-4, and 500 ng/ml LPS (from Escherichia coli 0111:B4, Sigma) for an additional 48 h.
Flow Cytometry
For immunophenotyping, cells were stained with FITC- or phycoerythrin-conjugated murine monoclonal antibodies for 20 min at 4 °C in PBS con5taining 1% FBS and 0.1 sodium azide.
Statistical Analyses
The data are presented as the mean ± S.D. All statistical analyses were performed using the Statistical Package for the Social Sciences (Korean version 14.0). Student's t test was used to compare parameters between two groups. Analysis of variance followed by Tukey's or Fisher's least significant difference post hoc test was used to compare the parameters of the three groups. p values less than 0.05 were considered statistically significant.
RESULTS
PGRN Is a Bona Fide Osteoclastogenic Factor That Is Dependent on the RANKL/RANK Axis
A series of expression profiling experiments conducted via pubic domains revealed a gene named granulin (GRN) that was highly expressed in human and mouse bone marrow. GRN encodes progranulin (PGRN), which was originally identified as a wound healing factor (18). To determine whether PGRN exerted any effects on bone homeostasis, MBMMs pretreated with M-CSF were stimulated with recombinant mouse PGRN in the presence of RANKL (19–22). We noticed a strong synergistic effect of these treatments on osteoclastogenesis in a RANKL-dependent manner (Fig. 1A). The results of an XTT assay suggested that PGRN treatment had no toxic effects on bone marrow cells (data not shown). PGRN treatment markedly enhanced the number of TRAP+ MNCs containing more than five nuclei in a dose-dependent manner (Fig. 1B). In RANKL-treated MBMMs, the levels of both PGRN mRNA and secreted PGRN protein were concomitantly increased in a time-dependent manner, suggesting that PGRN is an endogenous soluble factor induced by the RANKL/RANK axis (Fig. 1C).
FIGURE 1.

RANKL/RANK axis-dependent PGRN induction plays a key role in cell fusion and bone resorption. A, MBMMs stimulated with M-CSF (30 ng/ml) were cultured with or without RANKL (100 ng/ml) in the presence of the indicated concentrations of PGRN for 3 days. B, the number of TRAP+ MNCs was determined by counting MNCs with more than five nuclei. ***, p < 0.01, which was considered significant. C, PGRN mRNA expression and secreted PGRN were assessed by real time PCR and ELISA, respectively. D, MBMMs were transduced with retroviruses containing sh-mPGRNs or a control shRNA (sh-Con) and differentiated in the presence of RANKL for 3 days. PGRN mRNA expression was analyzed by real time PCR. TRAP+ MNCs containing more than five nuclei were counted. E, MBMMs were cultured in the presence of RANKL with or without PGRN for 4 days. Then the cells were fixed and permeabilized, and actin formation was visualized by staining with rhodamine-conjugated phalloidin. Fluorescence images were captured under a Leica fluorescent microscope (100× magnification). F, mature osteoclasts were seeded on hydroxyapatite-coated plates or dentin slices and were treated for 48 h with PGRN (500 ng/ml). The pit area was quantified with ImageJ software.
To obtain direct evidence for the involvement of PGRN in the RANKL/RANK axis, we designed three shRNAs targeting mouse PGRN (sh-mPGRNs) and a control shRNA, designated sh-Con (23). Whereas sh-mPGRN3 significantly suppressed the expression of PGRN mRNA, leading to the depletion of secreted PGRN (Fig. 1D), sh-mPGRN1 showed a modest knockdown effect (data not shown). Further, PGRN knockdown markedly reduced the numbers of TRAP+ MNCs (Fig. 1D), suggesting that the endogenous PGRN expressed in osteoclasts or other cell types in the bone micro niche is crucial for full maturation of OCs via the RANKL/RANK axis. When MBMMs were stimulated with increasing concentrations of mouse PGRN, enhanced F-actin formation was observed (Fig. 1E). However, as indicated by the alkaline phosphatase activity and alizarin red staining, OB formation was unaffected (data not shown). We next hypothesized whether PGRN could also enhance the bone resorption capability of MNCs. MNCs were cultured on hydroxyapatite or dentin scaffold in the presence of PGRN for 24 h, and pit formation was enumerated. As shown in Fig. 1F, potent bone resorption capability associated with PGRN stimulation was noted. To determine whether PGRN knockdown with siRNAs affected the expression of osteoclastogenic signature genes, real time PCR was performed. We found significant induction of PGRN mRNA 2 days after RANK activation, which was down-regulated by sh-mPGRN3 (Fig. 2A). The expression of several genes important for osteoclastogenesis, including TRAP, NFATc1, and OSCAR, was also attenuated by sh-mPGRN3 (Fig. 2, B–D).
FIGURE 2.
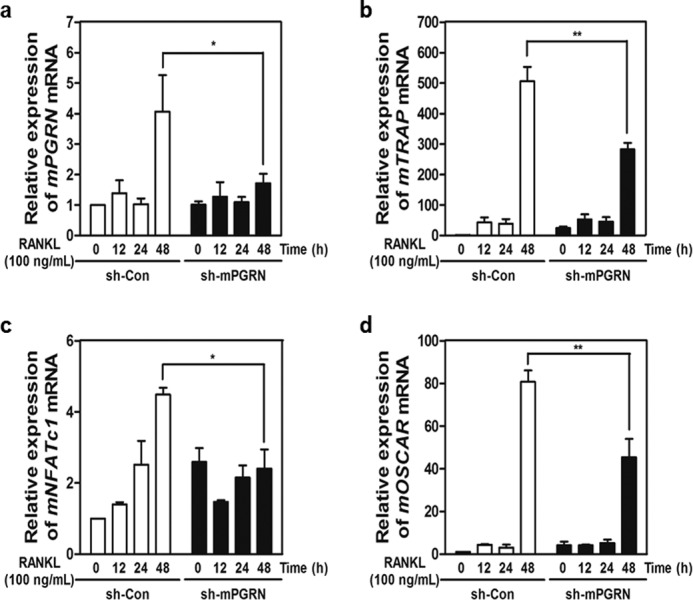
Induction of PGRN is essential for the regulation of key osteoclastogenic genes. A–D, fold induction of PGRN, TRAP, NFATc1, and OSCAR mRNAs at different stages of OC differentiation was measured by real time PCR. mRNA levels in sh-PGRN3- or sh-Con-infected OCs were compared.
Subsequently, HBMCs or peripheral blood mononuclear cells were stimulated with or without human PGRN in the presence of RANKL. PGRN treatment increased the number of TRAP+ MNCs (Fig. 3A). Finally, we tested whether knocking down PGRN in HBMCs decreased the formation of MNCs. Four sh-RNAs targeting human PGRN were transfected into cells, and in vitro osteoclastogenesis was assessed. As shown in Fig. 3B, transfection of two knockdown constructs, namely sh-hPGRN1 and sh-hPGRN3, exhibited decreased mRNA levels and significant decreases in TRAP+ MNCs. Taken together, these results strongly suggest that PGRN is a bona fide osteoclastogenic factor in vitro in both humans and mice.
FIGURE 3.

Effects of PGRN on the differentiation of human OCs and PGRN as a predictor of osteoporosis. A, HBMCs and PBMCs were differentiated with RANKL in the absence or presence of PGRN. TRAP+ MNCs were counted as described above. B, four sh-RNAs targeting human PGRN (sh-hPGRNs) plus a control sh-RNA (sh-Con) were transfected into HBMCs, and differentiation continued in the presence of RANKL with or without PGRN for 4 days. Knockdown efficiency was measured by real time PCR and TRAP+ MNCs were enumerated. p values less than 0.05 were considered significant. C, mouse sera or bone marrow fluids were prepared from sham-operated (SH) or OVX C57BL/6J mice.
PGRN Is Increased in Both Serum and Bone Marrow Fluid of OVX Mice
This finding prompted us to examine whether PGRN levels were elevated in a murine osteoporosis model. The concentration of PGRN in serum or bone marrow fluid in murine osteoporosis models using OVX mice was measured and compared with those in sham-operated mice (Fig. 3C). OVX mice exhibited significantly lower bone volume over 2-fold or density than sham control mice as determined by microcomputed tomography (data not shown). As shown in Fig. 3C, the concentrations of PGRN in both serum and bone marrow fluid in OVX mice were significantly higher than those in sham control mice, suggesting that PGRN is a biomarker of osteoporosis, at least, manifested in OVX mice model. Our initial screening using a limited number of human osteoporotic subjects in comparison with healthy individuals exhibited a modest increase in serum PGRN concentrations in these osteoporotic subjects (data not shown) but yet to be more extensively screened.
PIRO Is a PGRN-inducible Gene That Is Predicted to be a Five-TM Domain Receptor-like Gene
Because PGRN appeared to play a crucial role in regulating osteoclastogenesis through the RANKL/RANK axis, we hypothesized that PGRN may induce the expression of key factors that stimulate MNC formation. To identify the PGRN-induced genes, we carried out RNA-Seq analysis. We found 54 genes that were up-regulated by PGRN in the presence of RANKL. Among these genes, one designated GM10800 in ENSEMBL (ENSMUSG75014) was increased 20-fold in the presence of PGRN (p < 0.0001; data not shown). RANKL alone modestly induced the expression of GM10800, which could be due to stimulation by endogenous PGRN induced by the RANKL/RANK axis. Although GM10800 reached maximal induction 3 days after PGRN stimulation, TRAP mRNA expression was induced regardless of the presence or absence of PGRN (Fig. 4A). Because of its significant induction by PGRN, we named GM10800 mPIRO-1 (mouse PGRN-induced receptor-like protein during osteoclastogenesis 1). Knockdown of PGRN using sh-mPGRN3 significantly attenuated mPIRO-1 expression (Fig. 4B), again supporting mPIRO-1 as a PGRN-inducible gene. mPIRO-1 was only detected in our RNA-Seq analysis. The mPIRO-1 gene, which has been referred to as GM10800, is located on mouse chromosome 2 (98666547–98667301) and has a reverse gene orientation. We conclude that PGRN and mPIRO-1 are sequentially induced within the frame of the RANKL/RANK axis.
FIGURE 4.

PIRO is strongly inducible by PGRN and forms a supergene family. A, fold induction of GM10800 or TRAP mRNA at different stages of OC differentiation in the presence or absence of PGRN was measured by real time PCR. B, the mPIRO-1 mRNA levels in sh-PGRN3- or sh-Con-infected OCs were measured by real time PCR. C, the two PIRO domains are shown in red and green. SMART was utilized to predict the five transmembrane domain-containing structure of PIRO family proteins. The putative transmembrane domains are underlined. D, all mouse and human PIRO open reading frames were retrieved from ENSEMBL or the National Center for Biotechnology Information and were then used to create a phylogenetic tree with DNASTAR. E, the hPIRO open reading frame was assembled mainly via multiple blast searches of the human High Throughput Genomic Sequences with the mPIRO-1 cDNA or amino acid sequence. The three PIRO domains are shown in red, green, and blue.
mPIRO-1 was predicted to encode a five TM domain-containing molecule (TMs are underlined in Fig. 4C). mPIRO-1 contains two repeats called PIRO domains that are defined in the present study as consisting of 50–80 amino acids with highly conserved amino acids highlighted by two colors. Interestingly, the mouse genome contains 25 PIRO supergene family members, which include variants and potential noncoding RNAs scattered throughout chromosomes 2, 9, and 14. Massive gene duplication or rearrangement appears to have occurred on chromosome 9, which harbors 23 PIRO members (Fig. 4D). As seen in Fig. 4E, hPIRO contains three PIRO domains (highlighted in three colors) as well as a long C-terminal end. Like mPIRO-1, hPIRO was predicted to be a five TM domain-containing receptor-like molecule. mPIRO-1 and mPIRO-25 are highly homologous to hPIRO. The mPIRO-1 open reading frame was utilized in a protein blast search (BLASTP), leading to the finding of a partial cDNA sequence, referred to as CAD 70026, whose EMBL accession number is AX647823.1. Comparative genomic analysis of the mouse and human genomes suggests that the human homologs of mPIRO-1 are located on chromosomes 7 (clone RP11-776N22) and 18 (clone RP11-713H10), which likely arose as a result of gene duplication. hPIRO cDNAs were retrieved from the High Throughput Genomic Sequences, the human EST database, or through direct sequencing of the real time PCR products and assembled (data not shown). mPIRO-1 and hPIRO proteins shared greater than 80% amino acid identity (Fig. 5). We performed a set of experiments to determine the tissue distribution of PGRN or PIRO mRNA expression in C57BL6 via real time PCR, suggesting that bone marrow cells are the most adequate niche expressing both genes (Fig. 6). Further, when HBMCs were differentiated into osteoclasts in the presence or absence of PGRN, significant induction of hPIRO mRNA was observed 3 days after PGRN stimulation, recapitulating the induction of the mPIRO-1 mRNA under similar conditions (Fig. 7).
FIGURE 5.

Alignment of mPIRO-1 and hPIRO sequences. The 220-amino acid mPIRO-1 sequence was aligned with the 275-amino acid hPIRO sequence under optimal identity. Identical amino acids are denoted by the red circles.
FIGURE 6.
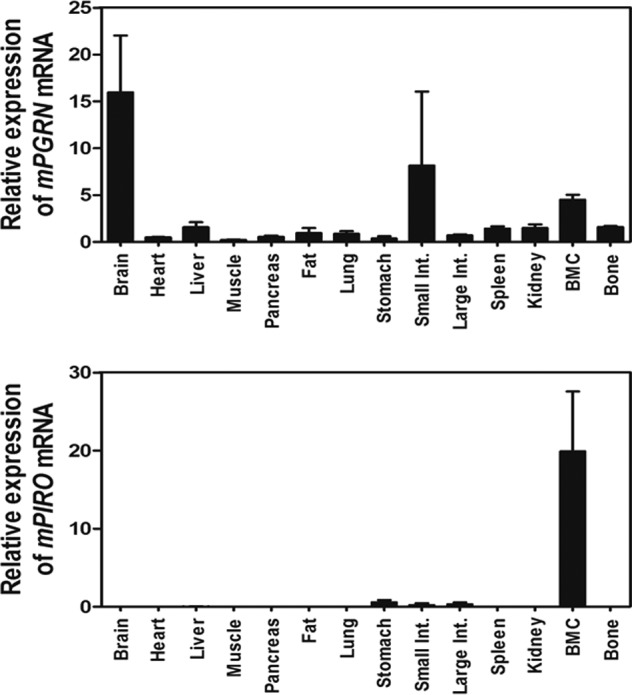
Tissue distribution of endogenous mouse PGRN and PIRO mRNA expression in C57BL6/J mice. The PGRN (A) and PIRO mRNA levels (B) in C57BL6/J mice tissues collected from the brain, heart, liver, muscle, pancreas, fat, lung, stomach, small intestine, large intestine, spleen, kidney, bone marrow cells (BMC), and bone were analyzed with real time RT-PCR. The data shown represent the relative mRNA levels normalized to mouse GAPDH.
FIGURE 7.
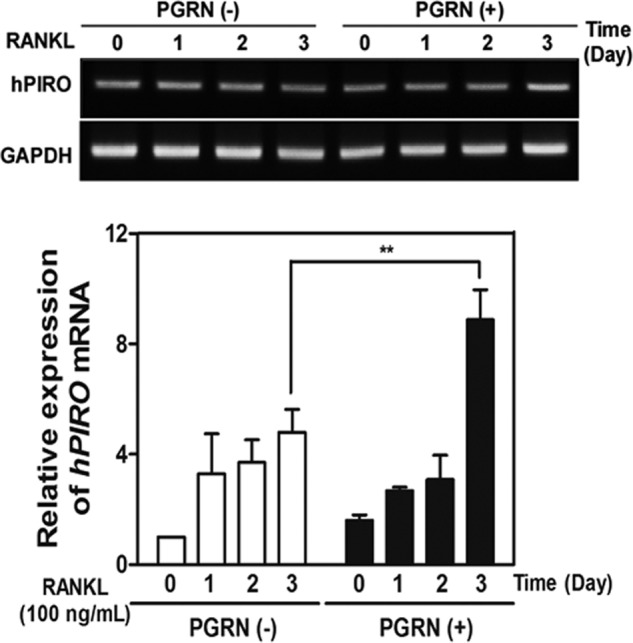
Effects of PGRN on hPIRO induction. Induction of hPIRO during the osteoclastogenesis from HBMCs. HBMCs were subjected to osteoclastogenesis in the presence of M-CSF and RANKL, and the temporal changes in the expression of hPIRO during the differentiation were analyzed by real time PCR. **, p < 0.02 was considered significant.
Knocking Down PIRO Markedly Inhibited the Formation of MNCs
Using a series of siRNAs to knockdown hPIRO or mPIRO-1, we tested whether silencing PIRO expression inhibited osteoclastogenesis. HBMCs were transfected with targeted or control siRNAs before stimulating them with RANKL. As shown in Fig. 8A, transfection of si-hPIRO2 markedly inhibited the formation of MNCs, suggesting that hPIRO is required for RANKL-stimulated formation of MNCs. Because si-hPIRO2 exhibited the strongest knockdown activity, we wanted to see whether generating sh-hPIRO2 via a retroviral vector would also show such knockdown efficiency to substantiate its specificity using another approach. As shown in Fig. 8B, indeed, sh-hPIRO2 markedly reduced the number of TRAP+ MNCs, as well as the levels of hPIRO mRNA. The same held true for knockdown of mPIRO-1 by si-mPIRO2 (Fig. 8C). More importantly, exogenous addition of recombinant mouse PGRN partially restored the formation of MNCs inhibited by si-mPIRO2 (Fig. 9).
FIGURE 8.

The PGRN-PIRO axis is an essential regulator for the formation of MNCs. A, HBMCs were transfected with the indicated siRNA oligonucleotides at a final concentration of 100 pmol. Transfected HBMCs were cultured in the presence of M-CSF for 1 or 2 days, and hPIRO mRNA expression was analyzed by real time PCR to verify knockdown efficiency. Other batches of transfected HBMCs were differentiated for an additional 4 days in the presence of M-CSF and RANKL. TRAP+ MNCs were enumerated. B, a si-hPIRO2 sequence-bearing retroviral vector called sh-hPIRO2 was generated. The si-hPIRO2 sequence was flanked by two viral-filler sequences with rotational symmetry. HMBCs were infected with sh-hPIRO2 and stimulated with RANKL for 4 days. TRAP+ MNCs were counted, and hPIRO mRNA levels were measured as described above. C, mouse bone marrow cells were transfected with siRNA oligonucleotides, and the same procedures were used as described above for the human PIRO siRNA-treated HBMCs. TRAP+ MNCs were counted. *, p < 0.05, which was considered significant.
FIGURE 9.
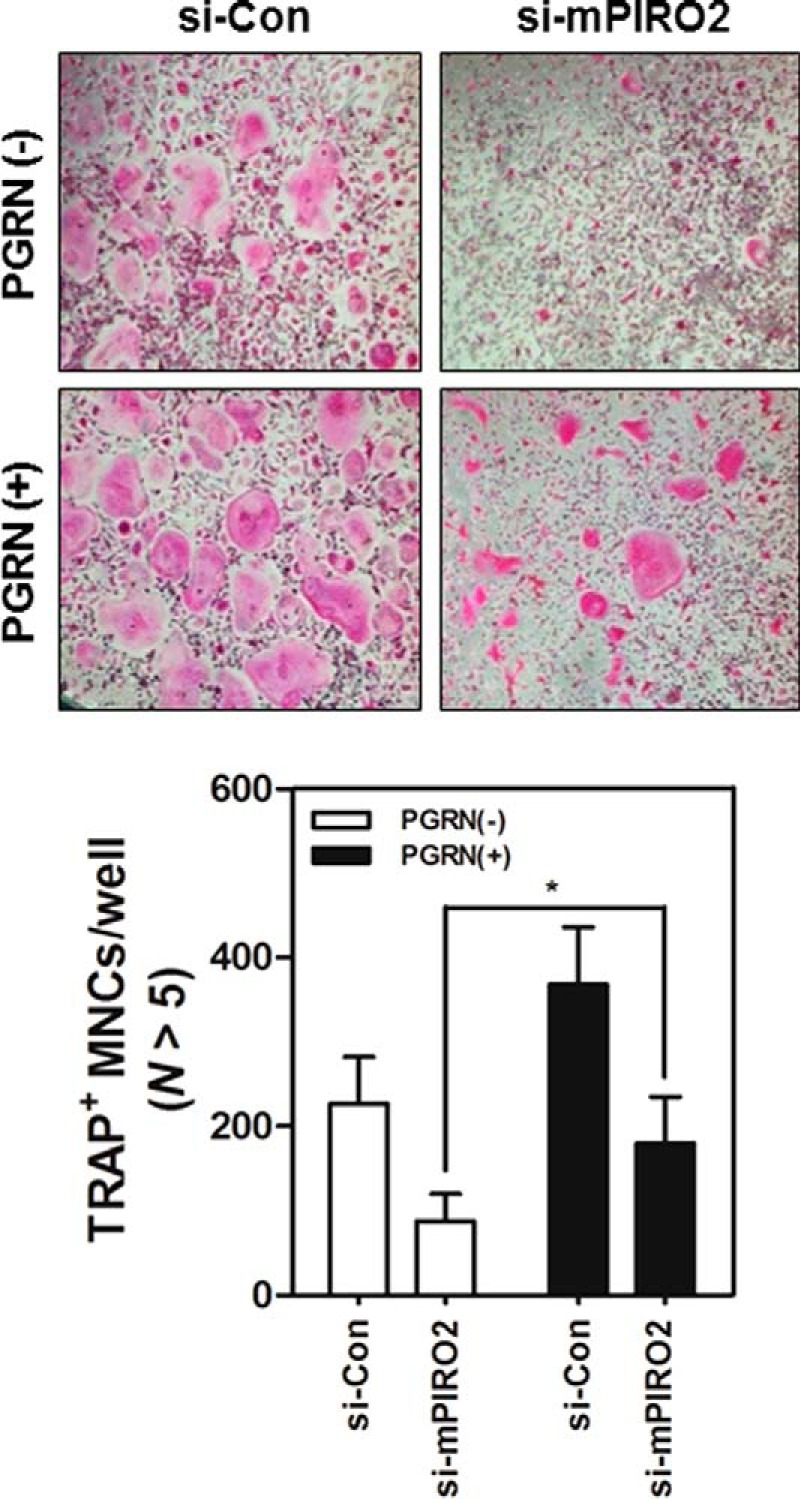
Restoration of the inhibitory effect of mPIRO-1 on the formation of MNCs by exogenous PGRN. MBMMs were transfected with si-mPIRO2 and were further differentiated in the presence of RANKL with or without recombinant mouse PGRN. TRAP+ MNCs were counted under a light microscope (100×). *, p < 0.05, which was considered significant.
DISCUSSION
Here, we reported a new unique cytokine/receptor axis, PGRN/PIRO, which is sequentially induced by the RANKL/RANK axis. These regulators appear to play a critical role in OC fusion at a later stage of osteoclastogenesis. An outstanding question is how RANK signaling induces the expression of PGRN. One possibility is that RANKL-treated OCs themselves produce and utilize PGRN in an autocrine manner (pathway I). Alternatively, other cell types in the bone niche, such as OBs, mesenchymal stem cells, or stromal cells, in the vicinity of the RANKL-treated OCs may produce PGRN, which then acts in a paracrine manner (pathway II). These possibilities are schematically presented in Fig. 10. OBs have been shown to produce PGRN (17). A recent study showed that PGRN is an essential regulator of chondrogenesis, and tissue-specific knockdown of PGRN in mice significantly inhibited chondrocyte differentiation, suggesting that PGRN might be an important regulator of cartilage formation (24). Although PGRN had no effect on dendritic cell maturation, it is likely that PGRN acts on macrophages or pre-osteoclasts (data not shown). An interesting but controversial feature associated with PGRN is whether or not this cytokine or metabolic hormone is inflammatory or anti-inflammatory. Exaggerated osteoclastogenesis is a feature of inflammation. IL-1β, TNFα, and LPS are known to drive osteoclastogenesis via activation of the inflammatory regulatory loop (19, 22). The more interestingly but controversial report that PGRN is a high affinity ligand for both TNFR1 and TNFR2 needs to be more rigorously tested because of the strong implication that PGRN has potent anti-inflammatory cytokine activity functioning as a decoy ligand (13). A few subsequent studies have negated this possibility; therefore, the answer is uncertain. Our study strongly suggests that PGRN is a potent osteoclastogenic factor in the RANK/RANKL axis. This feature is in contrast to the observation that TNFα induced osteoclastogenesis in a RANKL-independent manner (19). In any case, the PGRN receptor(s) that are expressed in differentiating osteoclasts remain to be identified.
FIGURE 10.

A working hypothesis for how the PGRN/PIRO axis regulates the formation of multinucleated osteoclasts. RANK downstream may induce endogenous PGRN expression on day 2 in differentiating osteoclasts (pathway I/autocrine), which would then act on an unknown PGRN receptor(s), triggering a second wave of downstream signaling, which culminates in the induction of PIRO on day 3 of differentiation. Up-regulation of PIRO is responsible for cell fusion. Pathway II, a paracrine way, depicts the provision of PGRN from other cell types stimulated by RANKL-engaged OCs, which leads to the activation of unknown PGRN receptor(s) expressed in differentiating osteoclasts to induce PIRO.
PIRO is part of a supergene family. There are 25 mouse homologs and at least two human PIRO genes. The singularity of these two species is that these genes may encode novel five-TM domain receptors that have not been previously reported. Interestingly, regarding the mouse PIRO genes, some appear to be noncoding RNAs because their initiator and/or termination codons are absent. For example, mPIRO-25 is situated 1.8 kb away from mPIRO-1, the gene we used in the present paper. These two genes are located on mouse chromosome 2. We had a great deal of difficulty trying to identify the human counterparts by using bioinformatics. It does not seem that the hPIRO supergene family differs from the mouse PIRO multigene family, suggesting a species difference. PIRO is also found in the rat genome; however, it appears to be different from the PIRO genes in mice. One thing that should be stressed is that mPIRO-1 was only detected in the OCs of 25 members. Its tissue distribution was restricted to mouse bone marrow and was not detected in any other organs. PGRN is a major factor contributing to frontotemporal dementia, and it seems to be involved in Alzheimer disease or Parkinsonism (25, 26). It is a potent neuronal growth factor (27). However, PIRO is not expressed at all in the mouse brain, indicating that PIRO may be a specific regulator of OCs. This also suggests that PIRO might be a good therapeutic target. Our observation that the mouse and human PIRO siRNAs were very powerful inhibitors of MNC formation strongly supports our hypothesis. Our classification of PIRO as a five transmembrane domain-containing receptor-like molecule relied on structural prediction, and its cellular localization and trafficking remain to be studied. We believe that the PIRO family is the first five-TM domain receptor-like one, which has never been discovered before but whose function is at least related to osteoclastogenesis. Osteoporosis is an aging-associated disease, and osteoclasts can cause tumor metastasis to bone or osteolysis by metastasized tumors that still requires new therapeutic interventions (28–30). The new PGRN/PIRO axis may be a useful therapeutic target for treating a broad range of bone-associated diseases.
Acknowledgment
We are deeply indebted to Dr. Hal E Broxmeyer for valuable comments and for proofreading this manuscript.
This work was supported by intramural funding from OsteoNeuroGen and Grant HI12C0110 from the Korean Health Technology R&D Project of the Ministry of Health & Welfare, Republic of Korea. B.-S.Y. has ownership in OsteoNeuroGen.
The nucleotide sequence(s) reported in this paper has been submitted to the DDBJ/GenBankTM/EBI Data Bank with accession number(s) AB968079.
- RANKL
- receptor activator of nuclear factor κB ligand
- MBMM
- mouse bone marrow-derived macrophage
- OVX
- ovariectomized
- OC
- osteoclast
- OB
- osteoblast
- TNFR
- tumor necrosis factor receptor
- PGRN
- progranulin
- m
- mouse
- h
- human
- M-CSF
- macrophage colony-stimulating factor
- HBMC
- human bone marrow cells
- PBMC
- peripheral blood mononuclear cell
- TM
- transmembrane
- MNC
- multinucleated cell.
REFERENCES
- 1. Wong B. R., Rho J., Arron J., Robinson E., Orlinick J., Chao M., Kalachikov S., Cayani E., Bartlett F. S., 3rd, Frankel W. N., Lee S. Y., Choi Y. (1997) TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J. Biol. Chem. 272, 25190–25194 [DOI] [PubMed] [Google Scholar]
- 2. Dougall W. C., Glaccum M., Charrier K., Rohrbach K., Brasel K., De Smedt T., Daro E., Smith J., Tometsko M. E., Maliszewski C. R., Armstrong A., Shen V., Bain S., Cosman D., Anderson D., Morrissey P. J., Peschon J. J., Schuh J. (1999) RANK is essential for osteoclast and lymph node development. Genes Dev. 13, 2412–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lems W. F., Geusens P. (2014) Established and forthcoming drugs for the treatment of osteoporosis. Curr. Opin. Rheumatol. 26, 245–251 [DOI] [PubMed] [Google Scholar]
- 4. Suda T., Takahashi N., Udagawa N., Jimi E., Gillespie M. T., Martin T. J. (1999) Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 20, 345–357 [DOI] [PubMed] [Google Scholar]
- 5. Capozzi A., Lello S., Pontecorvi A. (2014) The inhibition of RANK-ligand in the management of postmenopausal osteoporosis and related fractures: the role of denosumab. Gynecol. Endocrinol. 30, 403–408 [DOI] [PubMed] [Google Scholar]
- 6. Tokuyama N., Tanaka S. (2014) [Updates of denosumab, anti-RANKL antibody for osteoporosis]. Clin. Calcium 24, 85–91 [PubMed] [Google Scholar]
- 7. Yao Q., Ni J., Hou Y., Ding L., Zhang L., Jiang H. (2014) Expression of sclerostin scFv and the effect of sclerostin scFv on healing of osteoporotic femur fracture in rats. Cell Biochem. Biophys 69, 229–235 [DOI] [PubMed] [Google Scholar]
- 8. Anderson D. M., Maraskovsky E., Billingsley W. L., Dougall W. C., Tometsko M. E., Roux E. R., Teepe M. C., DuBose R. F., Cosman D., Galibert L. (1997) A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390, 175–179 [DOI] [PubMed] [Google Scholar]
- 9. Simonet W. S., Lacey D. L., Dunstan C. R., Kelley M., Chang M. S., Lüthy R., Nguyen H. Q., Wooden S., Bennett L., Boone T., Shimamoto G., DeRose M., Elliott R., Colombero A., Tan H. L., Trail G., Sullivan J., Davy E., Bucay N., Renshaw-Gegg L., Hughes T. M., Hill D., Pattison W., Campbell P., Sander S., Van G., Tarpley J., Derby P., Lee R., Boyle W. J. (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89, 309–319 [DOI] [PubMed] [Google Scholar]
- 10. Bhandari V., Bateman A. (1992) Structure and chromosomal location of the human granulin gene. Biochem. Biophys. Res. Commun. 188, 57–63 [DOI] [PubMed] [Google Scholar]
- 11. Zhou J., Gao G., Crabb J. W., Serrero G. (1993) Purification of an autocrine growth factor homologous with mouse epithelin precursor from a highly tumorigenic cell line. J. Biol. Chem. 268, 10863–10869 [PubMed] [Google Scholar]
- 12. Ahmed Z., Mackenzie I. R., Hutton M. L., Dickson D. W. (2007) Progranulin in frontotemporal lobar degeneration and neuroinflammation. J. Neuroinflammation 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tang W., Lu Y., Tian Q. Y., Zhang Y., Guo F. J., Liu G. Y., Syed N. M., Lai Y., Lin E. A., Kong L., Su J., Yin F., Ding A. H., Zanin-Zhorov A., Dustin M. L., Tao J., Craft J., Yin Z., Feng J. Q., Abramson S. B., Yu X. P., Liu C. J. (2011) The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 332, 478–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matsubara T., Mita A., Minami K., Hosooka T., Kitazawa S., Takahashi K., Tamori Y., Yokoi N., Watanabe M., Matsuo E., Nishimura O., Seino S. (2012) PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metab. 15, 38–50 [DOI] [PubMed] [Google Scholar]
- 15. Etemadi N., Webb A., Bankovacki A., Silke J., Nachbur U. (2013) Progranulin does not inhibit TNF and lymphotoxin-α signalling through TNF receptor 1. Immunol. Cell Biol. 91, 661–664 [DOI] [PubMed] [Google Scholar]
- 16. Chen X., Chang J., Deng Q., Xu J., Nguyen T. A., Martens L. H., Cenik B., Taylor G., Hudson K. F., Chung J., Yu K., Yu P., Herz J., Farese R. V., Jr., Kukar T., Tansey M. G. (2013) Progranulin does not bind tumor necrosis factor (TNF) receptors and is not a direct regulator of TNF-dependent signaling or bioactivity in immune or neuronal cells. J. Neurosci. 33, 9202–9213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Romanello M., Piatkowska E., Antoniali G., Cesaratto L., Vascotto C., Iozzo R. V., Delneri D., Brancia F. L. (2014) Osteoblastic cell secretome: a novel role for progranulin during risedronate treatment. Bone 58, 81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nguyen A. D., Nguyen T. A., Martens L. H., Mitic L. L., Farese R. V., Jr. (2013) Progranulin: at the interface of neurodegenerative and metabolic diseases. Trends Endocrinol. Metab. 24, 597–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takayanagi H., Kim S., Matsuo K., Suzuki H., Suzuki T., Sato K., Yokochi T., Oda H., Nakamura K., Ida N., Wagner E. F., Taniguchi T. (2002) RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-β. Nature 416, 744–749 [DOI] [PubMed] [Google Scholar]
- 20. Hayashi M., Nakashima T., Taniguchi M., Kodama T., Kumanogoh A., Takayanagi H. (2012) Osteoprotection by semaphorin 3A. Nature 485, 69–74 [DOI] [PubMed] [Google Scholar]
- 21. Negishi-Koga T., Shinohara M., Komatsu N., Bito H., Kodama T., Friedel R. H., Takayanagi H. (2011) Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat. Med. 17, 1473–1480 [DOI] [PubMed] [Google Scholar]
- 22. Koga T., Inui M., Inoue K., Kim S., Suematsu A., Kobayashi E., Iwata T., Ohnishi H., Matozaki T., Kodama T., Taniguchi T., Takayanagi H., Takai T. (2004) Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature 428, 758–763 [DOI] [PubMed] [Google Scholar]
- 23. Lima W. F., Prakash T. P., Murray H. M., Kinberger G. A., Li W., Chappell A. E., Li C. S., Murray S. F., Gaus H., Seth P. P., Swayze E. E., Crooke S. T. (2012) Single-stranded siRNAs activate RNAi in animals. Cell 150, 883–894 [DOI] [PubMed] [Google Scholar]
- 24. Feng J. Q., Guo F. J., Jiang B. C., Zhang Y., Frenkel S., Wang D. W., Tang W., Xie Y., Liu C. J. (2010) Granulin epithelin precursor: a bone morphogenic protein 2-inducible growth factor that activates Erk1/2 signaling and JunB transcription factor in chondrogenesis. FASEB J. 24, 1879–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sleegers K., Brouwers N., Van Broeckhoven C. (2010) Role of progranulin as a biomarker for Alzheimer's disease. Biomark Med. 4, 37–50 [DOI] [PubMed] [Google Scholar]
- 26. Brouwers N., Nuytemans K., van der Zee J., Gijselinck I., Engelborghs S., Theuns J., Kumar-Singh S., Pickut B. A., Pals P., Dermaut B., Bogaerts V., De Pooter T., Serneels S., Van den Broeck M., Cuijt I., Mattheijssens M., Peeters K., Sciot R., Martin J. J., Cras P., Santens P., Vandenberghe R., De Deyn P. P., Cruts M., Van Broeckhoven C., Sleegers K. (2007) Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch. Neurol. 64, 1436–1446 [DOI] [PubMed] [Google Scholar]
- 27. Gao X., Joselin A. P., Wang L., Kar A., Ray P., Bateman A., Goate A. M., Wu J. Y. (2010) Progranulin promotes neurite outgrowth and neuronal differentiation by regulating GSK-3β. Protein Cell 1, 552–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schramek D., Leibbrandt A., Sigl V., Kenner L., Pospisilik J. A., Lee H. J., Hanada R., Joshi P. A., Aliprantis A., Glimcher L., Pasparakis M., Khokha R., Ormandy C. J., Widschwendter M., Schett G., Penninger J. M. (2010) Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 468, 98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jones D. H., Nakashima T., Sanchez O. H., Kozieradzki I., Komarova S. V., Sarosi I., Morony S., Rubin E., Sarao R., Hojilla C. V., Komnenovic V., Kong Y. Y., Schreiber M., Dixon S. J., Sims S. M., Khokha R., Wada T., Penninger J. M. (2006) Regulation of cancer cell migration and bone metastasis by RANKL. Nature 440, 692–696 [DOI] [PubMed] [Google Scholar]
- 30. Morony S., Capparelli C., Sarosi I., Lacey D. L., Dunstan C. R., Kostenuik P. J. (2001) Osteoprotegerin inhibits osteolysis and decreases skeletal tumor burden in syngeneic and nude mouse models of experimental bone metastasis. Cancer Res. 61, 4432–4436 [PubMed] [Google Scholar]