

Background: The mode of action of PI(4,5)P2 in TRPV1 is controversial.
Results: Positively charged amino acids in the S4-S5 linker and in the TRP box form the PI(4,5)P2 binding site.
Conclusion: PI(4,5)P2 is a TRPV1 agonist and induces a conformational change of the internal gate.
Significance: The molecular nature of the PI(4,5)P2 binding site in TRPV1 is defined.
Keywords: electrophysiology; Ion Channel; Molecular Docking; Molecular Dynamics; Transient Receptor Potential Channels (TRP Channels); PI(4,5)P2; TRPV1; Multiscale Simulation
Abstract
Phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) has been recognized as an important activator of certain transient receptor potential (TRP) channels. More specifically, TRPV1 is a pain receptor activated by a wide range of stimuli. However, whether or not PI(4,5)P2 is a TRPV1 agonist remains open to debate. Utilizing a combined approach of mutagenesis and molecular modeling, we identified a PI(4,5)P2 binding site located between the TRP box and the S4-S5 linker. At this site, PI(4,5)P2 interacts with the amino acid residues Arg-575 and Arg-579 in the S4-S5 linker and with Lys-694 in the TRP box. We confirmed that PI(4,5)P2 behaves as a channel agonist and found that Arg-575, Arg-579, and Lys-694 mutations to alanine reduce PI(4,5)P2 binding affinity. Additionally, in silico mutations R575A, R579A, and K694A showed that the reduction in binding affinity results from the delocalization of PI(4,5)P2 in the binding pocket. Molecular dynamics simulations indicate that PI(4,5)P2 binding induces conformational rearrangements of the structure formed by S6 and the TRP domain, which cause an opening of the lower TRPV1 channel gate.
Introduction
Of the thermo-TRP channels known to date, the transient receptor potential vanilloid 1 (TRPV1),4 which was the first to be cloned, is activated by temperature increases in the noxious range and by capsaicin (CAP), which is the pungent ingredient of chili peppers (1, 2). TRPV1 channels, which are found in a subset of neurons in the dorsal root and trigeminal ganglia, have been implicated in thermal nociception, specifically in inflammation-induced thermal hyperalgesia (3–5). TRPV1 is a nonselective cation channel that works as a polymodal signal integrator of chemical and physical stimuli, such as activation by CAP, anandamide, heat, and protons. At the structural level, TRPV1 is a homotetramer, where each subunit has six transmembrane segments (i.e. S1-S6) with the N and C termini located in the intracellular region (6, 7). Recently, a three-dimensional structure of the TRPV1 channel has been determined at a resolution of 3.3 Å by cryo-electron microscopy. Moreover, the structure of TRPV1 in complex with two potent agonists, namely resiniferatoxin (RTX; Ref. 1) and the spider double-knot toxin (Ref. 8; DkTx), as well as TRPV1 in complex with CAP were determined at resolutions of 3.8 and 4.2 Å, respectively (9, 10).
With regard to phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2), while it is agreed that the C terminus contains a PI(4,5)P2 binding site (9, 11–13), there is still debate as to whether PI(4,5)P2 behaves as an activator (13, 14) or as a TRPV1 channel antagonist, as recently proposed by (15). Regardless of whether PI(4,5)P2 activates TRPV1 or exerts an inhibitory effect, it may be safe to assume that the negatively charged headgroups of PI(4,5)P2 should be in contact with positively charged regions of the TRPV1 channel. A molecular simulation showed that a PI(4,5)P2 headgroup made contact with positively charged residues Lys-694, Lys-698, and Lys-701 from the proximal TRPV1 C terminus, and with amino acids Arg-575 and Arg-579 located in the S4-S5 linker (11). This finding is consistent with the notion that the negatively charged PI(4,5)P2 headgroup should bind to a positively charged TRPV1 region in the vicinity of the bilayer surface. In fact, a deletion analysis of the distal C-terminal region of TRPV1 showed that this segment is not required for TRPV1 to be regulated by PI(4,5)P2, while, in contrast, the proximal C-terminal fragment was observed to interact directly with PI(4,5)P2 in vitro (13).
In the present study, by using electrophysiology, mutagenesis and by performing atomistic molecular dynamics (MD) and docking simulations, we observed that PI(4,5)P2 behaves as a channel agonist and we were able to unveil the binding site for PI(4,5)P2 in the TRPV1 channel. The molecular simulations also allowed us to identify the structural rearrangements that the channel undergoes upon PI(4,5)P2 binding.
EXPERIMENTAL PROCEDURES
Molecular Biology
cDNA coding for rat TRPV1 (GenBankTM accession no. NM031982) cloned in pcDNA3 vector was used. All mutants were generated by Quickchange mutagenesis (Stratagene, La Jolla, CA), according to the manufacturer's instructions, using the sense and antisense primers. All clones were confirmed by DNA sequencing.
Cell Culture and Transfection
HeLa and HEK 293 cells were transfected with pcDNA3 vector containing the wild-type or mutant coding sequence, so as to show that the effects are independent of the cell expression system. Transfection was carried out by using the FuGENE® 6 Transfection Reagent (Promega).
PI(4,5)P2
All phosphoinositides were the short-chain DiC8 versions. DiC8-PI(4,5)P2 solutions were solubilized in recording solution from 1 mm stock, frozen at −20 °C, and used the same day they were diluted from the stock.
Electrophysiology
For electrophysiological experiments, culture cells were grown on 12-mm cover slips and were directly mounted in the experimental chamber installed on the stage of an inverted microscope (Olympus). Inside-out excised patch recordings were performed at 20 °C using filamented borosilicate glass pipettes (o.d. = 1.5 mm, i.d. = 0.86 mm, Warner Instruments, Hamden, CT) that had been heat-polished with a microforge. Patch pipettes (tip size, 3–4 μm) were filled with saline solution (150 mm NaCl, 10 mm EGTA, 2 mm MgCl2, 10 mm HEPES, pH 7.4), and the same solution was also used as the external recording solution. All experiments were performed at 20 °C using the following voltage protocol: hp = 0 mV, and the membrane was pulsed to voltages between −100 and +220 mV in 20 mV increments for 30 ms, each followed by a step to −80 mV. Currents were recorded with an Axopatch 200B system (Axon Instruments), filtered at 1 kHz using a low-pass 10-pole Bessel filter, and transferred to a computer using a Digidata interface (Axon Instruments) at a sampling rate of 20 kHz. CAP was dissolved in ethanol to a final concentration of 1 mm in ethanol and then diluted to the desired concentrations on the day of the experiment.
Dose-response Curves
All electrophysiological experiments were repeated at least four times. Representative traces are shown in Fig. 1, A and B. Electrophysiological data were analyzed with Origin Pro (OriginLab Corp. Northampton). All of the results are presented as mean ± S.D. for each experimental condition. CAP responses were obtained at the beginning or end of the recordings for the purpose of TRPV1 expression control. PI(4,5)P2 enhanced the TRPV1-induced current channel activity when added to inside-out patches already treated with 1 μm CAP. However, PI(4,5)P2 concentrations over 15 μm was observed to activate endogenous current in HeLa cells, possibly due to the activation of TRPM4 (16). To compare the effect of PI(4,5)P2 before and after stimulation with CAP, we normalized our recordings to the maximum current obtained with CAP in the presence of 15 μm PI(4,5)P2 at steady state (Equation 1).
The experimental data of each patch were fitted (Fig. 1E, solid lines) as free parameters by using the following Hill Equation 2,
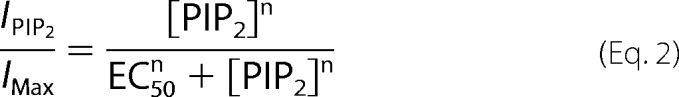 |
where n is the Hill coefficient and EC50 is the PI(4,5)P2 concentration producing 50% of the maximal effect. Statistical significance was assayed with the two-tailed Student's t test or a one-way ANOVA, as appropriate, with Statgraphics Plus 5.0 (GraphPad Software, Inc., San Diego, CA). Statistical significance was considered at p < 0.05.
FIGURE 1.

PI(4,5)P2 directly activates TRPV1 and enhances capsaicin-induced current. Macroscopic currents for TRPV1 induced by PI(4,5)P2 before (A) and after (B) the addition of 1 μm CAP obtained using inside-out patches. Current recordings were obtained in response to voltage pulses from 0 mV to 220 mV in 10 mV steps. This voltage protocol was applied for each concentration of PI(4,5)P2 before and after 1 μm of CAP in the same patch (n>4). No currents were detected in these voltage ranges in the absence of agonists. Temperature: 20 °C. C, current-voltage relationship obtained by adding the indicated PI(4,5)P2 concentration and adding capsaicin. Each point is the average of measurements on 4 patches. D, current-voltage relationship obtained by adding CAP prior to the PI(4,5)P2 addition. E, dose-response curves for TRPV1wild-type treated with PI(4,5)P2 before and after adding 1 μm CAP. EC50 values were 3.2 ± 0.2 μm and 5.6 ± 0.9 μm for respectively (nH = 1.7 and 1.5, n >4). F, allosteric model proposed for the activation of the TRPV1 channel by CAP and PI(4,5)P2. F, allosteric model for TRPV1 gating. The opening reaction is described by the equilibrium constant L and the binding of PI(4,5)P2, and CAP by the equilibrium constants Q and K, respectively. Allosteric factors F and C facilitate opening when PI(4,5)P2 and CAP, respectively bind to the channel. G, PI(4,5)P2 dose-response curves. The curves were identified by using the equation given in the figure with Q = 7 μm (Fig. 1E) and K = 0.6 μm (REF. 4) and C = 1,000 and D = 10,000.
Computational Methods
In this study, we employed a computational method to evaluate the interaction between PI(4,5)P2 molecules and the wild-type and mutated TRPV1 channels. First, molecular docking simulations of PI(4,5)P2 for the wild-type channel and for the R575A, R579A, K694A, and R701A mutants were performed. Then, by using the structure obtained from docking simulations as a starting point, the structural and energetic properties of the TRPV1-PI(4,5)P2, TRPV1-CAP, and TRPV1- PI(4,5)P2-CAP- complexes were analyzed through molecular dynamics simulations.
Atomistic Protein Systems Preparation
Cryo-electron microscopy structures of the wild-type closed (3J5P) (10) and open (3J5Q) (9) TRPV1 channels were obtained directly from the Protein Data Bank entry. Beginning from the atomic positions in the closed structure, point mutations were made in all four subunits of the TRPV1 channel. Molecular models of the R575A, R579A, R701A, and K694A mutants were thus created by using the plugin mutator1.3 implemented in the software VMD (17).
Molecular Docking of PI(4,5)P2 into the TRPV1 Channel
For the purpose of evaluating the most probable PI(4,5)P2 binding site in the wild-type TRPV1 structure (PDB id:3J5P) (10), we performed docking simulations with the program Molsoft ICM (Internal Coordinate Mechanics) software (18). ICM was used to draw the PI(4,5)P2 structure, assign bond orders, place explicit hydrogen atoms, identify hydrogen bonds using water sampling and minimize energy using the MMFF94 force field (19). We used a cubic box of 30 × 30 × 45 Å3. Default docking parameters were used. As docking progressed, we utilized a systematic search of PI(4,5)P2 conformations, followed by the insertion of PI(4,5)P2 into the wild-type or mutant TRPV1 structures. Subsequently, a minimization of the interaction energy between PI(4,5)P2 and TRPV1 was performed, as defined by the MMFF94 force field. Finally, the lowest energy configurations were subjected to a Biased Probability Monte Carlo procedure, so as to refine the TRPV1 structure and further reduce the conformational energy (18). This methodology and the same simulation conditions were used to dock PI(4,5)P2 and CAP into the APO- and CAP-TRPV1 structures (PDB ID:3J5P and 3J5R, respectively) (9, 10).
Molecular Dynamics Simulations of TRPV1 Systems
Beginning from the structures determined by cryo-electron microscopy, we built several TRPV1-membrane systems in atomistic detail. First, we performed 300 ns of simulation to verify that the APO-TRPV1 structure obtained from cryo-electron microscopy remained stable in simulation while embedded in a POPC lipid bilayer membrane. To determine whether we could observe the conformational changes of TRPV1 associated with PI(4,5)P2 binding, we modified the above system by adding PI(4,5)P2 (APO-TRPV1- PI(4,5)P2) in the most favorable configuration determined by docking. Thereafter, the system was unrestrained to reach a total simulation time of 300 ns. Finally, we added 4 CAP molecules (APO-TRPV1- PI(4,5)P2-CAP) to the APO-TRPV1- PI(4,5)P2 system, which subsequently was left unrestrained during 200 ns simulation. In addition, an independent system was constructed by inserting PI(4,5)P2 and CAP into the RTX/DkTx-TRPV1 structure determined experimentally.
The detailed protocols of the molecular dynamics simulations were as follows. All MD simulations were performed with the program NAMD 2.9 (20) and the CHARMM36 force field (21) and analyzed with the software VMD (17). A steady temperature was enforced with a Langevin thermostat, having a damping coefficient of 1 ps−1. A steady pressure of 1 atm was enforced using the Langevin piston method (22). Short-range electrostatic and van der Waals interactions were truncated smoothly from 10 to 12 Å, while long-range electrostatic forces were computed using the particle-mesh Ewald (PME) summation method (23). The parameters and charges for PI(4,5)P2 and CAP were determined from the ParamChem online server using the CGenFF force field (24). The POPC lipids were modeled using a united-atom modification of the CHARMM36 force field for lipids (25, 26). The POPC bilayer membrane had a lateral area of 160 × 160 Å2 and was solvated in a periodic box of pre-equilibrated TIP3P water molecules (27) using VMD (17). Potassium and chloride ions (0.11 m KCl) were added to the aqueous phase to mimic the experimental conditions and to guarantee electrical neutrality. The initial configurations of each system were optimized by means of an energy minimization algorithm. Soft harmonic restraints were applied to the protein backbone (0.5 kcal/(mol Å2)) in the first 70 ns of equilibration. Thereafter, the system was unrestrained to reach a total time of 300 ns. When CAP was added, harmonic restraints (2 kcal/(mol Å2)) were assigned to preserve the distance between CAP molecules and residues of the CAP binding site in the first 30 ns of the simulation. The system was then left unrestrained during a 200 ns trajectory. To understand the expansion of the lower gate, two distance restraints (10 kcal/(molÅ2)) were applied between the Cα carbon of the Ile-679 faced. The distance was applied to obtain a radius of 2.2 Å, which is observed into the lower gate in the RTX/DkTx TRPV1:PI(4,5)P2:CAP simulation. The last stage lasted for 30 ns trajectory.
RESULTS
PI(4,5)P2 Directly Activates TRPV1 Channels
To study whether PI(4,5)P2 activates TRPV1 and how this lipid modulates CAP-induced current, the current-voltage relationship was first determined by using inside-out membrane patches expressing TRPV1 channels. In the absence of CAP, no macroscopic current was detected in the voltage range tested. We found that adding PI(4,5)P2 to the internal solution activated outward current only in cells expressing TRPV1 channels (Fig. 1, A and B). Moreover, PI(4,5)P2 alone was observed to activate TRPV1 steady state current in the absence of all stimuli, such as CAP or temperature (Fig. 1, A and C). As was also reported by Ufret-Vincenty et al. (13), we found that PI(4,5)P2 further enhances TRPV1-induced currents when added to inside-out patches previously treated with 1 μm CAP (Fig. 1, B and D). PI(4,5)P2 activation data were fitted with a Hill equation (see under “Experimental Procedures”). Dose-response data were well fitted using an EC50 value of 3.2 μm and 5.6 μm as well as Hill coefficients of 1.7 and 1.5 for data obtained in the presence and absence of CAP, respectively (solid lines in Fig. 1E). PI(4,5)P2 dose-response data in the presence of CAP is in reasonable agreement with that obtained by Ufret-Vincenty et al. (13), and the rightward shift seen in the absence of CAP can be explained on the basis of a simple allosteric model (28, 29) (Fig. 1, F and G). The main assumption of this model, which is that CAP and PI(4,5)P2 sensors are contained in separate structures, is corroborated by the structural data (See below, Fig. 4E). In this model, PI(4,5)P2 binding, as described by the equilibrium constant Q, facilitated the opening transition through the allosteric factor F (LF, F ≫ 1). CAP addition was observed to further facilitate the opening, since the opening reaction was then determined by LFC (C ≫ 1), thus promoting a shift to the left of the PI(4,5)P2 dose-response curve (Fig. 1E). It is interesting to note that we consistently found that maximal currents were larger when CAP was added prior to the addition of PI(4,5)P2 (cf. Fig. 1, C and D). A possible explanation for this result is provided in the discussion section on the basis of our molecular simulations.
FIGURE 4.

Stability of PI(4,5)P2 in its binding site, electrostatic potentials and the effect of PI(4,5)P2 and CAP binding on the conformation of TRPV1. A, root-mean-square deviation of the atomic coordinates of each of the PI(4,5)P2 molecules during and MD simulation with respect to the positions identified by docking. B, distances between key residues of TRPV1 (Arg-575, Arg-579, Lys-694, and Arg-701) and each of the PI(4,5)P2 phosphate groups as function of time Phosphate groups are identified as in Fig. 2C. C, pore radius along TRPV1's channel axis for different experimental structures: RTX-TRPV1 (blue), CAP-TRPV1 (green), and APO-TRPV1 (red). C, radius of APO-TRPV1 (PDB:3J5P) pore in the absence of ligands (black line) and in the presence of CAP and PI(4,5)P2 (blue line). The black line was obtained from the last frame of the simulation of the complex APO-TRPV1+ PI(4,5)P2+CAP using distances restraints. D, projection of the calculated electrostatic potential onto the surface of the TRPV1 channel. E, spatial location of the putative binding site for PI(4,5)P2 (red) and CAP (cyan). Position of PI(4,5)P2 determined by docking into the APO-TRPV1 (F) and CAP-TRPV1 experimentally determined structures (G).
Computational Determination of the Binding Site
To explain the molecular characteristics of PI(4,5)P2 binding to the TRPV1 channel at the atomic level, molecular docking simulations of PI(4,5)P2 were performed on the atomistic structure of wild-type TRPV1 (9). The locations of four binding sites for PI(4,5)P2 in TRPV1 are shown in Fig. 2, A and B. Fig. 2D shows in detail the lowest energy conformation of the 50 docking simulations produced by the software ICM. In most (82%) of these configurations, strong interactions were present between the phosphate at position 5 of the PI(4,5)P2 headgroup (Fig. 2C) and the guanidine groups of residues Arg-575 and Arg-579. The distances between the phosphorus atom and the center of mass of the amino acid were 4 Å and 3 Å, respectively. Additionally, among the conformations produced by docking, the Lys-694 residue was seen to alternate between making contact with the phosphate located at positions four and five of PI(4,5)P2. These contacts appeared in ∼50% of the conformations. Furthermore, a hydrogen bond was formed between the ester group of one of the hydrocarbon chains of PI(4,5)P2 and the guanidine group of residue Arg-579 (∼2.9 Å) in most (76%) of the generated configurations (Fig. 2D). On the other hand, it was unlikely for the Arg-701 residue to form part of the binding pocket of PI(4,5)P2 because it was far (>12 Å) from PI(4,5)P2 in 100% of the conformations obtained by docking. In addition to the residues mutated in this work, Lys-571, Lys-688, and Gln-561 appeared to contribute to the binding of PI(4,5)P2. Moreover, it was possible to identify that the 50 lowest energy docking conformations of PI(4,5)P2 in the wild-type TRPV1 channel are quite similar, implying a single distinct bound state.
FIGURE 2.

Theoretical search for a putative PI(4,5)P2 binding site in the TRPV1 channel. A and B, top view and side view (with respect to the plane of the membrane) of the TRPV1 channel bound to four PI(4,5)P2 molecules. C, atomistic representation of the PI(4,5)P2 model. Numbers 1, 4, and 5 indicate the position of the phosphate groups. D, conformation of PI(4,5)P2 in the binding pocket of the wild-type TRPV1 channel. Inset (dotted green lines), magnified view of the atomic-level interactions between wild-type TRPV1 and PI(4,5)P2. Portions of two different subunits are shown. Amino acid residues Arg-575 and Arg-579 are located in the S4-S5 linker of the blue subunit and residue Lys-694 is in the TRP domain of the yellow subunit.
Cationic Residues Identified in Silico Are Involved in PI(4,5)P2 Activation of TRPV1 in Vivo
Toward the aim of testing the predictions forecasted by our structural models, the residues we identified in silico (Arg-575, Arg-579, Lys-694) were experimentally mutated to alanine. We also mutated residue Arg-701 to alanine as a negative control. All the neutralization mutants of TRPV1 were almost identically sensitive to CAP, just like wild-type TRPV1 (data not shown). However, neutralization of residues Arg-575, Arg-579, and Lys-694 (but not Lys-701) strongly affected PI(4,5)P2-induced activation (Fig. 3A) by shifting dose-response curves to the right along the concentration axis (Fig. 3B). Thus, mutations of positively charged residues at the proposed PI(4,5)P2 binding site decreased the apparent affinity of PI(4,5)P2 to this binding site (Fig. 3B). Our results showed that the proximal regions of the S4-S5 linker and the TRP domain show a high affinity for PI(4,5)P2 and induce TRPV1 activation in the absence of other agonists and at a temperature well below the threshold for this channel.
FIGURE 3.

Effects of mutations on the key residues that link PI(4,5)P2 to the TRPV1 channel. A, current-voltage relationships for the wild type TRPV1 and TRPV1 alanine mutants obtained in the presence of 10 μm PI(4,5)P2. B, Hill plots for wild-type TRPV1 and alanine mutants. EC50 and Hill coefficients were: wild-type: 5.6 μm and 1.5; R575A: 18.5 μm and 1.0; R579A: 73.6 and 1.0; K694A: 88.1 and 1.0; R701A: 5.5 and 1.4. C–G, an enlarged view of the lowest energy configurations attained by PI(4,5)P2 in the binding pocket in one of the 50 docking experiments performed using the wild type and mutant TRPV1 structures. H–L, fifty superimposed configurations generated by docking of PI(4,5)P2 into the wild-type and R575A, R579A, K694A, and R701A mutants (C, H: Wild-type TRPV1, score 0; D,I: R575A, score 12.5; E, J: R579A, score 7.5; F, K: K694A, score 11.5; G, L: R701A, score 2.3). The TRPV1 wild-type and alanine TRPV1 mutants are shown in a white cartoon representation, while PI(4,5)P2 is represented by a stick model in which carbon, oxygen, and phosphorus atoms are shown in cyan, red, and brown, respectively. A similar representation is used for amino acids of interest, except that the carbon atoms are shown in yellow.
To identify possible mutation effects on the molecular interactions between PI(4,5)P2 and its binding site, molecular docking simulations were performed on the structures of the TRPV1 mutants that were obtained in silico (R575A, R579A, K694A, and K701A). Fig. 3, C–L shows the lowest energy configurations produced by docking PI(4,5)P2 into the binding pocket of the wild-type and mutant proteins. By observing the data presented in Fig. 3, D and I (mutant R575A), we arrived at the general conclusion that PI(4,5)P2 shows a high convergence of configurations when bound to the binding pocket in wild-type TRPV1 (Fig. 3, C and H). On the other hand, PI(4,5)P2 was able to bind in a broad range of configurations in the alanine mutants. More specifically, R579A (Fig. 3, E and J), and K694A (Fig. 3, F and K) mutants showed a drastic difference in binding mode as compared with the wild-type system. Therefore, the positively charged amino acid residues described above were shown to be key residues for PI(4,5)P2 binding based on the mutant docking simulations of the mutant TRPV1 channels. In the negative control experiment, PI(4,5)P2-induced activation of TRPV1 channels was unaltered when a residue outside of the binding pocket (i.e. Arg-701) was changed to alanine. Correspondingly, the modeling showed little change in the bound configuration of PI(4,5)P2 for the R701A mutant (Fig. 3, G and L). In quantitative terms, the minimum binding energy score predicted by the docking software (ICM) was substantially lower for the wild-type than for any of the mutants, with the exception of the R701A mutant, which exhibited a very similar score to that of the wild-type system.
In summary, the fifty lowest energy configurations produced by docking for each TRPV1 system (Fig. 3, I–L) reveal a single strong binding mode for the wild-type and negative control mutant, but many distinct, weakly bound conformations for the other mutants. The single strong binding mode seen in the simulations is associated with PI(4,5)P2-induced activation of TRPV1 at low PI(4,5)P2 concentrations in electrophysiological experiments, while the weak binding corresponded to measured PI(4,5)P2-induced TRPV1 activation only at very high PI(4,5)P2 concentrations.
TRPV1-PI(4,5)P2 Complex Stability
To assess the stability of the configuration established by docking and further probe the interactions between PI(4,5)P2 and the protein, a 300 ns MD simulation was performed and started from the configuration shown in Fig. 2D. Fig. 4A shows the RMSD of the atomic positions as a function of time with respect to the starting configuration for each PI(4,5)P2 molecule at the four independent binding sites. These RMSDs generally showed that the molecular conformation slightly diverged in the first few nanoseconds for each PI(4,5)P2 molecule. This result was expected, due to the approximate nature of the binding energy estimated by the docking software and the fact that the protein was fixed during docking. After about 50 ns, the RMSD saturated at a relatively small value, while maintaining the qualitative features of the optimal configuration as per the docking algorithm. Fig. 4B shows that the distances to the Arg-575, Arg-579, and Lys-694 residues remained stable after 50 ns of MD simulation, suggesting that the TRPV1-PI(4,5)P2 complex was in conformational equilibrium. In particular, the phosphate group at position four of the PI(4,5)P2 polar headgroup formed a salt bridge with the Arg-575 (∼4.2 Å) and Arg-579 (∼4.6 Å) NH3+ groups (Fig. 2C). Additionally, the phosphate group at position five of the PI(4,5)P2 polar headgroup established another salt bridge with residue Lys-694 (∼3.8 Å). Therefore, the key interactions predicted by docking for residues Arg-575, Arg-579, and Lys-694 were maintained, and the side chains of these residues adapted their orientations, which were fixed during docking, to actually increase the strength of the electrostatic interactions.
The HOLE algorithm was used to determine whether changes in the internal radius of the TRPV1 channel pore caused by PI(4,5)P2 binding, could be observed during in MD simulation. Beginning with the experimentally determined APO-TRPV1 structure embedded in a membrane, we added PI(4,5)P2, and performed 300 ns of MD simulation. Then CAP was added to the system and the MD was extended 200 ns to stabilize the complex APO-TRPV1-PI(4,5)P2-CAP. As shown in Fig. 4C, the results showed that the pore diameter in the area named lower-gate by Cao et al. (9) gradually increased at the end of 530 ns of simulation, after apply a restrain on Ile-679 to accelerate the gating process (Fig. 4C). The diameter of the lower-gate obtained has a greater values than those observed for the experimentally determined CAP-TRPV1 open structure. This increased radius allows us to argue that PI(4,5)P2 directly leads to increased channel conductance, attributable to the displacement of the S6 transmembrane that “breaks” the hydrophobic seal formed by residues Thr-685 to Ile-696 (Fig. 4C).
To understand how the charge distribution of the amino acids surrounding the binding site may modulate interactions between PI(4,5)P2 and the wild-type TRPV1 channel, the three-dimensional electrostatic potential of the equilibrated complex was calculated and averaged over the trajectory. The resulting electrostatic potential was projected over the surface of the APO-TRPV1 channel (Fig. 4D). The residues surrounding the PI(4,5)P2 binding pocket created an overall positive potential in their vicinity. The residues that predominantly contributed to the electrostatic potential of the binding site were Gln-561, Lys-571, Arg-575, Arg-579, Lys-688, and Lys-694. Additionally, it was possible identify a hydrophobic area near the PI(4,5)P2 hydrocarbon chains, which may have also contributed to the stabilization of the TRPV1-PI(4,5)P2 complex.
Fig. 4E shows the PI(4,5)P2 position proposed here (red) and the position for CAP (cyan) proposed by Cao et al. (9). These two models suggest that simultaneous binding of PI(4,5)P2 and CAP is possible, with the two compounds separated by 4.3 Å at their closest contact. Fig. 4, F and G show a comparison between the PI(4,5)P2 docking calculations into the APO-TRPV1 and CAP-TRPV1 experimental structure, respectively. These conformations showed that the CAP-TRPV1 experimental structure (Fig. 4F) favored PI(4,5)P2 binding more than the APO-TRPV1 experimental structure (Fig. 4G). In the former case, the entrance of PI(4,5)P2 into the binding pocket was facilitated. The CAP-TRPV1 structure improved PI(4,5)P2 affinity energy by an ICM score of −2.7, presenting a distance of 4 Å between the phosphate at position 5 of the PI(4,5)P2 headgroup and residue Lys-694, as well as a distance of 6 Å in relation to residues Arg-575, Arg-579, and Gln-561, while a distance of 8 Å was found with respect to the Lys-688 residue. These results indicate that PI(4,5)P2 binds with greater affinity to the TRPV1 channel after the channel binds CAP.
The results obtained by docking simulations were used as a starting point for MD simulations. The TRPV1-PI(4,5)P2 configuration obtained by docking represents a hypothetical interaction before the conformational perturbation generated by PI(4,5)P2 because the protein was kept fixed during docking. In MD simulations, we observed a fast decrease in TRPV1-PI(4,5)P2 pair interaction energy (data not shown), due to relaxation of PI(4,5)P2 and the residues Arg-575, Arg-579, and Lys-694. Lys-694 was the amino acid found to be altered the most from its initial conformation, which could be related to the bending mechanism of the segment S6-TRP domain (see Discussion). The strong interaction between phosphates of PI(4,5)P2 and residues Arg-575, Arg-579, and Lys-694 is observed over 60% of time in APOTRPV1: PI(4,5)P2 (Fig. 5A) and RTX/DkTx:TRPV1:PI(4,5)P2 (Fig. 5B) simulations. The interaction between the residues Arg-575, Arg-579, and Lys-694 and PI(4,5)P2 is clearly seen, even if the initial conformation of TRPV1 in molecular dynamics simulation starts from “closed state” (APO TRPV1) (Fig. 5A) or “open state” (RTX-DkTx TRPV1) (Fig. 5B). The distances between PI(4,5)P2 and Arg-575, Arg-579, and Lys-694 residues located in the different subunits are less than 3 Å during most of the molecular dynamics simulation time (Fig. 5, A and B). Arg-575 shows the largest interaction time in both molecular dynamics simulations, reaching the 100% of time in contact with PI(4,5)P2. In agreement with the electrophysiological results (Fig. 3), the residue R701 there in no interaction with PI(4,5)P2 along the simulation. On the other hand, the absence (Fig. 5A) and presence (Fig. 5B) of Capsaicin does not affect the high percentage of interaction of phosphates groups of PI(4,5)P2 and Arg-575, Arg-579, and Lys-694 amino acids.
FIGURE 5.
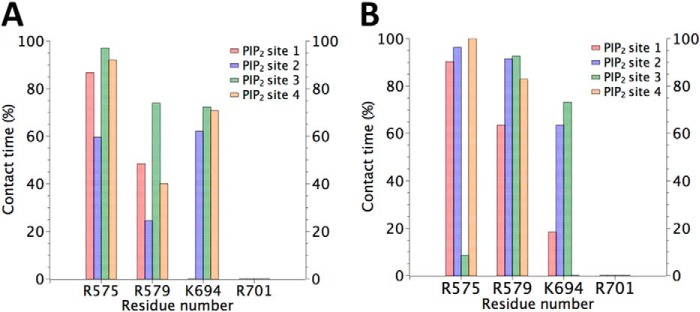
Statistical analysis of the residues in contact with the phosphates groups of PI(4,5)P2. The contact time is represented as the percentage of the time of the full molecular dynamics simulation in which the amino acid residue indicated in the abscissa interacts with the phosphate groups of PI(4,5)P2. A distance between the amino acid residue and PI(4,5)P2 less than 3 Å is considered as a contact. Four PI(4,5)P2 molecules were located in the four identical binding pockets designed Site 1, 2, 3, and 4. Panel A shows the data from simulation APO-TRPV1+ PI(4,5)P2 and B is the result from RTX-DkTx TRPV1 PI(4,5)P2+CAP trajectory. The negatively charged headgroup of PI(4,5)P2 shows a high frequency of interaction with the residues Arg-575, Arg-579, and Lys-694. Residue Arg-701 has negligible contact with PI(4,5)P2.
DISCUSSION
How Does PI(4,5)P2 Regulate TRP Channels?
Most TRP channels are modulated by PI(4,5)P2 (30, 31). However the detailed molecular mechanisms by means of which this lipid regulates channel activity are still unknown. All we can say a priori with a relatively high degree of certainty is that electrostatics plays a fundamental role in PI(4,5)P2 binding to proteins and all that is needed, in principle, is a cluster of basic residues that can serve as a site of attraction for multivalent negatively charged phosphoinositides (32, 33). In fact, basic residues in the TRP domain in the TRPM8 proximal region of the C terminus, as well as TRPV5 and TRPM5 are involved in protein-lipid interaction (34). PI(4,5)P2 also activates TRPM4 (35, 36). In the latter case, however, the basic residues involved in the interaction with PI(4,5)P2 were suggested to reside in a pleckstrin homology domain (i.e. [KR]-X(3–11)- [KR]-X-[KR]- [KR]) located toward the distal end of the C terminus (35). PI(4,5)P2 also potentiates TRPV4 responses to heat and hypotonic stimuli. However, the neutralization of basic residues located in a sequence, resembling a phosphoinositide-binding site in the N terminus, is that which renders the channel unresponsive to hypotonicity and heat (37).
The TRPV1- PI(4,5)P2 Conundrum
PI(4,5)P2 was initially proposed to have a TRPV1 inhibitory role and a putative PI(4,5)P2 binding site in the TRPV1 distal part of the C terminus was identified (38, 39). However, several reports have questioned the inhibitory effect of PI(4,5)P2 on TRPV1. First of all, lack of recovery from desensitization due to the inhibition of phosphatidylinositol-4-kinase was observed. More importantly, truncation of the distal C-terminal region containing the alleged PI(4,5)P2 binding site does not affect recovery from desensitization as compared with the wild-type (40). Second, the direct application of PI(4,5)P2 to the internal side of a patch expressing TRPV1 channels has been reported to lead to channel activation, and the sequestration of PI(4,5)P2 with polysine has been seen to inhibit channel activation (14, 41, 42). Finally, Ufret-Vincenty et al. (13) showed that PI(4,5)P2 is able to directly activate TRPV1 channels, even after the removal of the distal C terminus, which suggests that the proximal C terminus would be involved in PI(4,5)P2 binding.
Although the experiments performed in excised patches strongly support the role of PI(4,5)P2 as a TRPV1 channel activator, the debate still continues. Lukacs et al. (42) showed that PI(4,5)P2 may behave as an activator at high CAP concentrations, but as an inhibitor at low concentrations of the agonist. More recently, Cao et al. (15) were able to reconstitute the TRPV1 channel into liposomes and showed that if the liposomes were formed by a lipid mixture containing 4% PI(4,5)P2, TRPV1 activity would be inhibited compared with liposomes lacking the phosphoinositide. These reconstitution experiments have been questioned on the basis of a promiscuous activation of TRPV1 by negatively charged lipids (12) and an inhibitory effect of PI(4,5)P2 when present at high concentrations in the external lipid bilayer leaflet (43). It was further found that when TRPV1 was reconstituted into bilayers containing only neutral lipids, channel activation mediated by capsaicin depended on the presence of PI(4,5)P2 (12).
Inferences about the Structural Basis of PI(4,5)P2 Activation in the TRPV1 Channel
Binding to the C terminus is particularly interesting, due to its vicinity to the internal gate, as proposed by Cao et al. (9). However, the structural nature of the PI(4,5)P2 binding site(s) and the mechanisms involved in the modulation of TRP channel gating mediated by the lipid are at present unknown. It is important to note here that MacKinnon's group (44) reported the crystal structure of the inward rectifier, Kir2.2, in complex with a PI(4,5)P2 analog. In this case, PI(4,5)P2 binds at the interface between the transmembrane domain and the cytoplasmic domain and a comparison between the Kir2.2 (APO) crystal structure and the bound PI(4,5)P2 shows that phosphoinositide binding promotes a large (6 Å) displacement of the cytoplasmic domain, becoming joined to the transmembrane domain. We have compared the PI(4,5)P2 binding site described here to the crystallographic structure of the Kir2.2 channel in complex with PI(4,5)P2 molecules. Although the Kir2.2 channel is structurally distinct from TRPV1, it is similar to TRPV1 in that its crystallographic structure exhibits four PI(4,5)P2 binding sites located at the interface between the transmembrane domain (TMD) and the cytoplasmic domain (CTD), lined with positive residues (mainly Arg and Lys) in each of the four subunits (44). Both structures (i.e. TRPV1-PI(4,5)P2 and Kir2.2-PI(4,5)P2) have been superimposed with respect to the ionic pore. We found that both binding sites are lined with basic residues and are located in the headgroup region of the intracellular bilayer leaflet. Indeed, the PI(4,5)P2 headgroup's center of mass along the pore axis in the Kir2.2 experimental structure (44) differed by only 0.52 Å from the structure produced by docking PI(4,5)P2 into TRPV1.
In the TRPV1-binding pocket, phosphate groups of PI(4,5)P2 make contact with basic residues Arg-579 and Arg-575 from one subunit and Lys-694 from an adjacent subunit (Fig. 2D). Contrary to previous results, we show here that only Lys-694 in the TRP domain is part of the PI(4,5)P2 binding site and that Arg-701 plays no role in binding, but it may help in stabilizing the binding pocket once it is formed. It is interesting to note here that in KCNQ (Kv7.2) channels, as in TRPV1, PIP2 interacts with positive charges contained in the S4-S5 linker only when channel are open but with the S2-S3 linker in the closed state (45). In Kv channel the S4-S5 linker interacting with S6 (46) plays an important role in channel gating (47).
Fig. 6 shows the main difference between the internal TRPV1 gate (9, 10) in the closed and open configurations, namely the appearance (in the open configuration) of a pronounced bending of the α-helical constituting the S6 transmembrane domain and the TRP domain (Fig. 6A). This structure presents a 30.5° curvature at the level of residues Thr-685 to Ile-696 (max. bending at I689) when the internal gate is closed (APO, PDB: 3J5P; Fig. 6, B and E). The bending increases to 50.1° at the level of residues Asn-687 to Trp-697 (max. bending S693) when the upper and lower gates are open (RTX/DkTx, PDB: 3J5Q, Fig. 6, D and E). In the intermediate state of gating, and only when the lower gate is open (CAP, PDB: 3J5R), two maximum bending degrees in angles in residues Thr-685 (35.8°) and Ser-693 (25.3°) were observed (48) (Fig. 6, C and E). We performed a 300-ns molecular dynamics simulation, starting with the closed state of APO-TRPV1 in the presence of PI(4,5)P2. We found that after 140 ns of simulation, two of the α-helical attained curvatures very similar to what we found in the open state of RTX-TRPV1 (cf., Fig. 6D and Fig. 7, A and B). Interestingly, when CAP was added to this simulation, we found three bent chains after only 66 ns, after which the structure remained stable for the remaining 200 ns of molecular dynamic simulation (Fig. 7, C and D). At 133 ns of this simulation (APO-TRPV1+PI(4,5)P2 +CAP), the four subunits reached curvatures higher than 45° degrees, two of which approached 60° (S693) and the other two were close to 45° and showed a shoulder greater than 21° (Ile-689) (Fig. 6D). To compare this conformation with the experimental structure of the TRPV1 open state, we performed a simulation by using the TRPV1 structure with the upper and lower gate open (RTX/DkTx) in the presence of PI(4,5)P2 and CAP, both fitted to this structure by means of docking simulations. After 40 ns of simulation, the curvature of the four chains reached over 50° (Fig. 7, E and F), similar to the conformation reached by the APO-TRPV1+PI(4,5)P2+CAP) system.
FIGURE 6.

Internal gate curvature of cryo-EM structures A, curvature analysis of residues 656 to 712, in the S6-TRP domain, where the S6 segment is shown in purple and the TRP domain in cyan, as defined by Liao et al. (10). B, bottom view of the S6-TRP domain helix of the APO-TRPV1 structure (PDBid: 3J5P), (C) CAP-TRPV1 (PDBid: 3J5R) and (D) RTX-DkTx TRPV1 structures (PDBid: 3J5Q). Curvatures over 30° are highlighted in green and over 50° are shown in red. E, measurements of the bending angle in the S6-TRP domain of the cryo-microscopy structures were calculated using the VMD Bendix package (48).
FIGURE 7.

Bending angle analysis of the fragment S6-TRP domain (residue 656 to 712) after molecular dynamics simulations. A, simplified bottom view of the four subunits of the experimental APO-TRPV1structure after the addition of PI(4,5)P2 and 300 ns of subsequent molecular dynamics simulation in a phospholipid membrane. B, bending angles shown for each residue of the segment S6-TRP domain. Two of the four subunits reached the bending angle expected for the open state of TRPV1 (about 50°). C and D show the experimental APO-TRPV1structure after the addition of PI(4,5)P2 and CAP and a subsequent molecular dynamics simulation of 200 ns. At the end of this simulation, the four subunits reached the bending angle for the TRPV1open state. Finally, E and D show the final conformation obtained for the experimental RTX-DkTx TRPV1 structure after the addition of PI(4,5)P2 and CAP and 40 ns of subsequent molecular dynamics simulation. They reached a steady conformation after just 5 ns and maintained the open conformations during the entirety of the simulation.
Our experiments show differences in TRPV1 activation depending on whether PI(4,5)P2 is added before or after CAP (Fig. 1E). We found that the current induced by PI(4,5)P2 when CAP was added after PI(4,5)P2 was always smaller than the TRPV1-induced currents when CAP was added prior to PI(4,5)P2. In that context, when simulating the PI(4,5)P2 complexed to the experimentally determined APO-TRPV1 structure, we observed that one of the PI(4,5)P2 aliphatic chains strongly interacted with the CAP-binding pocket (Fig. 8, A–D). The presence of bound PI(4,5)P2 may therefore partially obstruct the CAP-binding pocket, slowing the kinetics of CAP binding and possibly explaining the difference in TRPV1 activation when PI(4,5)P2 is added before or after CAP.
FIGURE 8.

Bound PI(4,5)P2 can partially occlude the nearby CAP binding site. A, snapshot from an MD simulation in which PI(4,5)P2 is bound to the binding site determined in the present work. Here, in the absence of CAP, an aliphatic chain of PI(4,5)P2 occupies the hypothesized CAP binding site. Atoms of PI(4,5)P2 are shown as spheres, while residues of TRPV1 that may be in contact with both bound PI(4,5)P2 and bound CAP are shown by a stick model and highlighted by a yellow surface. B, percentage of time in which atoms of PI(4,5)P2 make contact with specific residues of the CAP binding site in the absence of CAP. Occupancy of the CAP binding site by the aliphatic chain of PI(4,5)P2 is only partial, and it frequently exits and reenters this site. This suggests that bound PI(4,5)P2 may partially, but not completely, block the access of CAP to its binding site. C, snapshot from an MD simulation where both CAP and PI(4,5)P2 occupy their respective binding sites. Atoms of CAP are shown as spheres, while PI(4,5)P2 is represented by thin lines. D, percentage of time in which atoms of CAP make contact with specific residues of its binding site while PI(4,5)P2 is bound nearby. CAP stably occupies the hypothesized binding site. E, another snapshot from the same simulation represented in panel C, highlighting contact between PI(4,5)P2 and CAP. Atoms of PI(4,5)P2 are shown as spheres, while CAP is represented by thin orange tubes. F, percentage of time in which atoms of PI(4,5)P2 make contact with specific residues of the CAP binding site while occupied by CAP. The presence of CAP substantially reduces the occupation of the site by the PI(4,5)P2 aliphatic chain, which can be clearly seen by comparing to panel B.
The TRPV1 structure shows that residue Ile-679 forms a hydrophobic seal (lower gate) that expands in the presence of the agonist resineferatoxin and the scorpion toxin double-knot (DkTx) to a diameter of 9.3 Å (9, 10). Our MD simulations showed that the interaction between PI(4,5)P2 and basic residues beneath the gate promotes the necessary structural changes to open it. The increase in curvature at the junction between the S6 and the TRP domain (the helical “elbow”) (9) promotes a displacement of S6 that “breaks” the hydrophobic seal formed by Ile-679 (Fig. 4, A–C). To understand the intermediate states that lead to the expansion of the lower gate, we performed an extension of our molecular dynamics simulation (30 ns) using soft restraints that move the Cα carbon atom of Ile-679 from the close to the open conformation. During this process we observed that the radius of the pore increase from 0.8 Å (APO, blue line) to 2.2 Å (black line, APO-TRPV1 +PIP2 +CAP) (Fig. 4C). During the first 10 ns the side chains of Ile-679 remain in contact. However, at the end of this period the Ile-679 side chains begin to collapse over the surface of the pore allowing the passage of water molecules through the hydrophobic seal (cf. Fig. 9, A and B).
FIGURE 9.
CAP and PI(4,5)P2 expand theTRPV1 pore radius at the internal gate level. A, structure of the hydrophobic seal formed by I679 observed in the MD of the complex APO-TRPV1+ PI(4,5)P2+CAP after 200 ns of MD simulation (before gate opening). C, at 40 ns of MD simulation after the addition of PI(4,5)P2 and CAP the hydrophobic seal brakes (the pore increases in radius) allowing water to permeate.
This work was supported by the Centro Interdisciplinario de Neurociencia de Valparaíso (CINV), a Millennium Institute supported by the Millennium Scientific Initiative of the Ministerio de Economía, Fomento y Turismo. This work was also supported by the Doctoral Program of Applied Sciences at Talca University, as well as CONICYT-Chile for a doctoral fellowship and Project FONDECYT 3140288 (to H. P.), the Project FONDECYT 1110430 (to R. L.) and 1131003 (to F. G. N.), and Anillo Cientifico ACT-1107 (to F. G. N.) and Anillo ACT-1104 (to C. G. L.), Grant RI-130006 (to R. L. and C. G.) and Project FONDECYT 11100047 (to P. O.).
- TRPV
- transient receptor potential vanilloid
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- CAP
- capsaicin
- MD
- molecular dynamics
- PME
- particle-mesh Ewald.
REFERENCES
- 1. Caterina M. J., Schumacher M. A., Tominaga M., Rosen T. A., Levine J. D., Julius D. (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824 [DOI] [PubMed] [Google Scholar]
- 2. Hayes P., Meadows H. J., Gunthorpe M. J., Harries M. H., Duckworth D. M., Cairns W., Harrison D. C., Clarke C. E., Ellington K., Prinjha R. K., Barton A. J., Medhurst A. D., Smith G. D., Topp S., Murdock P., Sanger G. J., Terrett J., Jenkins O., Benham C. D., Randall A. D., Gloger I. S., Davis J. B. (2000) Cloning and functional expression of a human orthologue of rat vanilloid receptor-1. Pain 88, 205–215 [DOI] [PubMed] [Google Scholar]
- 3. Caterina M. J., Leffler A., Malmberg A. B., Martin W. J., Trafton J., Petersen-Zeitz K. R., Koltzenburg M., Basbaum A. I., Julius D. (2000) Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288, 306–313 [DOI] [PubMed] [Google Scholar]
- 4. Davis J. B., Gray J., Gunthorpe M. J., Hatcher J. P., Davey P. T., Overend P., Harries M. H., Latcham J., Clapham C., Atkinson K., Hughes S. A., Rance K., Grau E., Harper A. J., Pugh P. L., Rogers D. C., Bingham S., Randall A., Sheardown S. A. (2000) Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405, 183–187 [DOI] [PubMed] [Google Scholar]
- 5. Moriyama T., Iida T., Kobayashi K., Higashi T., Fukuoka T., Tsumura H., Leon C., Suzuki N., Inoue K., Gachet C., Noguchi K., Tominaga M. (2003) Possible involvement of P2Y2 metabotropic receptors in ATP-induced transient receptor potential vanilloid receptor 1-mediated thermal hypersensitivity. J. Neurosci. 23, 6058–6062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baez-Nieto D., Castillo J. P., Dragicevic C., Alvarez O., Latorre R. (2011) Thermo-TRP channels: biophysics of polymodal receptors. Adv. Exp. Med. Biol. 704, 469–490 [DOI] [PubMed] [Google Scholar]
- 7. Gaudet R. (2008) TRP channels entering the structural era. J. Physiol. 586, 3565–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bohlen C. J., Priel A., Zhou S., King D., Siemens J., Julius D. (2010) A bivalent tarantula toxin activates the capsaicin receptor, TRPV1, by targeting the outer pore domain. Cell 141, 834–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cao E., Liao M., Cheng Y., Julius D. (2013) TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liao M., Cao E., Julius D., Cheng Y. (2013) Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brauchi S., Orta G., Mascayano C., Salazar M., Raddatz N., Urbina H., Rosenmann E., Gonzalez-Nilo F., Latorre R. (2007) Dissection of the components for PIP2 activation and thermosensation in TRP channels. Proc. Natl. Acad. Sci. U.S.A. 104, 10246–10251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lukacs V., Rives J.-M., Sun X., Zakharian E., Rohacs T. (2013) Promiscuous activation of transient receptor potential vanilloid 1 (TRPV1) channels by negatively charged intracellular lipids: the key role of endogenous phosphoinositides in maintaining channel activity. J. Biol. Chem. 288, 35003–35013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ufret-Vincenty C. A., Klein R. M., Hua L., Angueyra J., Gordon S. E. (2011) Localization of the PIP2 sensor of TRPV1 ion channels. J. Biol. Chem. 286, 9688–9698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stein A. T., Ufret-Vincenty C. A., Hua L., Santana L. F., Gordon S. E. (2006) Phosphoinositide 3-kinase binds to TRPV1 and mediates NGF-stimulated TRPV1 trafficking to the plasma membrane. J. Gen. Physiol. 128, 509–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cao E., Cordero-Morales J. F., Liu B., Qin F., Julius D. (2013) TRPV1 channels are intrinsically heat sensitive and negatively regulated by phosphoinositide lipids. Neuron 77, 667–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Simon F., Leiva-Salcedo E., Armisén R., Riveros A., Cerda O., Varela D., Eguiguren A. L., Olivero P., Stutzin A. (2010) Hydrogen peroxide removes TRPM4 current desensitization conferring increased vulnerability to necrotic cell death. J. Biol. Chem. 285, 37150–37158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–8 [DOI] [PubMed] [Google Scholar]
- 18. Abagyan R., Totrov M., Kuznetsov D. (1994) ICM?A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 15, 488–506 [Google Scholar]
- 19. Abagyan R., Totrov M. (1994) Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235, 983–1002 [DOI] [PubMed] [Google Scholar]
- 20. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 22. Feller S. E., Zhang Y., Pastor R. W., Brooks B. R. (1995) Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 103, 4613 [Google Scholar]
- 23. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577 [Google Scholar]
- 24. Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., Mackerell A. D. (2010) CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hénin J., Shinoda W., Klein M. L. (2008) United-atom acyl chains for CHARMM phospholipids. J. Phys. Chem. B 112, 7008–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D., Jr., Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926 [Google Scholar]
- 28. Brauchi S., Orio P., Latorre R. (2004) Clues to understanding cold sensation: thermodynamics and electrophysiological analysis of the cold receptor TRPM8. Proc. Natl. Acad. Sci. U.S.A. 101, 15494–15499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matta J. A., Ahern G. P. (2007) Voltage is a partial activator of rat thermosensitive TRP channels. J. Physiol. 585, 469–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gamper N., Rohacs T. (2012) Phosphoinositide sensitivity of ion channels, a functional perspective. Subcell. Biochem. 59, 289–333 [DOI] [PubMed] [Google Scholar]
- 31. Nilius B., Owsianik G., Voets T. (2008) Transient receptor potential channels meet phosphoinositides. EMBO J. 27, 2809–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McLaughlin S. (2006) Cell biology. Tools to tamper with phosphoinositides. Science 314, 1402–1403 [DOI] [PubMed] [Google Scholar]
- 33. McLaughlin S., Murray D. (2005) Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438, 605–611 [DOI] [PubMed] [Google Scholar]
- 34. Rohács T., Lopes C. M. B., Michailidis I., Logothetis D. E. (2005) PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 8, 626–634 [DOI] [PubMed] [Google Scholar]
- 35. Nilius B., Mahieu F., Prenen J., Janssens A., Owsianik G., Vennekens R., Voets T. (2006) The Ca2+-activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5-biphosphate. EMBO J. 25, 467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Z., Okawa H., Wang Y., Liman E. R. (2005) Phosphatidylinositol 4,5-bisphosphate rescues TRPM4 channels from desensitization. J. Biol. Chem. 280, 39185–39192 [DOI] [PubMed] [Google Scholar]
- 37. Garcia-Elias A., Mrkonjic S., Pardo-Pastor C., Inada H., Hellmich U. A., Rubio-Moscardó F., Plata C., Gaudet R., Vicente R., Valverde M. A. (2013) Phosphatidylinositol-4,5-biphosphate-dependent rearrangement of TRPV4 cytosolic tails enables channel activation by physiological stimuli. Proc. Natl. Acad. Sci. U.S.A. 110, 9553–9558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chuang H. H., Prescott E. D., Kong H., Shields S., Jordt S. E., Basbaum A. I., Chao M. V., Julius D. (2001) Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411, 957–962 [DOI] [PubMed] [Google Scholar]
- 39. Prescott E. D., Julius D. (2003) A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science 300, 1284–1288 [DOI] [PubMed] [Google Scholar]
- 40. Liu B., Zhang C., Qin F. (2005) Functional recovery from desensitization of vanilloid receptor TRPV1 requires resynthesis of phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 25, 4835–4843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Klein R. M., Ufret-Vincenty C. A., Hua L., Gordon S. E. (2008) Determinants of molecular specificity in phosphoinositide regulation. Phosphatidylinositol (4,5)-bisphosphate (PI(4,5)P2) is the endogenous lipid regulating TRPV1. J. Biol. Chem. 283, 26208–26216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lukacs V., Thyagarajan B., Varnai P., Balla A., Balla T., Rohacs T. (2007) Dual regulation of TRPV1 by phosphoinositides. J. Neurosci. 27, 7070–7080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Senning E. N., Collins M. D., Stratiievska A., Ufret-Vincenty C. A., Gordon S. E. (2014) Regulation of TRPV1 ion channel by phosphoinositide (4,5)-bisphosphate: the role of membrane asymmetry. J. Biol. Chem. 289, 10999–11006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hansen S. B., Tao X., MacKinnon R. (2011) Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477, 495–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zaydman M. A., Cui J. (2014) PIP2 regulation of KCNQ channels: biophysical and molecular mechanisms for lipid modulation of voltage-dependent gating. Front. Physiol. 5, 195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Long S. B., Campbell E. B., Mackinnon R. (2005) Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science 309, 903–908 [DOI] [PubMed] [Google Scholar]
- 47. Zhang Q., Zhou P., Chen Z., Li M., Jiang H., Gao Z., Yang H. (2013) Dynamic PIP2 interactions with voltage sensor elements contribute to KCNQ2 channel gating. Proc. Natl. Acad. Sci. U.S.A. 110, 20093–20098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dahl A. C. E., Chavent M., Sansom M. S. P. (2012) Bendix: intuitive helix geometry analysis and abstraction. Bioinformatics 28, 2193–2194 [DOI] [PMC free article] [PubMed] [Google Scholar]